
Abstract
Optic neuritis (ON) is the most common cause of acute optic neuropathy in patients younger than 50 years of age and is most frequently idiopathic or associated with multiple sclerosis. However, the discovery of aquaporin-4 immunoglobulin G (IgG) and myelin oligodendrocyte glycoprotein (MOG)-IgG as biomarkers for two separate central nervous system inflammatory demyelinating diseases has revealed that neuromyelitis optica spectrum disorder (NMSOD) and MOG-IgG-associated disease (MOGAD) are responsible for clinically distinct subsets of ON. NMOSD-ON and MOGAD-ON both demonstrate tendencies for bilateral optic nerve involvement and often exhibit a relapsing course with the potential for devastating long-term visual outcomes. Early and accurate diagnosis is therefore essential. This review will summarize the current understanding of the clinical spectra of NMOSD and MOGAD, the radiographic and serological findings which support their diagnoses, and the current evidence behind various acute and long-term therapeutic strategies for ON related to these conditions. A particular emphasis is placed on a number of recent multi-centre randomized placebo-controlled trials, which provide the first level I evidence for long-term treatment of NMOSD.
摘要
视神经炎 (ON) 是50岁以下患者急性视神经病变最常见的原因, 常表现为特发性或与多发性硬化相关。然而, 水通道蛋白-4免疫球蛋白G (IgG)和髓鞘少突胶质细胞糖蛋白(MOG)-IgG作为两种的中枢神经系统炎性脱髓鞘疾病的生物标志物的发现, 揭示了视神经脊髓炎谱系疾病(NMSOD)和髓鞘少突胶质细胞糖蛋白-IgG相关疾病(MOGAD)是ON临床。NMOSD-ON和MOGAD-ON双侧视神经受累, 并经常复发, 导致视力损伤。因此, 早期诊断至关重要。本文总结了目前对NMOSD和MOGAD临床谱的认识、支持其诊断的影像学和血清学结果、以及与这些疾病相关的各种短效和长效制剂方案背后的现有证据。并特别讨论了一些近期的多中心随机安慰剂对照试验, 这些试验为NMOSD的长效提供了一级证据。
Similar content being viewed by others

Introduction
Optic neuritis (ON) is a major cause of visual morbidity in children and younger adults, representing the most common cause of acute optic neuropathy in people under 50 years of age [1]. Literally defined, ON encompasses any infectious or non-infectious inflammatory process involving the optic nerve. However, the term is most commonly used to refer to an immune-mediated demyelination of the optic nerve. While most cases of ON are idiopathic or associated with multiple sclerosis (MS), it has become increasingly recognized that certain subsets of demyelinating ON represent wholly distinct clinical entities, with differing long-term prognoses and optimal management strategies.
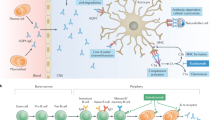
Our ability to classify ON has greatly benefited from advances in serological antibody testing, in particular live cell-based assays (Fig. 1). The most pivotal advance occurred in 2004 with the discovery of a serum autoantibody associated with neuromyelitis optica (NMO), identified 1 year later as immunoglobulin G (IgG) specific for the central nervous system (CNS) water channel aquaporin-4 (AQP4) [2, 3]. A number of inflammatory demyelinating disorders—a subset of which includes severe ON—now fall under the umbrella of NMO spectrum disorder (NMOSD), in which AQP4-IgG seropositivity is a major diagnostic criterion [4]. More recently, the improved specificity of serological testing for IgG specific for myelin oligodendrocyte glycoprotein (MOG) has identified a discrete class of demyelinating disease known as MOG-IgG-associated disorder (MOGAD). This review will summarize the current understanding of the clinical spectra of NMOSD and MOGAD and the evidence supporting various acute and long-term therapeutic strategies for ON related to these conditions.

Human embryonic kidney cells are transfected with a plasmid resulting in co-expression of green fluorescent protein (GFP; green triangle) and human AQP4 or MOG (blue rectangle) in their native transmembrane conformations (Step 1). Patient serum is added to the cells. If present, IgG specific for AQP4 or MOG will bind to its respective antigen (Step 2). Goat anti-human IgG conjugated to a fluorophore (Alexa 647) is then applied to label bound primary antibody (Step 3). The mixture of transfected and un-transfected cells is then analyzed by flow cytometry, with individual cells simultaneously interrogated for green and red fluorescence. The IgG binding index is calculated by dividing the median fluorescence intensity (MFI) for red signal in the GFP-positive (transfected) population by the MFI in the GFP-negative (un-transfected) population, the latter representing non-specific binding. An IgG binding index ≥2 or ≥2.5 is typically the cut-off for a positive test result. (Illustration by Paul Honermann, Mayo Clinic).
Typical idiopathic demyelinating and MS optic neuritis
Our conception of typical ON derives from the landmark optic neuritis treatment trial (ONTT), which enrolled patients with or without a history of MS presenting with acute unilateral ON in a previously unaffected eye [5,6,7]. The classic presentation is one of vision loss in a young adult, developing over hours or several days, reaching a nadir within 2 weeks, and associated with painful eye movements [5]. The degree of vision loss in typical idiopathic-ON or MS-ON is highly variable and in rare cases can be as severe as no-light perception. Frequently, fundus exam on initial presentation is unremarkable: only 35% of subjects in the ONTT manifested optic disc oedema, while the remainder suffered from retrobulbar ON with a normal optic disc. Although typical ON is inherently a clinical diagnosis, the ONTT demonstrated prognostic utility of magnetic resonance imaging (MRI), using the presence or absence of CNS white matter lesions to risk-stratify patients for future development of clinically definite MS [8]. Although a comprehensive discussion of typical idiopathic-ON and MS-ON is beyond the scope of this review, features of typical ON are highlighted in Table 1 and compared to NMOSD-ON and MOGAD-ON.
Current treatment standards for typical ON [9, 10] originate from the observation in the ONTT that intravenous methylprednisolone (IVMP) given as 1 g/day for 3 consecutive days followed by oral (PO) prednisone dosed at 1 mg/kg/day for 11 days and then tapered over 4 days hastened visual recovery, but did not influence final visual outcome when compared to PO placebo. In contrast, low-dose PO prednisone alone at a dose of 1 mg/kg/day for 14 days and then tapered over 4 days failed to speed visual recovery and was associated with a higher risk of recurrent ON [7]. However, the superiority of IVMP administration has recently been challenged, as new evidence suggests that the IV bioequivalent dose of PO prednisone (1250 mg/day) given over 3 days is non-inferior to IVMP (1 g/day) in hastening visual recovery, with no increased risk of ON recurrence [11]. It has also been posited that optic nerve axon preservation and final visual outcomes in ON may be improved by expeditious initiation of corticosteroids within 48 h of onset; however, this hypothesis awaits formal prospective testing [12].
NMOSD-optic neuritis
Pathophysiology and epidemiology
The term ‘neuro-myélite optique aiguë’ translated into the English ‘neuromyelitis optica’ was introduced by Eugène Devic in 1894 when he described a case of bilateral blindness and complete paraplegia, in which the post-mortem examination revealed demyelination of both optic nerves and longitudinally extensive demyelination and necrosis of the spinal cord [13]. Subsequent to this definition of NMO as combined ON and transverse myelitis, a longstanding controversy existed over whether NMO represented a geographically limited subtype of MS or an entirely distinct disease. Not until the first decade of the twenty-first century—with the discovery of AQP4-IgG as a novel pathogenic mediator—was NMO definitively proven to be an independent clinical entity [14]. It is now recognized that NMOSD represents a broad spectrum of clinical manifestations and MRI findings (Table 2) [4], unified by a high rate (73–90%) of AQP4-IgG seropositivity [15, 16].
AQP4 is a transmembrane water channel enriched in the end-feet of perivascular astrocytes of the CNS [17]. Pathological examination of early NMOSD lesions has revealed selective loss of astrocytes and deposition of activated complement, preceding demyelination and apoptosis of oligodendrocytes [18]. Intracerebral administration of purified IgG from AQP4-IgG seropositive NMOSD patients can produce demyelinating lesions in mice, but only when co-injected with human complement [19]. This observation highlights the critical roles of humoral immunity and the complement system in NMOSD pathogenesis, an insight with important therapeutic implications.
NMOSD is a rare disorder, with a reported incidence of 0.4 to 2 per 1,000,000 person-years and a prevalence of 0.5 to 4 per 100,000, largely dependent on the ethnicity of the study population [20]. The median age of onset is 39 years (roughly 10 years later than in MS) [21], and there is a striking female predominance (70–90% of cases) [22, 23]. Unlike MS, NMOSD is more common in people of Asian or African descent than whites [24, 25]. In a large international study, the most common initial manifestation of NMOSD was transverse myelitis (48%) followed closely by ON (42%), with area postrema syndrome (10%) and brainstem, diencephalic, or cerebral syndrome (14%) much less common [23]. The study also found that 63% of patients with NMOSD ultimately developed ON. Compared to MS, the risk of permanent disability is substantially higher in NMOSD [26]. While idiopathic-ON and MS-ON tend to have favourable visual outcomes [7], the natural history of NMOSD-ON is marked by multiple recurrences and significant long-term visual disability, with final visual acuity ≤20/200 in at least one eye in 50–70% of patients [27,28,29,30].
Clinical presentation
It can be clinically challenging to distinguish a first episode of NMOSD-ON from other forms of ON (Table 1). Although visual loss at its nadir tends to be quite severe in NMOSD-ON (median visual acuity 20/1250) [31], profound vision loss is also observed in typical ON (the ONTT reported initial visual acuity of 20/200 or worse in 35.9% of participants, including 3.1% with no-light perception) [5]. Furthermore, both MS-ON and NMOSD-ON tend to spare the anterior-most portion of the optic nerve, with optic disc oedema noted in only a minority of cases (5–33% for NMOSD-ON and 9.5–35% for idiopathic-ON/MS-ON) [5, 29, 32]. Although one retrospective series reported a low rate of painful eye movements associated with NMOSD-ON (27%) compared to patients in the ONTT (87%), it is questionable whether this symptom was adequately documented in the reviewed charts [5, 30]. Simultaneous or rapidly sequential bilateral ON are rare presentations of MS and should suggest an alternative aetiology such as NMOSD (or MOGAD; see below) [33, 34].
Assessment and diagnosis
The clinical suspicion for NMOSD-ON may be informed by in-office ancillary testing. Although automated perimetry may demonstrate any pattern of visual field loss, a bitemporal hemianopsia indicative of chiasmal involvement is considered a “red flag” for NMOSD-ON [35]. However, It should be noted that 5% of patients with ON exhibited a bitemporal hemianopsia in the ONTT [36]. Optical coherence tomography (OCT) is of minimal value in distinguishing NMOSD-ON from idiopathic-ON/MS-ON in the acute phase, as the peripapillary retinal nerve fibre layer (pRNFL) is normal or only mildly thickened in cases without optic disc oedema. However, in the chronic phase, OCT typically reveals more severe thinning of the pRNFL (Fig. 2A, B) and of the ganglion cell-inner plexiform layer (GCIPL) in NMOSD-ON than in idiopathic-ON/MS-ON, reflecting profound loss of retinal ganglion cell axons and somas, consistent with worse visual outcomes in NMOSD-ON [37,38,39].

The patient presented with acute vision loss to hand motions in the right eye, 3 months after having lost vision to no-light perception in the left eye. Visual acuity improved to 20/25 in the right eye following intravenous methylprednisolone and plasma exchange therapy. She was subsequently treated with intravenous rituximab infusions every 6 months. A The right optic disc (left panel) appeared normal on presentation, with minimal thickening of the peripapillary retinal nerve fibre layer (pRNFL) on optical coherence tomography (OCT; middle panel). Four months later, substantial pRNFL thinning had developed, particularly in the temporal quadrant (right panel). B The left optic disc exhibited considerable pallor, and pRNFL was severely thinned on initial presentation, consistent with profound optic atrophy. Some additional interval thinning was apparent 4 months later. C Coronal, post-contrast, T1-weighted magnetic resonance imaging (MRI) of the orbits demonstrated enhancement of the right optic nerve at the level of the orbital apex (blue arrow), while the left optic nerve did not enhance but appeared atrophic (red arrow). The length of optic nerve enhancement was 20.5 mm and involved the posterior orbital, intracanalicular and cisternal segments. D Sagittal, T2-weighted, MRI revealed abnormal hyperintense signal of the thoracic spinal cord spanning 3 vertebral segments (blue arrow).
Gadolinium-enhanced MRI reveals optic nerve enhancement in ~94% of cases of acute ON (Fig. 2C) [40]. The findings of longitudinally extensive optic nerve enhancement, chiasmal involvement, and/or bilateral optic nerve involvement are atypical for idiopathic-ON/MS-ON and should raise suspicion for NMOSD-ON [41,42,43]. In a retrospective analysis of patients with NMOSD-ON or MS-ON, optic nerve enhancement ≥17.6 mm was 80.8% sensitive and 76.9% specific for NMOSD-ON, and involvement of ≥3 optic nerve segments was 100% specific for NMOSD-ON [44]. Importantly, however, extensive optic nerve involvement does not distinguish NMOSD-ON from MOGAD-ON (see below). For those patients with MRI optic nerve abnormalities suspicious for NMOSD-ON, spine imaging should be performed to assess for longitudinally extensive transverse myelitis, defined as spinal cord involvement >3 vertebral segments (Fig. 2D) [4]. Brain MRI lesions are present in up to 85% of patients with NMOSD, commonly affecting structures with high AQP4 expression such as ependymal cells, the hypothalamus and brainstem [45, 46]. Most brain lesions in NMOSD are non-specific in appearance and rarely have the characteristic periventricular distribution seen in MS [47].
Serological testing for AQP4-IgG is critical in confirming the diagnosis of NMOSD [4]. Serum AQP4-IgG may be tested using a variety of methods, but live cell-based assays (Fig. 1) offer the highest sensitivity, yielding a positive result in three-quarters of NMOSD cases while achieving a specificity >99% [48]. Notably, AQP4-IgG was detected in 20% of 34 patients with recurrent ON who did not otherwise meet clinical criteria for NMOSD, with seropositivity associated with more severe visual loss on presentation, poor visual outcome and subsequent development of transverse myelitis [49].
In contrast to serological testing, cerebrospinal fluid (CSF) analysis is less helpful in NMOSD. Pleocytosis, with either a mononuclear or neutrophilic predominance, is more common in NMOSD than in MS, with CSF cell count >50 cells/µl seen in up to 35% of patients [45]. Oligoclonal bands may be identified in up to one-third of cases and thus their presence does not exclude NMOSD [45]. Detection of AQP4-IgG in the CSF is less sensitive than in the serum [50], although there are rare documented cases of positive CSF AQP4-IgG in seronegative patients with recurrent transverse myelitis [51].
Treatment
Acute visual loss
Treatment of acute flares of NMOSD-ON is imperative to reduce long-term visual morbidity. IVMP is the mainstay of treatment, and early treatment has been shown to correlate with preservation of pRNFL [52]. IVMP treatment typically consists of 1 g/day for 3–5 consecutive days with or without a PO prednisone taper. The use of high-dose (1250 mg) PO prednisone has not been evaluated in NMOSD-ON. Although largely limited to case series and retrospective cohort studies, there is evidence that plasma exchange (PLEX) or immunoadsorption apheresis may be beneficial in the acute treatment of NMOSD, including NMOSD-ON [53,54,55,56,57]. Administered every 48 h for 5–7 cycles, PLEX is often initiated in IVMP-refractory disease, with one series reporting average final visual acuity of 20/50 in NMOSD-ON patients receiving sequential IVMP and PLEX compared to 20/400 in those receiving IVMP alone [55]. In two non-randomized studies of acute NMOSD (including some ON cases), 40–50% of attacks treated with PLEX within 2 days of symptom onset experienced complete recovery, with efficacy decreasing over time and 0–5% recovering fully with PLEX initiation after 20 days [56, 57]. While specific visual outcomes were not reported, these findings support the use of combined IVMP and PLEX as first-line therapy.
Long-term management
The hallmark of NMOSD is a relapsing course (67–90% of patients) [26, 58, 59] with relentless, stepwise progression of neurological disability, including blindness. As such, all patients require long-term immunosuppression with the goal of reducing the frequency of relapses [45]. Chronic immunosuppressive treatment is typically continued indefinitely. Until very recently, therapeutic options were limited to immunosuppressant agents without class I or II evidence (see below).
It is important to correctly diagnose NMOSD and differentiate it from MS because NMOSD may worsen when treated with immunomodulatory therapies developed for MS, such as interferon-β, natalizumab, or fingolimod [60]. Instead, traditional immunosuppressant agents are used as maintenance therapy. Oral corticosteroids commonly serve as a bridge to steroid-sparing maintenance treatments following an NMOSD exacerbation. Table 3 summarizes the results of off-label use of azathioprine, mycophenolate mofetil (MMF) and rituximab for long-term management of NMOSD. Less commonly used agents include methotrexate, intravenous immunoglobulin (IVIG), mitoxantrone and cyclophosphamide [45].
A recent randomized double-blind, placebo-controlled trial demonstrated that rituximab significantly reduced relapses in NMOSD [61]. Two retrospective series demonstrated a greater reduction of the annualized relapse rate (ARR) with rituximab or MMF compared to azathioprine [62, 63]. Similarly, a prospective open-label randomized trial demonstrated superior reduction in ARR by rituximab compared to azathioprine combined with prednisone [64].
As of the time of this writing, three drugs have completed phase 2/3 or 3, randomized, double-blind placebo-controlled trials for the treatment of NMOSD (Table 3). All three have recently been awarded United States Food and Drug Administration (FDA) approval for long-term treatment of NMOSD; guidance from the National Institute for Health and Care Excellence in the United Kingdom is still under development. The first FDA-approved medication for adults with AQP4-IgG seropositive NMOSD was eculizumab in June 2019. Eculizumab is a humanized monoclonal antibody, which binds to the complement protein C5 and halts activation of the complement cascade. In a phase 3 study of NMOSD patients on stable doses of immunosuppressive therapy, the addition of eculizumab reduced the occurrence of adjudicated relapses to 3%, compared to 43% with placebo, and the adjudicated ARR was 0.02 in the eculizumab group compared to 0.35 in the placebo group [65].
The anti-CD19 monoclonal antibody inebilizumab was tested as monotherapy for NMOSD in a multi-centre, double-blind, randomized placebo-controlled phase 2/3 study [66]. Enrolment was terminated early due to a clear demonstration of efficacy, with the proportion of participants experiencing relapse decreased by more than threefold. Efficacy in seronegative participants could not be determined due to a low number enrolled. Inebilizumab received FDA approval for use in NMOSD in June 2020.
The second-generation anti-interleukin (IL)-6 receptor monoclonal antibody satralizumab was investigated in two separate randomized, double-blind, placebo-controlled phase 3 trials as adjunct therapy for NMOSD patients stable on immunosuppressant treatment [67] and as monotherapy [68]. Both trials demonstrated reductions in the percentage of patients experiencing relapse and the overall ARR, which was more pronounced when analyzing only AQP4-IgG seropositive NMOSD patients. Neither study demonstrated efficacy in AQP4-IgG seronegative patients. FDA approval for satralizumab was granted in August 2020. Finally, tocilizumab, an older, first-generation IL-6 receptor monoclonal antibody, also recently showed promising results in a phase 2 trial, reducing the number of relapses more effectively than azathioprine [69].
Myelin oligodendrocyte glycoprotein associated disorder (MOGAD)-optic neuritis
Pathophysiology and epidemiology
Autoantibodies against MOG, a cell surface protein expressed by oligodendrocytes, have long been implicated in demyelinating disease based on observations that experimental autoimmune encephalomyelitis can be induced in rodents by infusion of MOG-IgG [70]. In fact, MOG-IgG was incorrectly identified as a biomarker for MS in the early 2000s [71], principally due to lack of specificity of older enzyme-linked immunosorbent assays [72]. The advent of live cell-based assays using transfected human cell lines to express MOG in its native transmembrane conformation revealed that MOG-IgG is indeed relevant to human demyelinating conditions as a biomarker for MOGAD, a disorder that is distinct from both MS and NMOSD [73, 74].
MOGAD represents a diverse spectrum of clinical manifestations, including ON, transverse myelitis, acute disseminating encephalomyelitis (ADEM) and brainstem encephalitis [75, 76]. Because the current 2015 NMOSD criteria were created before the recognition of MOGAD as a distinct entity, 21–42% of patients with AQP4-IgG seronegative NMOSD will test positive for MOG-IgG [77, 78]. However, MOGAD has a broader clinical phenotype than NMOSD, and only about one-third of patients with MOGAD satisfy the diagnostic criteria for NMOSD [76, 79]. Indeed, the fact that most NMOSD series have not identified AQP4-IgG and MOG-IgG double-positive cases suggests that NMOSD and MOGAD are not mediated by a common pathobiology [77, 78, 80, 81]. Furthermore, MOGAD pathological specimens demonstrate demyelination without astrocyte degeneration, the opposite of NMOSD [82, 83].
The median age of onset of MOGAD in adults is in the early-to-mid 30s, closer to that of MS patients than to NMOSD [84,85,86]. Unlike AQP4-IgG, MOG-IgG seropositivity is common in children with demyelinating disease (~30% of cases) [87, 88]. In contrast to MS and NMOSD, it appears that there is minimal predilection for females in MOGAD [76, 77, 79]. The national incidence of MOGAD in the Netherlands was recently reported to be 0.16 per 100,000 per year [85].
Clinical presentation
ON is the most common clinical presentation and relapse manifestation of MOGAD in adults (Table 1). In a large United Kingdom study of MOGAD, isolated ON (with bilateral involvement in ~50%) was the initial presentation in 58% of patients, followed by transverse myelitis in 21%, simultaneous ON and transverse myelitis in 12% and an ADEM-like presentation in 9% [76]. Brain-stem involvement, encephalitis, and seizures can also occur in MOGAD [89]. MOGAD presenting at age <9 years is more likely to manifest as ADEM, while older children commonly present with ON or transverse myelitis [85, 90, 91].
The clinical presentation of MOGAD-ON can be atypical in a number of ways. As mentioned above, MOGAD-ON has been reported to be bilateral in 50 to 84% of cases (Fig. 3A, B) [76, 85, 92, 93]. The frequency of bilateral involvement is similar to that seen in NMOSD-ON, but 3–4 times greater than in MS-ON [33, 93,94,95]. In contrast to NMOSD-ON and MS-ON, MOGAD-ON is commonly (76–86%) associated with optic disc oedema, which can be severe enough to be associated with peripapillary haemorrhages [92, 96, 97]. This fundus appearance may lead to diagnostic confusion with ischaemic optic neuropathy or papilledema, highlighting the importance of eliciting distinguishing features in the patient history (such as painful eye movements [96]), correlating the degree of vision loss to the severity of optic disc oedema (typically worse vision loss in MOGAD-ON than in papilledema) and obtaining an MRI (see below).

The patient presented with acute loss of visual acuity to 20/50 in the right eye (OD) and hand motion in the left eye (OS). A Both optic discs exhibited mild optic disc oedema on initial presentation. B Axial, post-contrast, T1-weighted magnetic resonance imaging of the orbits revealed bilateral enhancement of the anterior optic nerves (blue arrows) with co-existing enhancement of the left optic nerve sheath (green arrowhead). The length of enhancement was 21 mm for the right optic nerve and 19 mm for the left, less than 50% of total optic nerve length in both cases. C Humphrey visual field testing on initial presentation revealed severe generalized depression of both eyes, while optical coherence tomography (OCT) revealed peripapillary retinal nerve fibre layer (pRNFL) thickening in both eyes, consistent with the presence of optic disc oedema. D The patient was treated with intravenous methylprednisolone for 5 days, followed by a prednisone taper over 2 months. At 6-month follow-up, visual acuity had improved to 20/20 in both eyes and visual fields had entirely normalized. OCT revealed the interval development of pathological pRNFL thinning, consistent with partial optic atrophy of both eyes.
Visual loss upon initial presentation with MOGAD-ON can be quite severe, with a median nadir visual acuity of hand motion [96]. While also encountered in MS-ON and NMOSD-ON, central scotomas were observed in a sizeable majority of eyes affected by MOGAD-ON (73%), with an additional 22% demonstrating complete visual field depression (Fig. 3C) [97]. Fortunately, substantial recovery of visual function is quite common in MOGAD-ON, and it is rare to develop permanent severe vision loss after a single episode. Similar to NMOSD-ON, MOGAD-ON has a strong tendency to be recurrent, with at least 50% of patients suffering relapses [76, 98, 99]. While MOGAD patients may enjoy intervals of months or years between ON recurrences, some may relapse during or shortly following completion of a steroid taper, reminiscent of chronic relapsing inflammatory optic neuropathy (CRION). In fact, MOG-IgG seropositivity has been reported in 67–92% of patients previously diagnosed with CRION [100, 101]. Long-term visual outcomes after MOGAD-ON relapses are variable but in general more favourable than NMOSD-ON: 5–20% of MOGAD-ON patients suffer permanent vision loss to 20/200 or worse in at least one eye [76, 92, 96], compared to at least 50% in NMOSD, as detailed above.
Assessment and diagnosis
MOGAD-ON cases presenting acutely with optic disc oedema will demonstrate pRNFL thickening on OCT, whereas all cases will demonstrate progressive pRNFL thinning as optic atrophy develops over the subsequent months (Fig. 3D). Similar degrees of pRNFL and GCIPL thinning have been observed in affected eyes of NMOSD-ON and MOGAD-ON patients; however, the number of attacks required to produce this amount of thinning has been found to be greater in MOGAD-ON than NMOSD-ON [102]. Chronic pRNFL thinning is most prominent in the temporal quadrant representing the papillomacular bundle, consistent with the propensity of MOGAD-ON to produce central scotomas [103].
MRI has great value in distinguishing MOGAD-ON from other aetiologies. Longitudinally extensive enhancement of at least one-half the length of the optic nerve has been observed in up to 80% of MOGAD-ON cases but is not a pathognomonic sign, as it can also be seen in NMOSD-ON [93, 96]. However, consistent with the high rate of optic disc oedema, MOGAD-ON tends to exhibit enhancement of the anterior portion of the optic nerve, in contrast to the posterior optic nerve/chiasmal involvement common in NMOSD-ON [92, 93, 96]. Nevertheless, MOGAD-ON may affect the optic chiasm in up to 15% of cases [92, 96]. Perhaps most diagnostically useful is the frequent observation of enhancement of the optic nerve sheath (i.e., perioptic neuritis) and adjacent orbital fat, reported in up to 50% of MOGAD-ON, which is not typically seen in NMOSD-ON or MS-ON [79, 96, 104].
MOGAD demyelinating lesions in the brain often have a fluffy appearance and commonly localize to the juxtacortical and deep white matter, brainstem and surrounding the 4th ventricle, unlike the classic ovoid lesions perpendicular to the lateral ventricles in MS [76, 104,105,106]. Longitudinally extensive transverse myelitis can be seen in MOGAD, but compared to NMOSD, MOGAD-transverse myelitis is more likely to be non-enhancing, multifocal, involving the conus medullaris, and restricted to the grey matter (producing an ‘H sign’) [107].
As in NMOSD, the CSF findings in MOGAD are not diagnostically confirmatory. CSF evaluation may reveal a normal nucleated cell count or demonstrate a pleocytosis (exceeding 100 cells/µl in 28% of cases), sometimes with a neutrophilic component; oligoclonal bands are seen in a minority of cases [108]. Testing for MOG-IgG in the CSF is rarely performed due to a low sensitivity of 60–70% (typically with low titres when positive, suggesting an extrathecal origin of these autoantibodies) [109, 110]. In contrast, autoantibody testing in serum samples is critical to the diagnosis of MOGAD. Based on the recommendations of an international expert panel [75], MOG-IgG testing should be performed exclusively using cell-based assays with either fluorescence microscopy or flow cytometry (Fig. 1). While a fluorescence microscopy-based assay using fixed cells is widely used due to its convenience, live cell-based assays appear to offer higher accuracy [111]. Determining the MOG-IgG titre can help determine the likelihood of a true-positive test, although high titres do not correlate with recurrence risk or final clinical outcome [112]. Serial testing to evaluate the persistence of MOG-IgG seropositivity may have some clinical utility. It has been reported that transient seropositivity is correlated with monophasic disease and persistent seropositivity with a relapsing course [76, 85, 113]. A recent prospective study in children, however, found that while conversion to seronegative status was correlated with a lower (but not zero) risk of relapse, persistent seropositivity had little predictive value for recurrent demyelinating episodes [114]. Whether serological testing should be performed on every patient presenting with acute ON is controversial, as patients with monophasic MOGAD-ON with good recovery do not require chronic immunosuppressive therapy. Many clinicians therefore limit MOG-IgG testing to patients with recurrent or steroid-dependent ON or additional CNS lesions suspicious for MOGAD.
Treatment
Acute visual loss
As the natural history of untreated MOGAD-ON is not well-defined, it is uncertain whether MOGAD-ON differs from typical idiopathic-ON/MS-ON in terms of visual outcome. Notably, a re-analysis of blood samples from the ONTT identified three patients who were MOG-IgG seropositive and recovered to 20/20 visual acuity despite none of them receiving IVMP (two received PO prednisone and the third placebo) [115]. However, a retrospective study of MOGAD from Germany reported that only 7 of 12 untreated MOGAD-ON patients experienced complete or near-complete visual recovery [79]. Another retrospective study reported better visual outcomes in both NMOSD-ON and MOGAD-ON patients in whom IVMP was initiated within 4 days of symptom onset [116]. Given the tendency for corticosteroid-dependence in MOGAD-ON, treatment of acute attacks would seem prudent in most cases.
Acute treatment of MOGAD-ON typically starts with IVMP administered as 1 g/day for 3–5 days, often resulting in rapid visual improvement. The utility of high-dose PO prednisone is not known at this time. Unlike idiopathic-ON/MS-ON, a slow PO prednisone taper over 2–6 months after IVMP may be warranted. This is because a sizeable fraction of patients who will relapse do so early after an attack, with the median time to recurrence of 5 months [108]. It appears that the risk of early recurrence is reduced by a prolonged steroid taper. In a series of 59 patients with relapsing MOGAD (the majority with ON), 146 episodes were treated with a PO prednisone taper, with 28% suffering relapse during the taper (at a median prednisone dose of 10 mg per day) and an additional 42% suffering relapse following the taper (median interval of 2 months) [117]. Notably, among patients with relapse, the median planned duration of the taper was 1.5 months, compared to 5 months in patients who did not relapse. Limited data exists on the efficacy of PLEX in IVMP-refractory MOGAD. In a German study of 25 MOGAD patients with ON or transverse myelitis treated with IVMP and subsequent PLEX or immunoadsorption, 40% experienced complete or near-complete recovery, 56% partial recovery and 4% no recovery [79]. IVIG has not been studied in acute MOGAD-ON, but in two randomized trials, it failed to improve the vision of patients with idiopathic-ON/MS-ON [118, 119].
Long-term management
The necessity of long-term immunosuppressive therapy for MOGAD is less clear-cut than in AQP4-IgG seropositive NMOSD. Roughly half of MOGAD patients experience a monophasic disease course [76] and those who recur often experience substantial recovery after the first relapse. Therefore, for those patients who recovered well after an initial attack of MOGAD, long-term therapy is not needed until relapses occur. Randomized prospective trials of long-term therapy for MOGAD are currently lacking, with only observational studies providing class IV evidence.
As in NMOSD, the disease-modifying MS medications interferon-β and glatiramer acetate were found to be ineffective in MOGAD, while data on natalizumab were ambiguous [79, 120]. MMF, azathioprine, IVIG and rituximab have all been associated with reductions in ARR from ~2 episodes per year pre-treatment to ≤1 with each agent [117, 120]. A recent prospective, non-randomized study found that MMF plus maintenance prednisone resulted in a lower relapse rate than maintenance prednisone alone [121]. IVIG administered every 3–4 weeks has been reported effective in paediatric and adult MOGAD patients [117, 120]. A recent multi-centre retrospective study of 70 MOGAD patients with pre-treatment ARR of 1.6 found only 2 of 10 patients relapsed (median ARR 0) after treatment with IVIG, while other agents were associated with relapse in >50% of patients, including azathioprine (13/22 relapsing; ARR 0.2), MMF (14/19 relapsing; ARR 0.67) and rituximab (22/36 relapsing; ARR 0.59) [122]. Finally, in a case report, tocilizumab was reported to stabilize a patient with recurrent MOGAD-ON [123].
Conclusion
The discovery of AQP4-IgG and MOG-IgG as biomarkers for CNS inflammatory demyelinating diseases distinct from MS represents a paradigm shift in the field of neuro-immunology. Both NMOSD-ON and MOGAD-ON have the potential for devastating long-term visual disability. Autoantibody seropositivity, clinical manifestations and MRI findings are invaluable in establishing the correct diagnosis of each condition. IVMP is the mainstay of acute treatment for both NMOSD-ON and MOGAD-ON, with PLEX likely beneficial as rescue therapy and potentially as first-line treatment for NMOSD-ON (Table 4). While three new drugs targeting the pathogenic mechanisms underlying NMOSD appear poised to dramatically reduce visual morbidity from recurrent AQP4-IgG seropositive NMOSD-ON, optimal long-term therapy for MOGAD-ON awaits future prospective trials.
References
Rizzo JF 3rd, Lessell S. Optic neuritis and ischemic optic neuropathy. Overlapping clinical profiles. Arch Ophthalmol. 1991;109:1668–72.
Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202:473–7.
Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364:2106–12.
Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015;85:177–89.
The clinical profile of optic neuritis. Experience of the Optic Neuritis Treatment Trial. Optic neuritis study group. Arch Ophthalmol. 1991;109:1673–8.
Beck RW. The optic neuritis treatment trial. Arch Ophthalmol. 1988;106:1051–3.
Beck RW, Cleary PA, Anderson MM Jr., Keltner JL, Shults WT, Kaufman DI, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. The Optic Neuritis Study Group. N Engl J Med. 1992;326:581–8.
Optic Neuritis Study G. Multiple sclerosis risk after optic neuritis: final optic neuritis treatment trial follow-up. Arch Neurol. 2008;65:727–32.
Gal RL, Vedula SS, Beck R. Corticosteroids for treating optic neuritis. Cochrane Database Syst Rev. 2015;8:CD001430.
Kaufman DI, Trobe JD, Eggenberger ER, Whitaker JN. Practice parameter: the role of corticosteroids in the management of acute monosymptomatic optic neuritis. Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000;54:2039–44.
Morrow SA, Fraser JA, Day C, Bowman D, Rosehart H, Kremenchutzky M, et al. Effect of treating acute optic neuritis with bioequivalent oral vs intravenous corticosteroids: a randomized clinical trial. JAMA Neurol. 2018;75:690–6.
Petzold A, Braithwaite T, van Oosten BW, Balk L, Martinez-Lapiscina EH, Wheeler R, et al. Case for a new corticosteroid treatment trial in optic neuritis: review of updated evidence. J Neurol Neurosurg Psychiatry. 2020;91:9–14.
Jarius S, Wilderman B. The history of neuromyelitis optica. J Neuroinflammation. 2013;10:1–12.
Hinson SR, Lennon VA, Pittock SJ. Autoimmune AQP4 channelopathies and neuromyelitis optica spectrum disorders. Handb Clin Neurol. 2016;133:377–403.
Hamid SH, Elsone L, Mutch K, Solomon T, Jacob A. The impact of 2015 neuromyelitis optica spectrum disorders criteria on diagnostic rates. Mult Scler. 2017;23:228–33.
Hyun JW, Jeong IH, Joung A, Kim SH, Kim HJ. Evaluation of the 2015 diagnostic criteria for neuromyelitis optica spectrum disorder. Neurology. 2016;86:1772–9.
Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci. 1997;17:171–80.
Parratt JD, Prineas JW. Neuromyelitis optica: a demyelinating disease characterized by acute destruction and regeneration of perivascular astrocytes. Mult Scler. 2010;16:1156–72.
Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain 2010;133:349–61.
Pandit L, Asgari N, Apiwattanakul M, Palace J, Paul F, Leite MI, et al. Demographic and clinical features of neuromyelitis optica: a review. Mult Scler. 2015;21:845–53.
Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6:805–15.
Wingerchuk DM. Neuromyelitis optica: effect of gender. J Neurol Sci. 2009;286:18–23.
Kim SH, Mealy MA, Levy M, Schmidt F, Ruprecht K, Paul F, et al. Racial differences in neuromyelitis optica spectrum disorder. Neurology. 2018;91:e2089–99.
Flanagan EP, Cabre P, Weinshenker BG, Sauver JS, Jacobson DJ, Majed M, et al. Epidemiology of aquaporin-4 autoimmunity and neuromyelitis optica spectrum. Ann Neurol. 2016;79:775–83.
Marignier R, Cobo Calvo A, Vukusic S. Neuromyelitis optica and neuromyelitis optica spectrum disorders. Curr Opin Neurol. 2017;30:208–15.
Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology. 1999;53:1107–14.
Mealy MA, Wingerchuk DM, Greenberg BM, Levy M. Epidemiology of neuromyelitis optica in the United States: a multicenter analysis. Arch Neurol. 2012;69:1176–80.
Lin CW, Lin IH, Chen TC, Jou JR, Woung LC. Clinical course and treatment response of neuromyelitis optica spectrum disease: an 8-year experience. Asia Pac J Ophthalmol. 2019;8:206–10.
Merle H, Olindo S, Bonnan M, Donnio A, Richer R, Smadja D, et al. Natural history of the visual impairment of relapsing neuromyelitis optica. Ophthalmology. 2007;114:810–5.
Papais-Alvarenga RM, Carellos SC, Alvarenga MP, Holander C, Bichara RP, Thuler LC. Clinical course of optic neuritis in patients with relapsing neuromyelitis optica. Arch Ophthalmol. 2008;126:12–16.
Masuda H, Mori M, Uzawa A, Muto M, Uchida T, Ohtani R, et al. Recovery from optic neuritis attack in neuromyelitis optica spectrum disorder and multiple sclerosis. J Neurol Sci. 2016;367:375–9.
Liu H, Zhou H, Wang J, Sun M, Teng D, Song H, et al. The prevalence and prognostic value of myelin oligodendrocyte glycoprotein antibody in adult optic neuritis. J Neurol Sci. 2019;396:225–31.
Srikajon J, Siritho S, Ngamsombat C, Prayoonwiwat N, Chirapapaisan N.Siriraj Neuroimmunology Research Group Differences in clinical features between optic neuritis in neuromyelitis optica spectrum disorders and in multiple sclerosis. Mult Scler J Exp Transl Clin. 2018;4:2055217318791196.
Chen JJ, Bhatti MT. Clinical phenotype, radiological features, and treatment of myelin oligodendrocyte glycoprotein-immunoglobulin G (MOG-IgG) optic neuritis. Curr Opin Neurol. 2020;33:47–54.
Nakajima H, Hosokawa T, Sugino M, Kimura F, Sugasawa J, Hanafusa T, et al. Visual field defects of optic neuritis in neuromyelitis optica compared with multiple sclerosis. BMC Neurol. 2010;10:45.
Keltner JL, Johnson CA, Spurr JO, Beck RW. Visual field profile of optic neuritis. One-year follow-up in the Optic Neuritis Treatment Trial. Arch Ophthalmol. 1994;112:946–53.
Ratchford JN, Quigg ME, Conger A, Frohman T, Frohman E, Balcer LJ, et al. Optical coherence tomography helps differentiate neuromyelitis optica and MS optic neuropathies. Neurology. 2009;73:302–8.
Syc SB, Saidha S, Newsome SD, Ratchford JN, Levy M, Ford E, et al. Optical coherence tomography segmentation reveals ganglion cell layer pathology after optic neuritis. Brain. 2012;135:521–33.
Merle H, Olindo S, Donnio A, Richer R, Smadja D, Cabre P. Retinal peripapillary nerve fiber layer thickness in neuromyelitis optica. Invest Ophthalmol Vis Sci. 2008;49:4412–7.
Kupersmith MJ, Alban T, Zeiffer B, Lefton D. Contrast-enhanced MRI in acute optic neuritis: relationship to visual performance. Brain. 2002;125:812–22.
Khanna S, Sharma A, Huecker J, Gordon M, Naismith RT, Van, et al. Magnetic resonance imaging of optic neuritis in patients with neuromyelitis optica versus multiple sclerosis. J Neuroophthalmol. 2012;32:216–20.
Storoni M, Davagnanam I, Radon M, Siddiqui A, Plant GT. Distinguishing optic neuritis in neuromyelitis optica spectrum disease from multiple sclerosis: a novel magnetic resonance imaging scoring system. J Neuroophthalmol. 2013;33:123–7.
Pula JH, Kattah JC, Keung B, Wang H, Daily J. Longitudinally extensive optic neuritis in neuromyelitis optica spectrum disorder. J Neurol Sci. 2014;345:209–12.
Mealy MA, Whetstone A, Orman G, Izbudak I, Calabresi PA, Levy M. Longitudinally extensive optic neuritis as an MRI biomarker distinguishes neuromyelitis optica from multiple sclerosis. J Neurol Sci. 2015;355:59–63.
Sellner J, Boggild M, Clanet M, Hintzen RQ, Illes Z, Montalban X, et al. EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol. 2010;17:1019–32.
Pittock SJ, Weinshenker BG, Lucchinetti CF, Wingerchuk DM, Corboy JR, Lennon VA. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol. 2006;63:964–8.
Pittock SJ, Lennon VA, Krecke K, Wingerchuk DM, Lucchinetti CF, Weinshenker BG. Brain abnormalities in neuromyelitis optica. Arch Neurol. 2006;63:390–6.
Waters PJ, McKeon A, Leite MI, Rajasekharan S, Lennon VA, Villalobos A, et al. Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology. 2012;78:665–71.
Matiello M, Lennon VA, Jacob A, Pittock SJ, Lucchinetti CF, Wingerchuk DM, et al. NMO-IgG predicts the outcome of recurrent optic neuritis. Neurology. 2008;70:2197–200.
Majed M, Fryer JP, McKeon A, Lennon VA, Pittock SJ. Clinical utility of testing AQP4-IgG in CSF: Guidance for physicians. Neurol Neuroimmunol Neuroinflamm. 2016;3:e231.
Klawiter EC, Alvarez E 3rd, Xu J, Paciorkowski AR, Zhu L, Parks BJ, et al. NMO-IgG detected in CSF in seronegative neuromyelitis optica. Neurology. 2009;72:1101–3.
Nakamura M, Nakazawa T, Doi H, Hariya T, Omodaka K, Misu T, et al. Early high-dose intravenous methylprednisolone is effective in preserving retinal nerve fiber layer thickness in patients with neuromyelitis optica. Graefes Arch Clin Exp Ophthalmol. 2010;248:1777–85.
Kleiter I, Gahlen A, Borisow N, Fischer K, Wernecke KD, Wegner B, et al. Neuromyelitis optica: Evaluation of 871 attacks and 1,153 treatment courses. Ann Neurol. 2016;79:206–16.
Song W, Qu Y, Huang X. Plasma exchange: an effective add-on treatment of optic neuritis in neuromyelitis optica spectrum disorders. Int Ophthalmol. 2019;39:2477–83.
Merle H, Olindo S, Jeannin S, Valentino R, Mehdaoui H, Cabot F, et al. Treatment of optic neuritis by plasma exchange (add-on) in neuromyelitis optica. Arch Ophthalmol. 2012;130:858–62.
Kleiter I, Gahlen A, Borisow N, Fischer K, Wernecke KD, Hellwig K, et al. Apheresis therapies for NMOSD attacks: a retrospective study of 207 therapeutic interventions. Neurol Neuroimmunol Neuroinflamm. 2018;5:e504.
Bonnan M, Valentino R, Debeugny S, Merle H, Ferge JL, Mehdaoui H, et al. Short delay to initiate plasma exchange is the strongest predictor of outcome in severe attacks of NMO spectrum disorders. J Neurol Neurosurg Psychiatry. 2018;89:346–51.
Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, Geis C, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation. 2012;9:14.
Jiao Y, Fryer JP, Lennon VA, Jenkins SM, Quek AM, Smith CY, et al. Updated estimate of AQP4-IgG serostatus and disability outcome in neuromyelitis optica. Neurology. 2013;81:1197–204.
Kowarik MC, Soltys J, Bennett JL. The treatment of neuromyelitis optica. J Neuroophthalmol. 2014;34:70–82.
Tahara M, Oeda T, Okada K, Kiriyama T, Ochi K, Maruyama H, et al. Safety and efficacy of rituximab in neuromyelitis optica spectrum disorders (RIN-1 study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2020;19:298–306.
Mealy MA, Wingerchuk DM, Palace J, Greenberg BM, Levy M. Comparison of relapse and treatment failure rates among patients with neuromyelitis optica: multicenter study of treatment efficacy. JAMA Neurol. 2014;71:324–30.
Jeong IH, Park B, Kim SH, Hyun JW, Joo J, Kim HJ. Comparative analysis of treatment outcomes in patients with neuromyelitis optica spectrum disorder using multifaceted endpoints. Mult Scler. 2016;22:329–39.
Nikoo Z, Badihian S, Shaygannejad V, Asgari N, Ashtari F. Comparison of the efficacy of azathioprine and rituximab in neuromyelitis optica spectrum disorder: a randomized clinical trial. J Neurol. 2017;264:2003–9.
Pittock SJ, Berthele A, Fujihara K, Kim HJ, Levy M, Palace J, et al. Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N Engl J Med. 2019;381:614–25.
Cree BAC, Bennett JL, Kim HJ, Weinshenker BG, Pittock SJ, Wingerchuk DM, et al. Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet. 2019;394:1352–63.
Yamamura T, Kleiter I, Fujihara K, Palace J, Greenberg B, Zakrzewska-Pniewska B, et al. Trial of satralizumab in neuromyelitis optica spectrum disorder. N Engl J Med. 2019;381:2114–24.
Traboulsee A, Greenberg BM, Bennett JL, Szczechowski L, Fox E, Shkrobot S, et al. Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: a randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol. 2020;19:402–12.
Zhang C, Zhang M, Qiu W, Ma H, Zhang X, Zhu Z, et al. Safety and efficacy of tocilizumab versus azathioprine in highly relapsing neuromyelitis optica spectrum disorder (TANGO): an open-label, multicentre, randomised, phase 2 trial. Lancet Neurol. 2020;19:391–401.
Schluesener HJ, Sobel RA, Linington C, Weiner HL. A monoclonal antibody against a myelin oligodendrocyte glycoprotein induces relapses and demyelination in central nervous system autoimmune disease. J Immunol. 1987;139:4016–21.
Berger T, Rubner P, Schautzer F, Egg R, Ulmer H, Mayringer I, et al. Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N Engl J Med. 2003;349:139–45.
Ramanathan S, Dale RC, Brilot F. Anti-MOG antibody: the history, clinical phenotype, and pathogenicity of a serum biomarker for demyelination. Autoimmun Rev. 2016;15:307–24.
Chan A, Decard BF, Franke C, Grummel V, Zhou D, Schottstedt V, et al. Serum antibodies to conformational and linear epitopes of myelin oligodendrocyte glycoprotein are not elevated in the preclinical phase of multiple sclerosis. Mult Scler. 2010;16:1189–92.
Ketelslegers IA, Van Pelt DE, Bryde S, Neuteboom RF, Catsman-Berrevoets CE, Hamann D, et al. Anti-MOG antibodies plead against MS diagnosis in an Acquired Demyelinating Syndromes cohort. Mult Scler. 2015;21:1513–20.
Jarius S, Paul F, Aktas O, Asgari N, Dale RC, de Seze J, et al. MOG encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflammation. 2018;15:134.
Jurynczyk M, Messina S, Woodhall MR, Raza N, Everett R, Roca-Fernandez A, et al. Clinical presentation and prognosis in MOG-antibody disease: a UK study. Brain. 2017;140:3128–38.
Sato DK, Callegaro D, Lana-Peixoto MA, Waters PJ, de Haidar Jorge FM, Takahashi T, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology. 2014;82:474–81.
Hamid SHM, Whittam D, Mutch K, Linaker S, Solomon T, Das K, et al. What proportion of AQP4-IgG-negative NMO spectrum disorder patients are MOG-IgG positive? A cross sectional study of 132 patients. J Neurol. 2017;264:2088–94.
Jarius S, Kleiter I, Ruprecht K, Asgari N, Pitarokoili K, Borisow N, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 3: Brainstem involvement - frequency, presentation and outcome. J Neuroinflammation. 2016;13:281.
Kitley J, Woodhall M, Waters P, Leite MI, Devenney E, Craig J, et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology. 2012;79:1273–7.
Kunchok A, Chen JJ, McKeon A, Mills JR, Flanagan EP, Pittock SJ. Coexistence of myelin oligodendrocyte glycoprotein and aquaporin-4 antibodies in adult and pediatric patients. JAMA Neurol. 2020;77:257–9.
Spadaro M, Gerdes LA, Mayer MC, Ertl-Wagner B, Laurent S, Krumbholz M, et al. Histopathology and clinical course of MOG-antibody-associated encephalomyelitis. Ann Clin Transl Neurol. 2015;2:295–301.
Hoftberger R, Guo Y, Flanagan EP, Lopez-Chiriboga AS, Endmayr V, Hochmeister S, et al. The pathology of central nervous system inflammatory demyelinating disease accompanying myelin oligodendrocyte glycoprotein autoantibody. Acta Neuropathol. 2020;139:875–92.
Cobo-Calvo A, Ruiz A, Maillart E, Audoin B, Zephir H, Bourre B, et al. Clinical spectrum and prognostic value of CNS MOG autoimmunity in adults: The MOGADOR study. Neurology. 2018;90:e1858–69.
de Mol CL, Wong Y, van Pelt ED, Wokke B, Siepman T, Neuteboom RF, et al. The clinical spectrum and incidence of anti-MOG-associated acquired demyelinating syndromes in children and adults. Mult Scler. 2020;26:806–14.
Zhao G, Chen Q, Huang Y, Li Z, Sun X, Lu P, et al. Clinical characteristics of myelin oligodendrocyte glycoprotein seropositive optic neuritis: a cohort study in Shanghai, China. J Neurol. 2018;265:33–40.
Hennes EM, Baumann M, Schanda K, Anlar B, Bajer-Kornek B, Blaschek A, et al. Prognostic relevance of MOG antibodies in children with an acquired demyelinating syndrome. Neurology. 2017;89:900–8.
Duignan S, Wright S, Rossor T, Cazabon J, Gilmour K, Ciccarelli O, et al. Myelin oligodendrocyte glycoprotein and aquaporin-4 antibodies are highly specific in children with acquired demyelinating syndromes. Dev Med Child Neurol. 2018;60:958–62.
Reindl M, Waters P. Myelin oligodendrocyte glycoprotein antibodies in neurological disease. Nat Rev Neurol. 2019;15:89–102.
Hacohen Y, Rossor T, Mankad K, Chong W, Lux A, Wassmer E, et al. ‘Leukodystrophy-like’ phenotype in children with myelin oligodendrocyte glycoprotein antibody-associated disease. Dev Med Child Neurol. 2018;60:417–23.
Mao L, Yang L, Kessi M, He F, Zhang C, Wu L, et al. Myelin oligodendrocyte glycoprotein (MOG) antibody diseases in children in central south china: clinical features, treatments, influencing factors, and outcomes. Front Neurol. 2019;10:868.
Zhao Y, Tan S, Chan TCY, Xu Q, Zhao J, Teng D, et al. Clinical features of demyelinating optic neuritis with seropositive myelin oligodendrocyte glycoprotein antibody in Chinese patients. Br J Ophthalmol. 2018;102:1372–7.
Ramanathan S, Prelog K, Barnes EH, Tantsis EM, Reddel SW, Henderson AP, et al. Radiological differentiation of optic neuritis with myelin oligodendrocyte glycoprotein antibodies, aquaporin-4 antibodies, and multiple sclerosis. Mult Scler. 2016;22:470–82.
Jarius S, Frederikson J, Waters P, Paul F, Akman-Demir G, Marignier R, et al. Frequency and prognostic impact of antibodies to aquaporin-4 in patients with optic neuritis. J Neurol Sci. 2010;298:158–62.
Burman J, Raininko R, Fagius J. Bilateral and recurrent optic neuritis in multiple sclerosis. Acta Neurol Scand. 2011;123:207–10.
Chen JJ, Flanagan EP, Jitprapaikulsan J, Lopez-Chiriboga ASS, Fryer JP, Leavitt JA, et al. Myelin oligodendrocyte glycoprotein antibody-positive optic neuritis: clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol. 2018;195:8–15.
Ishikawa H, Kezuka T, Shikishima K, Yamagami A, Hiraoka M, Chuman H, et al. Epidemiologic and clinical characteristics of optic neuritis in Japan. Ophthalmology. 2019;126:1385–98.
Ramanathan S, Reddel SW, Henderson A, Parratt JD, Barnett M, Gatt PN, et al. Antibodies to myelin oligodendrocyte glycoprotein in bilateral and recurrent optic neuritis. Neurol Neuroimmunol Neuroinflamm. 2014;1:e40.
Chalmoukou K, Alexopoulos H, Akrivou S, Stathopoulos P, Reindl M, Dalakas MC. Anti-MOG antibodies are frequently associated with steroid-sensitive recurrent optic neuritis. Neurol Neuroimmunol Neuroinflamm. 2015;2:e131.
Lee HJ, Kim B, Waters P, Woodhall M, Irani S, Ahn S, et al. Chronic relapsing inflammatory optic neuropathy (CRION): a manifestation of myelin oligodendrocyte glycoprotein antibodies. J Neuroinflammation. 2018;15:302.
Liu H, Zhou H, Wang J, Xu Q, Wei S. Antibodies to myelin oligodendrocyte glycoprotein in chronic relapsing inflammatory optic neuropathy. Br J Ophthalmol. 2019;103:1423–8.
Pache F, Zimmermann H, Mikolajczak J, Schumacher S, Lacheta A, Oertel FC, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 4: afferent visual system damage after optic neuritis in MOG-IgG-seropositive versus AQP4-IgG-seropositive patients. J Neuroinflammation. 2016;13:282.
Havla J, Kumpfel T, Schinner R, Spadaro M, Schuh E, Meinl E, et al. Myelin-oligodendrocyte-glycoprotein (MOG) autoantibodies as potential markers of severe optic neuritis and subclinical retinal axonal degeneration. J Neurol. 2017;264:139–51.
Kim SM, Woodhall MR, Kim JS, Kim SJ, Park KS, Vincent A, et al. Antibodies to MOG in adults with inflammatory demyelinating disease of the CNS. Neurol Neuroimmunol Neuroinflamm. 2015;2:e163.
Salama S, Khan M, Shanechi A, Levy M, Izbudak I. MRI differences between MOG antibody disease and AQP4 NMOSD. Mult Scler. 2020:1352458519893093. [Epub ahead of print].
Shahriari M, Yousem DM, Sotirchos ES, Newsome SD. MOG antibody associated disease: how it differs from and resembles other neuroinflammatory disorders. Am J Roentgenol. 2020. https://doi.org/10.2214/AJR.20.24061 [Epub ahead of print].
Dubey D, Pittock SJ, Krecke KN, Morris PP, Sechi E, Zalewski NL, et al. Clinical, radiologic, and prognostic features of myelitis associated with myelin oligodendrocyte glycoprotein autoantibody. JAMA Neurol. 2019;76:301–9.
Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. J Neuroinflammation. 2016;13:280.
Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 1: Frequency, syndrome specificity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J Neuroinflammation. 2016;13:279.
Mariotto S, Gajofatto A, Batzu L, Delogu R, Sechi G, Leoni S, et al. Relevance of antibodies to myelin oligodendrocyte glycoprotein in CSF of seronegative cases. Neurology. 2019;93:e1867–72.
Reindl M, Schanda K, Woodhall M, Tea F, Ramanathan S, Sagen J, et al. International multicenter examination of MOG antibody assays. Neurol Neuroimmunol Neuroinflamm. 2020;7:e674.
Cobo-Calvo A, Sepulveda M, d’Indy H, Armangue T, Ruiz A, Maillart E, et al. Usefulness of MOG-antibody titres at first episode to predict the future clinical course in adults. J Neurol. 2019;266:806–15.
Hyun JW, Woodhall MR, Kim SH, Jeong IH, Kong B, Kim G, et al. Longitudinal analysis of myelin oligodendrocyte glycoprotein antibodies in CNS inflammatory diseases. J Neurol Neurosurg Psychiatry. 2017;88:811–7.
Waters P, Fadda G, Woodhall M, O’Mahony J, Brown RA, Castro DA, et al. Serial anti-myelin oligodendrocyte glycoprotein antibody analyses and outcomes in children with demyelinating syndromes. JAMA Neurol. 2020;77:82–93.
Chen JJ, Tobin WO, Majed M, Jitprapaikulsan J, Fryer JP, Leavitt JA, et al. Prevalence of myelin oligodendrocyte glycoprotein and aquaporin-4-IgG in patients in the optic neuritis treatment trial. JAMA Ophthalmol. 2018;136:419–22.
Stiebel-Kalish H, Hellmann MA, Mimouni M, Paul F, Bialer O, Bach M, et al. Does time equal vision in the acute treatment of a cohort of AQP4 and MOG optic neuritis? Neurol Neuroimmunol Neuroinflamm. 2019;6:e572.
Ramanathan S, Mohammad S, Tantsis E, Nguyen TK, Merheb V, Fung VSC, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. J Neurol Neurosurg Psychiatry. 2018;89:127–37.
Noseworthy JH, O’Brien PC, Petterson TM, Weis J, Stevens L, Peterson WK, et al. A randomized trial of intravenous immunoglobulin in inflammatory demyelinating optic neuritis. Neurology. 2001;56:1514–22.
Roed HG, Langkilde A, Sellebjerg F, Lauritzen M, Bang P, Morup A, et al. A double-blind, randomized trial of IV immunoglobulin treatment in acute optic neuritis. Neurology. 2005;64:804–10.
Hacohen Y, Wong YY, Lechner C, Jurynczyk M, Wright S, Konuskan B, et al. Disease course and treatment responses in children with relapsing myelin oligodendrocyte glycoprotein antibody-associated disease. JAMA Neurol. 2018;75:478–87.
Li S, Ren H, Xu Y, Xu T, Zhang Y, Yin H, et al. Long-term efficacy of mycophenolate mofetil in myelin oligodendrocyte glycoprotein antibody-associated disorders: a prospective study. Neurol Neuroimmunol Neuroinflamm. 2020;7:e705.
Chen JJ, Flanagan EP, Bhatti MT, Jitprapaikulsan J, Dubey D, Lopez Chiriboga ASS, et al. Steroid-sparing maintenance immunotherapy for MOG-IgG associated disorder. Neurology. 2020;95:e111–20.
Hayward-Koennecke H, Reindl M, Martin R, Schippling S. Tocilizumab treatment in severe recurrent anti-MOG-associated optic neuritis. Neurology. 2019;92:765–7.
Costanzi C, Matiello M, Lucchinetti CF, Weinshenker BG, Pittock SJ, Mandrekar J, et al. Azathioprine: tolerability, efficacy, and predictors of benefit in neuromyelitis optica. Neurology. 2011;77:659–66.
Jacob A, Matiello M, Weinshenker BG, Wingerchuk DM, Lucchinetti C, Shuster E, et al. Treatment of neuromyelitis optica with mycophenolate mofetil: retrospective analysis of 24 patients. Arch Neurol. 2009;66:1128–33.
Montcuquet A, Collongues N, Papeix C, Zephir H, Audoin B, Laplaud D, et al. Effectiveness of mycophenolate mofetil as first-line therapy in AQP4-IgG, MOG-IgG, and seronegative neuromyelitis optica spectrum disorders. Mult Scler. 2017;23:1377–84.
Damato V, Evoli A, Iorio R. Efficacy and safety of rituximab therapy in neuromyelitis optica spectrum disorders: a systematic review and meta-analysis. JAMA Neurol. 2016;73:1342–8.
Chen JJ, Pittock SJ, Flanagan EP, Lennon VA, Bhatti MT. Optic neuritis in the era of biomarkers. Surv Ophthalmol. 2020;65:12–7.
Acknowledgements
The authors thank Paul Honermann (Mayo Clinic, Rochester, Minnesota) for providing Fig. 1 illustration.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gospe, S.M., Chen, J.J. & Bhatti, M.T. Neuromyelitis optica spectrum disorder and myelin oligodendrocyte glycoprotein associated disorder-optic neuritis: a comprehensive review of diagnosis and treatment. Eye 35, 753–768 (2021). https://doi.org/10.1038/s41433-020-01334-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-020-01334-8
This article is cited by
-
Visualization and analysis of mapping knowledge domains for optic neuritis: a bibliometric research from 2013 to 2022
International Ophthalmology (2024)
-
The neuro-ophthalmological manifestations of NMOSD and MOGAD—a comprehensive review
Eye (2023)
-
Update on glial antibody-mediated optic neuritis
Japanese Journal of Ophthalmology (2022)
-
Emerging concepts in the treatment of optic neuritis: mesenchymal stem cell-derived extracellular vesicles
Stem Cell Research & Therapy (2021)
-
Bortezomib: a proteasome inhibitor for the treatment of autoimmune diseases
Inflammopharmacology (2021)
