
Abstract
Two non-inbred mouse lines, phenotypically selected for maximal (AIRmin) and minimal (AIRmax) acute inflammatory response, show differential susceptibility/resistance to the development of several chemically-induced tumor types. An intercross pedigree of these mice was generated and treated with the chemical carcinogen dimethylhydrazine, which induces lung and intestinal tumors. Genome wide high-density genotyping with the Restriction Site-Associated DNA genotyping (2B-RAD) technique was used to map genetic loci modulating individual genetic susceptibility to both lung and intestinal cancer. Our results evidence new common quantitative trait loci (QTL) for those phenotypes and provide an improved understanding of the relationship between genomic variation and individual genetic predisposition to tumorigenesis in different organs.
Similar content being viewed by others

Introduction
This study aimed to map chromosomal regions involved in the regulation of susceptibility/resistance of mice in developing colon and lung tumors, induced by treatment with chemical carcinogen. AIRmax and AIRmin mouse strains, produced by bidirectional phenotypic selection on the basis of a high (AIRmax) or low (AIRmin) acute inflammatory response (AIR) were used as experimental model [1]. The selection process to obtain the two lines started from a population consisting of the balanced crossing of 8 inbred strains. The mixture of strains of different origins ensured a wide genetic variability in this population, thus bringing the model closer to the heterogeneity found in human populations. In addition, the progenitor strains have divergent sensitivities to colon and lung tumorigenesis reviewed in [2, 3] and thus genetic components related to organ-specific carcinogenesis were present in the background of the foundation population. Dimethylhydrazine (DMH) and its metabolite Azoximethane (AOM) are pro-carcinogens with tropism to colon and induce the appearance of tumors molecularly similar to non-familial colon cancers in humans. Previous mapping studies using several inbred susceptible and resistant mouse lines and crosses, as well as recombinant congenic strains (CcS/Dem), identified about 20 regions distributed across the chromosomes containing candidate modifier genes of colon carcinogenesis. These QTL are named Scc (susceptibility to colon cancer) and their large number is evidence of the polygenic nature of susceptibility/resistance to this type of cancer [4, 5]. Numerous mapping studies in humans have demonstrated the existence of at least 20 genomic regions and candidate genes in non-family colon cancer [6]. AIRmax and AIRmin lines are widely divergent in susceptibility to the development of tumors induced by chemical carcinogenesis regardless of the carcinogen employed and the affected organs [7,8,9,10,11,12]. AIRmin are resistant and AIRmax mice are very sensitive in developing colon tumors caused by DMH while under the same treatment, multiple lung tumors appeared only in the AIRmin mice [11]. There are no reports of induction of lung tumors by DMH in laboratory mice and it should be noted that even in the most sensitive strains to develop colon tumors, no invasive tumors and metastasis appear. The susceptibility of AIRmax and AIRmin mice to develop tumors in the colon and lungs after treatment with the same carcinogen (DMH) makes the model suitable for the research of common genes involved in their regulation. For this we produced, an (AIRmax x AIRmin) F2 population and treated all F2 animals with (DMH). At the end of the experiment we recorded the incidence, number, and size of colon and lung tumors. High-density genotyping was carried out for all individuals with the 2B- RAD technique (Restriction Site-Associated DNA genotyping) [13,14,15], to map the genetic loci that modulate individual susceptibility to colon and lung cancer.
Results
Colon and lung carcinogenesis
The phenotypic characterization was performed in the pedigree consisting of AIRmax, AIRmin grandparents (n = 22), (AIRmax x AIRmin)F1 parents (n = 20), and (AIRmax x AIRmin)F2 (n = 180) animals to evaluate the association between the transmitted alleles and the phenotypes presented by the individuals. The onset of colon and lung tumors in AIRmax, AIRmin and (AIRmax x AIRmin)F1 (Fig. 1) showed a clear inverse susceptibility between the two strains, confirming previous results [11]. Resistance to colon carcinogenesis is dominant in (AIRmax x AIRmin)F1 animals, similar to what occurs in crosses between inbred strains. On the other hand, the appearance of lung tumors was dominant in this group, in disagreement with the conditions of codominance found among inbred strains [16].

AIRmax, AIRmin, and AIRmax × AIRmin F1 hybrids were injected ip with DMH (185 mg/kg. Colon (A) and lung (B) tumors were analyzed at different times after treatment. N2_colon sqrt = number of colon tumors with diameter >2 mm (square-root transformed values). N2_lung sqrt = number of lung tumors with diameter >2 mm (square root transformed values). Mean values and SD of 8–10 animals per group.
Table 1 shows the tumor incidence in the (AIRmax x AIRmin)F2 population at 300 days after treatment with the carcinogen. The frequencies of mice bearing colon or lung tumors were similar (28 and 25%, respectively), whereas about 18% of mice had both colon and lung tumors.
Figures 2–4 shows the macroscopic aspect and microscopic features of DMH-induced colon and lung tumors from (AIRmax x AIRmin)F2 mice at 280 days after treatment.

a spherical mass (arrow) growing at the apex and between the right (arrowhead) and left lung lobes. b Pediculate (dotted arrow) and sessile polyps distributed in the colon-rectal epithelium.

A and B Shows, at lower magnification, multiple in situ lesions arising in the large airways and some small invasive carcinomas characterizing the bronchial-alveolar progression of lung tumors. C Greater resolution of tumor cellularity showing large, pleomorphic cells with abundant eosinophilic cytoplasm containing some vacuoles suggestive of small cell lung carcinoma (SCLC) combined with focal areas of non-small cell lung carcinoma (NSCLC) and poorly differentiated consisting mainly of cells with degenerative changes in the nuclei and cytoplasm (arrowhead) and in the periarteriolar area a modern plasmacytic leukocyte infiltrate. D shows tumor cells progressing to alveoli forming neoplastic mats and tumor nodule composed of cells arranged in clusters and nodular aggregates (asterisk) and an intra-alveolar neoplastic embolus (arrow). E, F mucin production (magenta-colored substance) in smaller and larger increases. H&E and PAS; Bar 20–100.

A, B Pediculated polyp in smaller and larger magnification showing well-differentiated carcinoma, with level 4 invasion, invading the submucosa of the intestinal wall, without compromising the muscle layer classified by the Haggitt Classification System. This example shows a complex growth pattern indicating high-grade dysplasia shown in detail in C (*) leukocyte infiltrate, (#) desmoplastic reaction, and (arrow) presence of neoplastic emboli in the small vessel lumen of the pedicle submucosa. D, E progression of the leukocyte and neoplastic infiltrate from the base to the apex of the villi. F shows in greater resolution the invasion of the submucosa by neoplastic and leukocyte cell mats with dysplasia and vacuolization with a deposit of amorphous substance at the apex of the villi. G, H increased mucin production (magenta colored substance) shown in polyp and surrounding villi H&E and PAS; Bar 20–100.
Histopathological diagnosis: The pulmonary tumor model (Fig. 2a) gave rise to central tumors resulting from invasive lesions originating in the large bronchi, although occasional tumors have arisen in the peripheral lung of the bronchioles airways or alveolar ducts confirmed by microscopy analysis. In the pulmonary histopathologic analysis (Fig. 3), pre-invasive lesions consisted of cells with similar morphology to the invasive component and were considered carcinoma in situ lesions. Tumors occupy up to 70% of the lung volume, with peribronchial and perivascular invasion. The histopathological diagnosis of colorectal polyp (Fig. 2b) shown and described in Fig. 4 is suggestive of colon-rectal carcinoma arising from a malignant adenomatous polyp. Although PAS staining appears, tumors cannot be considered mucinous because staining occurs in less than 50% of the tumor area.
Linkage analysis in (AIRmax x AIRmin) F2 mice
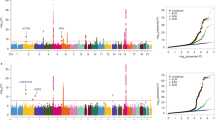
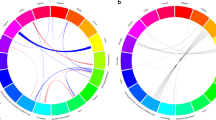
A total of 31913 SNPs were used for linkage mapping after quality control filtering. Parameters considered for tumorigenesis were the total number of tumors in each organ, the total number of tumors with mean diameter > 2 mm, and the total volume, by the sum of the volume of the tumors, calculated by the formula 4/3ΠR3. Thus, we have values to assess the incidence, multiplicity, and tumor development. Representative results are presented in Fig. 5. We observed clear signs of association with tumorigenesis phenotypes throughout the genome with several SNPs reaching statistical significance. In Fig. 6 we present the coordinates of regions containing SNPs showing the most significant association in some chromosomes. In addition to the specific regions for each phenotype, we observed coincident regions modulating colon and lung tumors, as shown in the distal portion of chromosomes 8, 17, and chromosome 18 (Table 2, Figs. 5 and 6).

Genomic wide association analysis of DMH-induced colon (A) and lung (B) tumor multiplicity in (AIRmax x AIRmin) F2 mice. Manhattan plot of observed P-values in −log scale for 31,913 SNPs. The horizontal line indicates the genome-wide thresholds of significant association for colon and lung tumor phenotypes.

The regions with higher significance of association and with similar positions for two phenotypes were highlighted considering an approximate 10 Mb interval.
Discussion
This study aimed to map genetic risk factors for lung and colon DMH-induced carcinogenesis. The AIRmax and AIRmin mouse lines are particularly powerful for this type of research since they differ widely in resistance/susceptibility to development of lung and colon tumors. The highly controlled conditions of the experimental design (breeding, treatment, animal facilities condition) minimize the influence of environmental variables, thereby reducing the confounding effects of gene-environmental interactions, a challenge for studies in human populations. High-density genotyping across the entire genome of the intercrossed F2 population of AIRmax/AIRmin animals using the 2B-RAD technique allowed for the high-resolution mapping of loci that modulate susceptibility to lung and intestinal cancer. DMH is a pro-carcinogen that is metabolized in the liver, producing reactive intermediates that are excreted through bile into the intestine or fall into the blood circulation; the ultimate metabolite methyl diazonium binds covalently to DNA, producing the pro-mutagenic O6 methyl guanine (O6Meg) lesion that induces GC to AC transitions in epithelial cells. These alterations induce activating mutations in oncogenes such as Kras, which are found in initial aberrant crypt foci lesions and in colon cancers, as well as in lung tumors in the mouse. Mutated Kras forms play a dominant role in driving metabolic reprogramming in several cancers [12, 17].
Tumor multiplicity in (AIRmax x AIRmin)F1 hybrids revealed dominance of resistance to DMH-induced colon cancer, and dominance of susceptibility to lung cancer. Nevertheless, in the (AIRmax x AIRmin)F2 population the incidence of colon or lung tumors was similar (28 and 25%). There was significant, albeit low, individual inverse correlation (r = −0.15 p = 0.045) in the F2 population between the incidence and progression of lung and colon tumors. Concordant susceptibility to both tumors was found in 18% of F2 mice, pointing to a link or identity in some genes controlling the development of both tumors. Histopathological analysis classified pulmonary tumors as carcinomas in situ and colon tumors as adenocarcinomas. Colon and lung derive from the foregut, the anterior part of the digestive tract; therefore some genes should regulate molecular pathways in both organs. However, lungs and digestive tract have different physiological functions and tissue-specific effects might play major influences in the group of genes acting in tumor onset and progression in each organ.
In the present genome screening, most SNPs that reached high statistical significance localize in non-coding regions of the genome, however, several significant SNPs are concentrated in specific regions of some chromosomes (Fig. 5). Nongenic cancer-risk SNPs are also usually found in human GWS studies [18, 19]. These genetic variants might affect the expression of nearby genes mapping to these regions, which might play a role in disease mechanisms in that tissue. Furthermore, we detected co-localization of chromosomal regions that control colon and lung tumor development. This may result from the presence of multiple closely linked genes, possibly controlling carcinogen processing or common carcinogenic pathways. Some of the several genes that physically map closely to the peak for association at these locus regions, highlighted in Fig. 6 and in Table 2, which are known to play roles in human cancers, are listed below.
For lung tumor multiplicity and progression phenotypes, the major association peaks were detected in chromosomes 1, 8, and 17. The region we mapped approximately at 50 Mb in chromosome 1, overlaps the region that spans the Scc20 QTL which harbors the transcription factors Stat1 and Stat4 involved in the transcription of cytokines and angiogenic factors in lung tumors [20]. These mediators might also contribute to inflammation and colon tumor development, as observed by the significant association of SNPs mapping in this region with colon tumors; the region in chromosome 8 at 54 Mb harbors the Vegf-C (vascular endothelial growth factor C) gene which is the main regulator of angiogenesis in the process of tumor growth and metastasis. Some variants of this gene in humans have been associated with the risk of developing lung tumors [21, 22]. The chemokines genes Ccl17 and Ccl22 are localized close to the highest peak of association at 98 Mb. Ccl17 and Ccl22 are important for cell recruitment in inflammatory processes, including cells that infiltrate tumors, and the genes coding for metallothioneins (MT), a group of low molecular weight cysteine-rich proteins involved in protection against DNA damage, oxidative stress, and apoptosis are also located in that region. Increased expression of these genes correlated with development of some types of tumors including those of colon and lung in humans [23]. Significant linkage with colon tumor development was observed at this same chromosomal location; in Chromosome 17, the region near 74 Mb harbors the Alk gene (anaplastic lymphoma kinase) that has been targeted for the therapy of some types of lung cancer [24] and the Xdh (xanthine dehydrogenase) gene. Interestingly xanthine oxidase (XO), the oxidized form of xanthine dehydrogenase, forms uric acid and ROS that activate the NLRP3 inflammasome complex for the release of IL-1β, a central mediator in inflammatory processes [25]. Another neighboring gene, Lpin2 (Lipin 2) is also involved in the activation of inflammasome by the purinergic receptor P2rX7 activation pathway [26]. Accordingly, we found significant associations of SNPs mapping in this interval with colon tumors.
The highest significant associations with colon tumors localized at chromosomes 3, 8, and 18. The distal portion of chromosome 3, between 131 to 146 Mbp colocalizes with Ccs3 (colon cancer susceptibility 3) QTL. This region harbors some candidate genes including Sgms2 (Sphingomyelin synthase 2S), which has a role in inflammation-mediated tumorigenesis, such as colon cancer originating from colitis [27]; Egf (epidermal growth factor) whose product is secreted by tumor cells of the colon and contributes to the M2 polarization of macrophages associated with the tumor [28]; NF-κB1 (nuclear factor-kappa B), that promotes cell proliferation, regulates immunological and inflammatory responses. Studies indicate that this gene is activated constitutively in malignant tumors and the action of some molecules with anti-tumor activity is attributed to the inactivation of this factor [29, 30]; a group of genes encoding alcohol dehydrogenases (Adh1, Adh5, Adh6a, Adh6b, Adh7) maps at 138 Mb. The activity of these enzymes which is dependent on polymorphisms, is implicated in the risk of colon cancer [31]. Mcc (mutated in colorectal cancers), localized at 44 Mb in chromosome 18 is involved in colon carcinogenesis. In colon cancer, the gene is silenced by methylation of the promoter, leading to a failure in repairing inflammation-induced DNA damage [32]. Decreased expression of this gene has been described in mouse lung tumors [33]. The region between 61 and 63 Mb contains Apcdd1 (Adenomatosis polyposis coli down-regulated 1), which is regulated by the beta-catenin/TCF complex and its high expression contributes to the colorectal tumorigenesis [34], as well as Il17b (interleukin 17B), involved in lung tumors and metastases [35, 36] and Csf1r (colony-stimulating factor 1 receptor) that has been considered as a target in cancer therapy [37]. Finally, the region around 80 Mb harbors the gene encoding the CD226 receptor that regulates NK cells function, which is important in tumor immunity [38, 39]. On chromosomes 17 and 18, significant association was detected at previously described loci that regulate colon and lung cancer in the mouse, i.e., the QTLs Susceptibility to lung cancer 32 (Sluc32) and Susceptibility to colon cancer 4 (Scc4) on chromosome 17 (78 Mb) and Sluc41 and Scc5 on chromosome 18 (20–57 Mb). Our results are in agreement with previous publications, which indicate a subset of genes that are functionally or genetically related presenting pleiotropic effects in different organs [40].
Several studies have been carried out in humans, to quantify the degree of shared genetic basis between different cancer types. Large scale cancer genetic epidemiological consortia such as the Genetic Associations and Mechanisms in Oncology (GAME ON) and Genetic Epidemiology of Colorectal Cancer Consortium (GECCO) have conducted cross-cancer genomic analysis based on numerous publicly available GWAs datasets. Genome-wide genetic correlations have been evaluated between pairs of cancer types and in one of these studies, a correlation of 0.31 (p = 0.001) was found between lung and colorectal cancers, involving genetic variations in inflammation-associated components [41,42,43,44,45].
As a general summary, the present study suggests the participation of genes encoding transcription and angiogenic factors, DNA damage protectors, genes that participate in the activation of the inflammasome complex, as well as genes coding for mediators such as chemokines and interleukins that modulate the activity of immunocompetent cells on the resistance or susceptibility to the development of colon and lung tumors. Functional polymorphisms in these candidate genes or, more likely in their regulatory regions could have accumulated differentially in the AIRmax and AIRmin strains during the bidirectional selection process based on inflammatory reactivity.
The polygenic risk scores described in other GWAS studies have contributed to the genetic profiling of individuals with higher risk of non-familiar or sporadic cancers. The genome-scale study presented here supports the modeling of multigene panels for understanding the complex genetics of cross- lung and colon cancer individual risk, and indicates pathways for the study of therapeutic targets linked to inflammation control.
Material and Methods
Mice and treatments
AIRmax and AIRmin mice, F1 hybrids (AIRmax x AIRmin) F1 and the resulting F2 population of F1 x F1 intercross, (AIRmax x AIRmin) F2 were inoculated ip with 1,2-dimethylhydrazine (DMH) (Sigma Aldrich Chemicals) at a dose of 26.4 mg/Kg bw, beginning at 2 months of age and repeated weekly for another 6 weeks, totaling the dose of 185 mg/kg bw. Groups of AIRmax, AIRmin, and (AIRmax x AIRmin) F1 were sacrificed at various times after treatment, to verify the temporal evolution of the onset of tumors in internal organs, especially in the lungs and colon. The F2 animals were sacrificed 300 days after the last dose of the carcinogen. All experiments followed the national guidelines for the care and use of animals and were approved by the Committee for the Use of Animals (CEUA) of Instituto Butantan, São Paulo, Brazil, Protocol number: 6754030915
DNA extraction
Tail tip DNA was extracted with E.Z.N.A. columns, according to the manufacturer’s instructions (Omega Biotech Inc. USA). Purity and concentration were determined in the Nanovue apparatus (GE) by the ratio of λ260/280 readings and integrity was analyzed in 1% agarose gel electrophoresis.
Restriction site-associated DNA genotyping (2B-RAD technique)
In this technique, only DNA fragments adjacent to sites of recognition of restriction endonucleases are sequenced. In our essay we used the enzyme CspCl, which has 283.329 cleavage sites in mice,(value obtained by in silico digestion through the genome). This enzyme recognizes an invariant sequence of 7 nucleotides, divided into 3 and 4 nucleotides separated by 5 variable nucleotides. It cleaves the genomic DNA upstream and downstream of the target site and, in total, CspCl produces 36 nucleotide-long fragments. All nucleotides within the DNA 36-mer fragments, but which are not part of cleavage sites, may vary and can be polymorphic. The 2B-RAD technique usually produces from 3 million to 5 million reads, whose distribution in all sites recognized by the enzyme is expected to yield 20–30X coverage to make the data reliable. The standard protocol consists of 6 phases: enzyme digestion, adaptor link, amplification, purification with beads, quantification, and sequencing. The 2B-RAD libraries were grouped and sequenced (single-end) in Illumina HiSeq 2500 apparatus. Sequencing data were analyzed using a custom computational pipeline (Bash script) and the following tools: Trimmomatic (trimming), BWA (alignment), Picard, Bamtools, GATK (haplotype caller). We used the CeGen-ISCIII Genotyping Service at Universidad de Compostela, Spain. Data are available upon request.
Statistical analysis of the association between genotypes and phenotypes
Statistical analysis and quality of genotyping data were performed with the PLINK program [46]. To verify the significance of the associations between genotypes and phenotypes, each SNP genotype received a code (0, 1, or 2) according to the number of the minor alleles present, in order to represent their additive effects in that variable. Associations with quantitative variables (multiplicity and volume of tumors) were tested with linear regression, considering variables of sex and family. Due to the large number of statistical tests, the correction for multiplicity was made by the Benjamini-Hochberg false discovery rate (FDR) method [47].
Histopathological examination
Fragments of lung and colon-rectal tissue were collected and fixed in 10% formalin buffer for 24 h. After fixation, the samples were dehydrated in a series of alcohol of ascending concentration (70%, 80%, 90%, and 100%), embedded in paraffin and sectioned blocks with a thickness of 5 µm, mounted on slides and stained with hematoxylin -eosin (H&E) and by special staining with Periodic Acid Schiff (PAS). The slides were blindly analyzed by a veterinary pathologist.
References
Ibanez OM, Stiffel C, Ribeiro OG, Cabrera WK, Massa S, de Franco M, et al. Genetics of nonspecific immunity: I. Bidirectional selective breeding of lines of mice endowed with maximal or minimal inflammatory responsiveness. Eur J Immunol. 1992;22:2555–63.
Rosenberg DW, Giardina C, Tanaka T. Mouse models for the study of colon carcinogenesis. Carcinogenesis. 2009;30:183–96.
Bissahoyo AC, Xie Y, Yang L, Scott Pearsall R, Lee D, Elliot RW, et al. A new polygenic model for nonfamilial colorectal cancer inheritance based on the genetic architecture of the Azoxymethane-induced mouse model. Genetics. 2020;214:691–703.
Demant P. Cancer susceptibility in the mouse: genetics, biology and implications for human cancer. Nat Rev Genet. 2003;4:721–34.
Flint J, Eskin E. Genome-wide association studies in mice. Nat Rev Genet. 2012;13:807–17.
MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, et al. The new NHGRIEBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017;45:D896–D901.
Maria DA, Manenti G, Galbiati F, Ribeiro OG, Cabrera WH, Barrera RG, et al. Pulmonary adenoma susceptibility 1 (Pas1) locus affects inflammatory response. Oncogene 2003;22:426–32.
Jensen JR, Galvan A, Borrego A, Cabrera WH, Ribeiro OG, Starobinas N, et al. Genetic control of renal tumorigenesis by the mouse Rtm1 locus. BMC Genomics. 2013;14:724–32.
de Carvalho LR, Borrego A, Jensen JR, Cabrera WHK, Santos AM, Ribeiro OG, et al. Genetic predisposition to hepatocarcinogenesis in inbred and outbred mouse lines selected for high or low inflammatory response. J Immunol Res. 2019;2019:5298792. Mar 31
De Souza VR, Cabrera WK, Galvan A, Ribeiro OG, De Franco M, Vorraro F, et al. Aryl hydrocarbon receptor polymorphism modulates DMBA-induced inflammation and carcinogenesis in phenotypically selected mice. Int J Cancer. 2009;124:1478–82.
Di Pace RF, Massa S, Ribeiro OG, Cabrera WH, De Franco M, Starobinas N, et al. Inverse genetic predisposition to colon versus lung carcinogenesis in mouse lines selected based on acute inflammatory responsiveness. Carcinogenesis 2006;27:1517–25.
Borrego A, Cabrera WHK, Jensen JR, Correa M, Ribeiro OG, Starobinas N, et al. Germline control of somatic Kras mutations in mouse lung tumors. Mol Carcinog. 2018;57:745–51. Jun
Wang S, Meyer E, McKay JK, Matz MV. 2b-RAD: a simple and flexible method for genome-wide genotyping. Nat Meth. 2012;9:808–10.
Davey JW, Cezard T, Fuentes-Utrilla P, Eland C, Gharbi K, Blaxter ML. Special features of RAD sequencing data: implications for genotyping. Mol Ecol 2013;22:3151–64.
Etter PD, Bassham S, Hohenlohe PA, Johnson EA, Cresko WA. SNP discovery and genotyping for evolutionary genetics using RAD sequencing. Methods Mol Biol. 2011;772:157–78.
Manenti G, Acevedo A, Galbiati F, Gianni Barrera R, Noci S, Salido E, et al. Cancer modifier alleles inhibiting lung tumorigenesis are common in inbred mouse strains. Int J Cancer. 2002;99:555–9.
La Vechia S, Sebastian C. Metabolic pathways regulating colorectal cancer initiation and progression. Semin Cell Dev Biol. 2020;98:63–70.
Tak YG, Farnham PJ. Making sense of GWAS: using epigenomics and genome engineering to understand the functional relevance of SNPs in non-coding regions of the human genome. Epigenetics Chromatin. 2015;8:57–75.
Fagny M, Platig J, Kuijjer ML, Lin X, Quackenbush J. Nongenic cancer-risk SNPs affect oncogenes, tumour-suppressor genes, and immune function. Br J Cancer. 2020;122:569–77.
Zhang J, Wang F, Liu F, Xu C. Predicting STAT1 as a prognostic marker in patients with solid cancer. Ther Adv Med Oncol. 2020;12:1758835920917558.
Huang Q, Duan L, Qian X, Fan J, Lv Z, Zhang X, et al. IL-17 promotes angiogenic factors IL-6, IL-8, and VEGF production via Stat1 in Lung Adenocarcinoma. Sci Rep. 2016 6:36551 Erratum : Sci Rep. 2017;7:39566.
Deng ZC, Cao C, Yu YM, Ma HY, Ye M. Vascular endothelial growth factor–634G/C and vascular endothelial growth factor–2578C/A polymorphisms and lung cancer risk: a case-control study and meta-analysis. Tumour Biol. 2014;35:1805–11.
Cherian MG, Jayasurya A, Bay BH. Metallothioneins in human tumors and potential roles in carcinogenesis. Mutat Res. 2003;533:201–9.
Lin E, Li L, Guan Y, Soriano R, Rivers CS, Mohan S, et al. Exon array profiling detects EML4-ALK fusion in breast, colorectal, and non-small cell lung cancers. Mol Cancer Res. 2009;7:1466–76.
Ives A, Nomura J, Martinon F, Roger T, LeRoy D, Miner JN, et al. Xanthine oxidoreductase regulates macrophage IL1β secretion upon NLRP3 inflammasome activation. Nat Commun. 2015;6:6555–66.
Lordén G, Sanjuán-García I, de Pablo N, Meana C, Alvarez-Miguel I, Pérez-García MT, et al. Lipin-2 regulates NLRP3 inflammasome by affecting P2X7 receptor activation. J Exp Med. 2017;214:511–28.
Ohnishi T, Hashizume C, Taniguchi M, Furumoto H, Han J, Gao R, et al. Sphingomyelin synthase 2 deficiency inhibits the induction of murine colitis-associated colon cancer. FASEB J. 2017;31:3816–30.
Lian G, Chen S, Ouyang M, Li F, Chen L, Yang J. Colon cancer cell secretes EGF to promote M2 polarization of TAM through EGFR/PI3K/AKT/mTOR pathway. Technol Cancer Res Treat. 2019;18:1533033819849068.
Plewka D, Plewka A, Miskiewicz A, Morek M, Bogunia E. Nuclear factor-kappa B as potential therapeutic target in human colon cancer. J Cancer Res Ther. 2018;14:516–20.
Zheng J, Lee HL, Ham YW, Song HS, Song MJ, Hong JT. Anti-cancer effect of bee venom on colon cancer cell growth by activation of death receptors and inhibition of nuclear factor kappa B. Oncotarget 2015;6:44437–51.
Jelski W, Zalewski B, Chrostek L, Szmitkowski M. The activity of class I, II, III, and IV alcohol dehydrogenase isoenzymes and aldehyde dehydrogenase in colorectal cancer. Alcohol intake, ADH1B and ADH1C genotypes, and the risk of colorectal cancer by sex and subsite in the Netherlands Cohort Study. Dig Dis Sci. 2004;49:977–81.
Currey N, Jahan Z, Caldon CE, Tran PN, Benthani F, De Lacavalerie P, et al. Mouse model of mutated in colorectal cancer gene deletion reveals novel pathways in inflammation and cancer. Cell Mol Gastroenterol Hepatol. 2019;7:819–39.
Oreffo VI, Robinson S, You M, Wu MC, Malkinson AM. Decreased expression of the adenomatous polyposis coli (Apc) and mutated in colorectal cancer (MCC) genes in mouse lung neoplasia. Mol Carcinog. 1998;21:37–49.
Takahashi M, Fujita M, Furukawa Y, Hamamoto R, Shimokawa T, Miwa N, et al. Isolation of a novel human gene, APCDD1, as a direct target of the beta-Catenin/T-cell factor 4 complex with probable involvement in colorectal carcinogenesis. Cancer Res. 2002;62:5651–6.
Liao T, Fan J, Lv Z, Xu J, Wu F, Yang G, et al. Comprehensive genomic and prognostic analysis of the IL-17 family genes in lung cancer. Mol Med Rep. 2019;19:4906–18.
Yang YF, Lee YC, Lo S, Chung YN, Hsieh YC, Chiu WC, et al. A positive feedback loop of IL-17B-IL-17RB activates ERK/β-catenin to promote lung cancer metastasis. Cancer Lett. 2018;422:44–55.
Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Rüttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. 2017;5:53–66.
Pauken KE, Wherry EJ. TIGIT and CD226: tipping the balance between costimulatory and coinhibitory molecules to augment the cancer immunotherapy toolkit. Cancer Cell. 2014;26:785–7.
Chan CJ, Martinet Ludovic, Gilfillan Susan, Souza-Fonseca- Guimaraes Fernando, Chow MelvynT, Town Liam. et al. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol. 2014;15:431
Quan L, Stassen APM, Ruivenkamp CAL, van Wezwe T, Fihneman RJA, Hutson A, et al. Most lung and colon cancer suseptibility genes are pair-wise linked in mice, humans and rats. PloS ONE. 2011;6:e14727.
Rashkin SR, Graff RE, Kachuri L, Thai KK, Alexeeff SE, Blatchins MA, et al. Pan-cancer study detects genetic risk variants and shared genetic basis in two large cohorts. Nat Commun. 2020;11:4423–37.
Hung RJ, Ulrich CM, Goode EL, Brhane Y, Muir K, Chan AT, et al. Cross cancer genomic investigation of inflammation pathway for five common cancers: lung, ovary, prostate, breast, and colorectal cancer. J Natl Cancer Inst. 2015;107:djv246.
Choi J, Jia G, Wen W, Long J, Zhen W. Evaluating polygenic risk scores in assessing risk of nine solid and hematologic cancers in European descendants. Intern J Cancer. 2020;147:3416–23.
Li X, Timofeeva M, Spiliopoulou A, McKeigue P, He Y, Zhang X, et al. Prediction of colorectal cancer risk based on profiling with common genetic variants. Intern J Cancer. 2020;147:3431–7.
Fehringer G, Kraft P, Pharoah PD, Eeles RA, Chatterjee N, Schumacher FR, et al. Cross-cancer genome-wide analysis of lung, ovary, breast, prostate and colorectal cancer reveals novel pleiotropic associations. Cancer Res. 2016;76:5103–14.
Purcell S, Neale B, Todd Brown K, Thomas L, Ferreira MA, Bender D. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser. 1995; B 57:289
Acknowledgements
This work was funded in part by a grant from Associazione Italiana Ricerca Cancro (AIRC) to GM (IG no. 15797). Animal Health and Welfare Unit, Department of Applied Research and Technical Development, Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and by a grant from São Paulo Research Foundation (FAPESP) São Paulo, Brazil (Process number 2017/50407-0). OMI and MDF were supported in part by National Council for Research (CNPq) Brazil, OMI, OGR, NS, AB, and JRJ were supported in part by Fundação Butantan (Brazil).
Author information
Authors and Affiliations
Contributions
AB participated in the preparation of the project, prepared the DNA samples, and performed the genomic analysis assay; JRJ analyzed the data of wide genome screening and assisted in the revision of the manuscript; WHKC crossed the animals, prepared the DNA samples, and carried out carcinogenesis experiments; SM carried out the carcinogenesis experiments; OGR is responsible for the selected strains and was involved in the analysis of genome screening data; NS assisted in the phenotypic analysis of the animals in the carcinogenesis experiments; MDeF analyzed the genomic data and assisted in the writing of the article, SFE performed the histopathological analysis of tumors, GM, TAD and OMI conceived the study and obtained funding; TAD and OMI wrote the article. All authors made a critical revision of the text and approved the final version submitted.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Borrego, A., Jensen, J.R., Cabrera, W.H.K. et al. Mapping of novel loci involved in lung and colon tumor susceptibility by the use of genetically selected mouse strains. Genes Immun 23, 23–32 (2022). https://doi.org/10.1038/s41435-021-00159-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41435-021-00159-z
