
Abstract
ARL13B encodes for the ADP-ribosylation factor-like 13B GTPase, which is required for normal cilia structure and Sonic hedgehog (Shh) signaling. Disruptions in cilia structure or function lead to a class of human disorders called ciliopathies. Joubert syndrome is characterized by a wide spectrum of symptoms, including a variable degree of intellectual disability, ataxia, and ocular abnormalities. Here we report a novel homozygous missense variant c.[223G>A] (p.(Gly75Arg) in the ARL13B gene, which was identified by whole-exome sequencing of a trio from a consanguineous family with multiple-affected individuals suffering from intellectual disability, ataxia, ocular defects, and epilepsy. The same variant was also identified in a second family. We saw a striking difference in the severity of ataxia between affected male and female individuals in both families. Both ARL13B and ARL13B-c.[223G>A] (p.(Gly75Arg) expression rescued the cilia length and Shh defects displayed by Arl13b hennin (null) cells, indicating that the variant did not disrupt either ARL13B function. In contrast, ARL13B-c.[223G>A] (p.(Gly75Arg) displayed a marked loss of ARL3 guanine nucleotide-exchange factor activity, with retention of its GTPase activities, highlighting the correlation between its loss of function as an ARL3 guanine nucleotide-exchange factor and Joubert syndrome.
Similar content being viewed by others


Introduction
The ARL13B gene (OMIM 608922) encodes the ADP-ribosylation factor-like protein 13B [1, 2], a regulatory GTPase involved in diverse cellular functions. ARL13B localizes to primary cilia and plays roles in cilia length regulation, cilia architecture, and cilia-dependent signaling [3,4,5,6]. Loss of Arl13b, as in the Arl13b hnn mouse [7], causes aberrant Sonic hedgehog (Shh) signaling and abnormal specification of cell fate in the developing spinal cord [1, 2, 6]. Additionally, Arl13b is critical during brain development, both in the initial formation of a polarized radial glial scaffold and for interneuron migration. Disruption of these processes may lead to altered neurodevelopment and brain abnormalities [7,8,9]. GTPase-activating proteins (GAP) or guanine nucleotide-exchange factors (GEF) interact with other GTPases and increase the rate of GTP hydrolysis or the dissociation of GDP from GTPase, respectively [10, 11]. ARL13B is active as a GEF for another ARF family GTPase, ARL3 [5].
Disruption in the structure or function of cilia can cause a wide spectrum of disorders known as ciliopathies. Variants in ARL13B cause a ciliopathy known as Joubert syndrome (JS) [1, 3], a genetically heterogeneous autosomal recessive or X-linked disorder characterized by ataxia, developmental delay, hypotonia, ocular motor apraxia, and a variable degree of intellectual disability [1, 3, 12, 13]. A malformation of the midbrain–hindbrain junction, known as the “molar tooth sign,” is a characteristic neuroimaging hallmark of JS [13, 14]. Variable features include retinal dystrophy, coloboma, polydactyly, cystic renal disease, hepatic fibrosis, and other brain malformations [15]. Previous studies reported compound heterozygous (c.[246G>A] (p.(Trp82Ter); c.[598C>T] (p.(Arg200Cys)) and homozygous (c.[65T>G] (p.(Val22Gly); c.[236G>A] (p.(Arg79Gln); c.[246G>A] (p.(Trp82Ter); c.[257A>G] (p.(Tyr86Cys); c.[259A>G] (p.(Ile87Val); c.[461A>G] (p.(Asn154Ser); c.[599G>A] (p.(Arg200His); and c.[765T>A] (p.(Asn255Lys)) variants in the ARL13B gene in patients with Joubert syndrome [1, 3, 16,17,18]. Here we identify a novel homozygous variant c.[223G>A] (p.(Gly75Arg) in five individuals with intellectual disability, epilepsy, and ataxia from two Pakistani families and, using cell culture and biochemical analysis, reveal the functional relevance of this novel variant.
Materials and methods
Phenotype of the patients
We describe two consanguineous families from Baluchistan, Pakistan. Patients from both families have intellectual disability (ID) and ocular abnormalities.
Family A
Patient IV:2 is a 24-year-old male with a severe form of ID and epilepsy (Fig. 1a). He started to sit at the age of 5 years and walked at the age of 9 years. Epileptic seizures began at the age of 9 years, with episodes that lasted for around 10 min and came along with urination. He is able to walk independently but does not have complete control of his movement. The second affected individual (IV:6; Fig. 1a) is a 13-year-old female with profound ID and epilepsy. She started sitting at the age of 6 years but was still unable to walk at the age of 13 years. Epileptic seizures started at the age of 9 years, lasting for around 10 min, and she also urinated during seizures. She behaved aggressively toward other family members. The third patient (IV:7; Fig. 1a) of this family is an 8-year-old male. He suffered from a severe form of ID but so far no epileptic seizures have been noticed. He was unable to sit until the age of 5 years. He was able to walk at 8 years of age. All affected individuals had normal birth and showed delayed motor skills and delayed developmental milestones and ataxia. They were unable to speak. They had no control of their eye movements and were completely dependent on others for food, dressing, cleaning, and other basic needs.

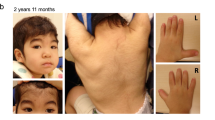
Pedigrees, pictures, and CT scan of the families studied. a Family A with three affected individuals. Whole-exome sequencing of the trio was performed, indicated by red arrows. b Family B with two affected individuals. The asterisk indicates individuals who could be analyzed; squares indicate males and circles females. Affected individuals are marked by filled symbols, and symbols crossed through indicate deceased individuals. c Conservation of the ARL13B across different species in the region adjacent to the Gly75 variant. The sequence alignment was obtained using the Clustal Omega protein sequence alignment (http://www.ebi.ac.uk/tools/msa/clustalo/). The sequence alignment spans amino acid positions 70–87 of ARL13B NCBI, Protein Accession number AAI04037.1. d CT scans are available from patients IV:2 and IV:6 of Family A. From left to right, the arrows in the first column (1) indicate a vermal cleft as a sign of a vermis defect. In (2), the arrows indicate the umbrella-shaped fourth ventricle that is typical in Joubert syndrome (JS). In (3), the molar tooth sign is encircled (anterior deep interpeduncular fossa and splayed elongated superior cerebellar peduncles). The asterisk in (4) marks an enlarged supravermian cistern (only visible in the female)
Family B
Both patients (V:4 and V:6; Fig. 1b) had a mild to moderate form of ID and epilepsy. After a normal birth, they showed delayed developmental milestones and ataxia. At the time of patient recruitment, they were able to speak only a few words. They had no control over their eye movements. Patient V:4 (Fig. 1b) is a 17-year-old male. He started sitting at the age of 2 years and walked independently at the age of 5 years. Patient V:6 (Fig. 1b) is a 15-year-old female. She started sitting at the age of 5 years and is unable to walk at the age of 15 years.
The patients from our study have no diagnosis for retinopathy, renal anomalies, or hepatic fibrosis, but these additional symptoms cannot be completely ruled out due to the limited availability of the patients for further diagnostic tests.
Whole-exome sequencing (WES) and Sanger sequencing
WES was performed on the genomic DNA as described previously [19]. Reads were mapped to 1000 genome reference GRCh37 using BWA 0.6.2 [20]. The proband (IV:2) had an on-target average coverage of 206.64× and parents III:5 and III:6 had an on-target coverage of 106.10× and 123.45×, respectively. SNVs and indels were called using SAM [21] tools and Platypus [22], respectively, and annotated with Gencode exonic classing using ANNOVAR [23].
In silico prediction programs (Mutation taster, SIFT, PolyPhen2, PROVEAN, and CADD obtained from dbNSFP [24]) were used to analyze a putative functional effect of variants. Selected variants were further considered and validated by Sanger sequencing using ABI3730xl, as described previously [19].
Homology modeling
Both Arl13b orthologs of Chlamydomonas reinhardtii (CrArl13) and Homo sapiens (hARL13B) consist of a GTP-binding domain, a coiled-coil (CC) domain, and a proline–arginine–arginine (PRR) repeat region. CrArl13 and human ARL13B share 42% sequence identity between amino acid residue 20–217 [25, 26]. The crystal structure of CrArl13 (PDB ID 5DI3, chain B) was used as a template for modeling the three-dimensional structure of the human ARL13B. The 5DI3 structure was crystallized in a hetero-oligomeric form, displaying interactions of CrArl13 with Arl3 protein (5DI3, chain A). Modeling of the wild-type human proteins was performed using MODELLER 9v10 [27, 28]. The c.[223G>A] (p.(Gly75Arg) variant of hARL13B was generated using the PyMOL Mutagenesis tool (Schrödinger, LLC, http://pymol.org/). The ligand GppNHp (a non-hydrolysable GTP analog) was imported from the template structure. The model of the human complex was generated by superposition of the individual proteins on the crystal structure of the complex of CrArl13 and CrArl3.
Animals
The cerebella of CD1 mice were dissected at embryonic stage (E) 17.5 and postnatal stages (P) P1.5 and P7.5.
Cell culture
Human neuroblastoma (SH-SY5Y) cells were grown in 75-cm [2] flasks in Dulbecco’s modified Eagle’s medium (Thermo Fisher Scientific) supplemented with 10% fetal calf serum, 1% non-essential amino acids, and 1% penicillin–streptomycin at 37 °C in a humidified environment with 5% CO2. Cells were split at 80–90% confluency and plated into six-well plates in phenol red-free medium containing 10% charcoal dextran-treated calf serum for 24 h. Cells were treated with either mock or 100 nM dihydrotestosterone (DHT) (Sigma-D-073-1ML) and mock plus 1 µM flutamide (Sigma F9397 – 1G) or 100 nM DHT together with 1 µM flutamide and harvested after 4 h of treatment.
Generation of stable mouse embryonic fibroblast lines
Arl13b hnn immortal mouse embryonic fibroblasts (MEFs) [7] were stably transfected with ARL13B wildtype (a gift from Eva Anton [9]) or c.[223G>A] (p.(Gly75Arg) variant (generated by the Emory Integrated Genomics Core) plasmid. Stable clonal populations were tested by western blot for ARL13B expression as described [29], and two clones from each construct were chosen for further analysis.
Western blots
Cell lysis was performed using modified RIPA buffer plus SIGMAFAST protease inhibitors (S8820) and western blots were performed as described previously [29]. Briefly, proteins (20 µg/sample) were resolved using Mini-PROTEAN TGX Stain-Free Precast Gels (Bio-Rad 4568034) and imaged after activation using the ChemiDoc Touch Imaging System (Bio-Rad). After transfer to nitrocellulose membrane and antibody incubation, the blot was imaged for chemiluminescence using the ChemiDoc. Bands visible on this second image were normalized to total protein as measured on the first (gel) image and analysis was performed using ImageLab softward (Bio-Rad).
Immunofluorescence staining
Cells were grown on coverslips and fixed in 4% paraformaldehyde for 10 min at room temperature, followed by fixation in 100% methanol for 15 min at −20°C, and then blocked for 1 h in 10% heat-inactivated goat serum containing 0.1% Triton X-100 and 1% bovine serum albumin. Cells were labeled with primary antibodies against acetylated alpha-tubulin (Sigma T6793, 1:1000) and anti-ARL13B (Proteintech 17711-1-AP, 1:400) for 80 min at room temperature, followed by three times 5 min washes with blocking buffer. Secondary antibodies (anti-mouse Alexa Fluor-568, 1:500, and anti-rabbit Alexa Fluor-488, 1:200, Thermo Fisher Scientific) were applied with Hoechst nuclear stain (1:3000) to counterstain nuclei for 45 min at room temperature, followed by three times 5 min washes with blocking buffer. Coverslips were mounted with ProLong Gold Antifade Reagent (Thermo Fisher Scientific).
Fluorescent micrographs were acquired with a ×40 objective on a Leica DM6000B microscope using SimplePCI software. For experiments quantifying the number of cilia, microscopy fields for imaging were chosen based only on Hoechst staining to ensure unbiased sampling of ciliated versus non-ciliated cells.
Images were analyzed using the Fiji distribution of the NIH ImageJ software [30]. For experiments testing the ciliogenesis rate of different MEF lines, nuclei were counted based on Hoechst staining, and cilia were counted based on acetylated α-tubulin staining.
Quantitative real-time PCR for analysis of mouse embryonic fibroblasts
MEFs were plated in six-well plates for 24 h at densities of 3 × 105 cells/well and were treated with 0.5% serum control medium or Shh-conditioned medium [2, 31, 32]. After 24 h, MEFs were harvested. RNA was isolated and cDNA was synthesized as described previously [29]. qRT-PCR was performed on a CFX96 cycler (Bio-Rad) using SsoAdvanced reagent (Bio-Rad) to amplify Shh target genes Gli1 and Ptch1 and reference gene Pold3, as described previously [29]. Primers sequences are given in Supplementary Table 1.
Quantitative real-time PCR for expression analysis of Arl13b in mouse cerebellum and SH-SY5Y cells
RNA was isolated using peqGOLD TriFastTM (PEQLAB-Life Science). First-strand cDNA synthesis was performed with either 2 µg (CD1 mice) or 1 µg (SH-SY5Y cells) of RNA using SuperScript II and Oligo (dT)12-18 primer (Thermo Fisher Scientific) according to the manufacturer’s instructions. Quantitative real-time PCR (RT-PCR) was performed with the qTOWER (Analytik Jena) using the Sensi FAST SYBR No-ROX Kit (Bioline). In mice, Hprt1 and Sdha1 and in human SH-SY5Y cells, HPRT1, SHDA1, GAPDH, HSPD1, and 18 S were selected as reference genes. Primers sequences are given in Supplementary Table 1.
Mammalian cell human embryonic kidney 293T expression and purification
The open reading frame encoding human ARL13B (NM_182896.2, 428 residues), the N-terminal 19-residue truncation mutant Δ19-ARL13B, or Δ19-ARL13B-c.[223G>A] (p.(Gly75Arg) were inserted into the pLEXm-GST expression vector, for expression of each as N-terminal GST fusion proteins. This vector includes a TEV cleavage site just upstream of the inserted open reading frame, which allows removal of the GST tag. The inserted open reading frames were completely sequenced to confirm they were correct.
Human embryonic kidney 293T (HEK) cells were grown, transfected and proteins were purified as described [29].
Arl13b guanine nucleotide-activating protein (GAP) assay
The intrinsic and GAP-stimulated GTPase activities of purified, recombinant murine Arl13b proteins were determined using the GAP assay described previously for ARL2 [29, 33, 34]. Briefly, GST-ARL13B, GST- Δ19-ARL13B, or GST-Δ19-ARL13B-c.[223G>A] (p.(Gly75Arg) was preloaded with [γ-32P]GTP at 30 oC in loading buffer. Intrinsic GTPase and GAP-stimulated activities were determined as described [29]. The experiments were repeated at least twice with at least two different preparations of each protein, performed in triplicate.
Arl13b guanine nucleotide-exchange factor (GEF) assay
The ability of purified, recombinant human GST-ARL13B, GST-Δ19-ARL13B, or GST-Δ19-ARL13B-c.[223G>A] (p.(Gly75Arg) to serve as a GEF for ARL3 was determined using a modification of the assay described in refs. [5, 29]. Purified recombinant human ARL3 was incubated along with 1 µM GST-ARL13B, GST-Δ19-ARL13B, or GST-Δ19-ARL13B-c.[223G>A] (p.(Gly75Arg). The rate of release of preloaded [3H]GDP from ARL3 was determined after stopping the reaction by dilution of 10 µl reaction cocktail into 2 ml of ice-cold buffer, followed promptly by filtration through BA85 nitrocellulose filters (0.45 μm, 25 mm (Whatman)), as described previously [35]. Binding was quantified using a liquid scintillation counter. The experiments were repeated at least twice with at least two different preparations of each protein, performed in duplicate, and no differences were observed between preparations with and without removal of the GST tag. To monitor the effects of activation (GTP-binding) of ARL13B on its ARL3 GEF activity, the ARL13B protein preparations were preincubated with GTP (final concentration, 100 µM) prior to addition into the GEF assay.
Statistics
The expression of Arl13b was analyzed using Microsoft Office Excel software and IBM SPSS Statistics. Prism 6 (GraphPad software) was used to analyze the MEFs results and the biochemical assays. Outliers in the data were determined by IBM SPSS Statistics and excluded from further analysis. Two-tailed Student’s t test, Dunnett’s comparison test, and one-way ANOVA or two-way ANOVA served to compare differences between the two groups. P values of ≤0.05 were considered significant. All data are presented as mean ± standard error.
Results
Genetic analysis
We investigated a large consanguineous Pakistani family with three individuals presenting with ID, ataxia, developmental impairment, epileptic seizures, and ocular abnormalities. To elucidate the underlying genetic cause of the disorder in this family, we performed WES on a selected trio (Family A, Fig. 1a). After filtering, we obtained 59 variants. Analysis with different in silico prediction programs revealed 33 variants that putatively affect function.
We prioritized variants in genes that were (a) expressed in brain (UniGene, Human Protein Atlas, Allen Brain Atlas), (b) associated with ID and other neurological disorders, or (c) variants residing in highly conserved protein regions for further analysis. According to these criteria, we selected seven homozygous variants from seven different genes for further validation by Sanger sequencing.
To clarify a putative contribution of the variants to disease, we Sanger sequenced three affected individuals (IV:2, IV:6, and IV:7), their healthy parents (III:5 and III:6), and unaffected siblings (IV:4 and IV:5) from Family A (Fig. 1a). Only the homozygous missense variant c.[223G>A] (p.(Gly75Arg) in the ARL13B gene co-segregated with the phenotype https://databases.lovd.nl/shared/individuals/00105223 (variant IDs 0000171265 and /0000171266). We sequenced additional individuals with intellectual disability and developmental delay from another 21 consanguineous Pakistani families and found the identical variant c.[223G>A] (p.(Gly75Arg) in two affected individuals of an apparently unrelated second family (Family B, Fig. 1b). The variant also co-segregated with the phenotype in this family.
In total, we identified the homozygous c.[223G>A] variant in five affected individuals from two families. This missense variant c.[223G>A] (p.(Gly75Arg) was predicted to affect protein function by all in silico programs used, likely due to its location in a highly conserved region of the protein (Fig. 1c). Moreover, the variant is not present in dbSNP (URL: http://www.ncbi.nlm.nih.gov/SNP/) (accessed June 2016) and the gnomAD browser (URL: http://gnomad.broadinstitute.org/) (accessed July 2017) with more than 15,000 South Asian individuals including more than 10,000 Pakistani individuals [36, 37]. A SNP analysis in the 27 Mbp region of homozygosity around the ARL13B gene argues in favor of a founder mutation (data not shown).
Because the ARL13B gene is known to be mutated in patients with JS [1, 3], we examined the clinical phenotype of our patients. Two affected individuals from Family A (IV:2 and IV:6; Fig. 1a) were available for a CT scan of the brain. We observed malformations of the cerebellum: a vermal cleft, an umbrella-shaped fourth ventricle, and the molar tooth sign, the diagnostic criterion for JS (Fig. 1d). In addition, we detected an enlarged supravermian cistern in the female patient only (Fig. 1d). Patients from Family B were unavailable for CT scan.
ARL13B expression analysis
As we noted phenotypic (ataxia) and anatomical (brain) differences between male and female patients, we decided to investigate whether Arl13b gene expression differs during brain development in male and female mice. First, we compared the gene expression levels in the cerebella of male and female mice at embryonic (E) and postnatal (P) stages, i.e., E17.5, P1.5, and P7.5, as the cerebellum is the most affected brain region in JS. We detected an almost 20% higher expression of Arl13b at E17.5 and P1.5 in female mice compared to males, but the values were not statistically significant (Supplementary Fig. 1A). Second, we analyzed whether ARL13B expression is influenced by the sex hormone dihydrotestosterone (DHT). Using human neuroblastoma SH-SY5Y cells, we analyzed ARL13B expression after 4 h of DHT stimulation or mock treatment. ARL13B was significantly downregulated in DHT-treated cells (Supplementary Fig. 1B), indicating a regulatory effect on ARL13B expression. We also treated SH-SY5Y cells with either mock and flutamide (androgen receptor (AR) antagonist) or flutamide together with DHT for 4 h, which abolished the DHT effect on ARL13B gene expression (Supplementary Fig. 1C). These data point to an androgen receptor-dependent regulation of ARL13B gene expression by DHT.
ARL13B 3D structure analysis
The domain architecture of ARL13B is shown in Fig. 2a. We used the crystal structure of CrArl13 from Chlamydomonas reinhardtii as a template to model the three-dimensional (3D) structure of the human ortholog. The 3D model of the ARL13B G-domain is shown in Fig. 2b. The regions of the protein which are directly involved in protein–protein interactions (e.g., to effectors or GAPs) are highlighted in color and are the P-loop, Switch I, and Switch II. The previously reported ARL13B variants (p.(Arg79Gln), p.(Trp82Ter), and p.(Try86Cys)) [1, 3] and our newly identified variant p.(Gly75Arg) are located in Switch II (Fig. 2c). In fact, residue p.Gly75 is located right at the boundary of the G-4 consensus GTP-binding motif (DXXGQ71 in the ARF family, but DXXGG75 in ARL13B) and the guanine nucleotide-sensitive Switch II region. ARL13B forms a hetero-dimer by interacting with ARL3. The modeled structure of this complex (Fig. 2d) indicates that ARL13B facilitates the release of GDP nucleotide from ARL3 through allosteric modulation. Replacing small amino acid glycine by the positively charged arginine residue at position 75 may result in an increased polar interaction with the negatively charged aspartic acid residue (D30), which could result in a conformational change in Switch II and the P-loop. These conformational changes may in turn lead to a chain of effects that may result in the loss of polar contacts between Arg79 and Glu106, as well as between Arg103 and Asp30. Thus, the c.[223G>A] (p.(Gly75Arg) variant might disrupt the binding of ARL13B to ARL3 and result in the loss of the allosteric effect caused by ARL13B binding. Another possible mechanism could be that, compared to CrArl13 the c.[223G>A] (p.(Gly75Arg) mutant contains two positively charged residues, Arg75 and Arg77 which are located at the interface (Fig. 2e) and create a positive electrostatic potential, that may disturb binding with ARL3, leading to the loss of GEF activity.

Illustration of the ARL13B domain architecture (Q3SXY8 UniProt) and 3D model of the entire ARL13B G-domain, to which the non-hydrolysable GTP analog GppNHp is bound. a The GTP binding-, coiled-coiled, and proline-rich domains are indicated in black. Previously reported variants in patients with Joubert syndrome are indicated in blue; the variant identified in the current study is indicated in red. b The GTP-binding region is highlighted in colors: Switch I in pink, Switch II and the α2-helix in red, and the P-loop in green. c Illustration of all known Joubert syndrome variant sites in the G-domain of ARL13B. Red sticks represent previously reported mutation sites, whereas yellow sticks indicate the position of our new variant p.(Gly75Arg). d The superimposition of mutant p.(Gly75Arg) of human ARL13B (gray) and hARL3 (green) on the crystal structure of the CrArl13 complex is shown. The GTP analog is shown in gray. Residues forming polar contacts are shown in yellow stick representation and polar contacts are shown by yellow dashed lines. e Close-up of the protein–protein interface showing Arg75 (white stick) and polar contacts between α1-helix, α2-helix, and P-loop residues in hARL13B in yellow stick representation. The important residues in CrArl13 in the Switch II and Arg77 in α2-helix region are shown in green stick representation
Functional analysis of the p.(Gly75Arg) variant in Arl13b hnn mouse embryonic fibroblasts MEFs
To determine the functional consequences of ARL13B-c.[223G>A] (p.(Gly75Arg) in vitro, we performed rescue experiments using MEFs from the protein-null Arl13b hennin(hnn) mouse model [6]. Arl13b hnn MEFs display defects in ciliation, cilia length, and in the transcriptional response to Shh [2]. We stably transfected human wild-type ARL13B or ARL13B-p.(Gly75Arg) into Arl13b hnn MEFs and obtained cell lines displaying low or high levels of proteins for each construct (Supplementary Fig. 2A). We compared the percentage of ciliated cells and cilia length between wild-type ARL13B and ARL13B-p.(Gly75Arg)-expressing cells. We found no statistically significant differences in cilia length or percentage of ciliated cells between the wild type and mutant-expressing lines regardless of construct expression level (Supplementary Fig. 2B, C). Next, we analyzed the Shh response by quantifying the expression of two Shh transcriptional targets, Gli1 and Ptch1, in the MEF lines with and without Shh stimulation. As expected, the control untransfected Arl13b hnn MEFs did not respond to Shh stimulation, consistent with the known role of ARL13B in regulating Shh response. Both wild-type ARL13B and ARL13B-p.(Gly75Arg) rescued the Shh-dependent stimulation of Gli1 and Ptch1 transcription. Interestingly, the high-expressing ARL13B-p.(Gly75Arg) MEFs had significantly higher Gli1 expression compared to high-expressing wild-type ARL13B MEFs, indicating a stronger Shh response for the mutant. In contrast, Ptch1 expression did not show any difference between wild-type and mutant ARL13B (Supplementary Fig. 2D). Taken together, these data indicate that the c.[223G>A] (p.(Gly75Arg) variant does not interfere with MEF ciliation, cilia length, or Shh response.
Analysis of Arl13b GTPase and GEF function
To assess whether the p.(Gly75Arg) variant had any impact on a specific biochemical protein function, we expressed recombinant versions of human ARL13B as GST fusion proteins in human embryonic kidney (HEK293T) cells and affinity purified the proteins for biochemical analyses, as described previously [38]. We generated both full-length human ARL13B and the N-terminal 19-residue truncation mutants (Δ19-ARL13B and Δ19-ARL13B-c.[223G>A] (p.(Gly75Arg) based on results from previously published biochemical analyses [39]. Based upon data from ARF1 [40,41,42], the prototype of the ARF family, it was predicted that deletion of the N-terminal 19 residues would promote phospholipid-independent exchange of guanine nucleotides, though it later was found to have no impact on ARL13B biochemical properties [43]. Each of these three protein preparations were similar in purity after affinity purification on glutathione-Sepharose beads, as previously described [29, 38] (Fig. 3a). First, we determined the intrinsic and GAP-stimulated GTPase activity of each of these preparations using well-established assays in the GTPase field. We observed no differences in the rates of intrinsic or ARL13B GAP-stimulated GTP hydrolysis among any of the analyzed proteins (Fig. 3b).

ARL13B-p.(Gly75Arg) is inactive as a GEF for ARL3, while retain the GTPase activities. a GST-ARL13B, GST-Δ19ARL13B, and GST-Δ19ARL13B-p.(Gly75Arg) were expressed in and purified from HEK cells. Each preparation (2 µg) was analyzed with SDS-PAGE by Coomassie blue (Sigma B0149) staining. Molecular weight standards (Precision Plus; Bio-Rad 161-0373) are shown on the left with indicated sizes. b No differences in intrinsic or GAP-stimulated GTPase activities were detected among GST-RL13B, GST-Δ19ARL13B, and GST-Δ19ARL13B-p.(Gly75Arg). c ARL13B is active as a GEF for ARL3, while ARL13B-p.(Gly75Arg) is inactive. ARL3 (1 µM) was loaded with [3H]GDP prior to use as substrate in the GEF assay, with the 1 µM GST-ARL13B, GST-Δ19ARL13B, or GST-Δ19ARL13B-p.(Gly75Arg) preloaded with 100 µM of GTP. Each analysis was performed in duplicate and repeated at least twice using different ARL13B protein preparations
Second, the same proteins were assayed for GEF activity against human ARL3, as previously described [29]. GST-ARL13B and GST-Δ19-ARL13B displayed indistinguishable activities in the ARL3 GEF assay (Fig. 3c), and these activities were not altered when GST was removed, indicating that neither deletion of the N-terminal 19 residues nor the GST fusion at the N-terminal interfered with ARL3 GEF activity. In contrast, the GST-Δ19-ARL13B-p.(Gly75Arg) displayed a marked loss in ARL3 GEF activity. Together, these results demonstrate the selective loss of one specific ARL13B function, ARL3 GEF activity, with retention of two other functions: the intrinsic and the GAP-stimulated GTP hydrolysis [43].
Discussion
Using whole-exome and Sanger sequencing, we identified a novel homozygous missense variant c.[223G>A] (p.(Gly75Arg) in the ARL13B gene in five affected individuals from two consanguineous Pakistani families, thereby increasing the number of reported Joubert syndrome patients with variants in ARL13B. Consistent with previously described individuals with JS who carry ARL13B variants [1, 3], the patients from our study showed global developmental delay, intellectual disability, and abnormal eye movements (Supplementary Table 2); additional phenotypic features included epilepsy and strikingly, the two affected females from both families presented with severe ataxia and were still unable to walk at the ages of 13 and 15 years, whereas the affected males started to walk at the ages of 5, 8, and 9 years. The enlarged supravermian cistern detected in the CT scan of one female, which was not visible in her affected brother, points to a more pronounced cerebellar malformation in the female and might explain the more pronounced ataxia problems. Although Arl13b did not show a significant sex-dimorphic expression pattern in the mouse cerebellum at early stages of development, we uncovered a regulatory effect by the sex hormone DHT in a human neuroblastoma cell line (SH-SY5Y). As testosterone levels differ markedly during early development of the male and female brain [44], this might also influence Arl13b expression or other downstream mechanisms. Previous studies of JS with ARL13B variants did not report on severity differences between male and female patients [1, 3], however the elder sister of the two affected females of the Pakistani family MTI-001 died for unknown reasons [1]. The sex difference we saw may be considered a coincidental finding due to the low number of individuals investigated (two females, three males), but we also cannot rule out that this is a gene- or variant-related finding meriting follow-up.
The c.[223G>A] (p.(Gly75Arg) variant in ARL13B is located directly at the boundary of the G-4 motif and Switch II, an important protein region for GTP binding and hydrolysis in all regulatory GTPases. The mutated protein, however, behaved in our biochemical assays like wild-type protein in terms of intrinsic and GAP-stimulated GTPase activity, indicating that the mutated protein is not altered in these in vitro assays of ARL13B functionality. Moreover, the c.[223G>A] (p.(Gly75Arg) variant showed a normal Shh response in our assays of ciliary functions in MEF cells, which is consistent with other JS variants in ARL13B [29]. As the complete loss of Arl13b is embryonic lethal [7], we would expect that only subtle alterations in Shh signaling are compatible with life. We know that in vivo, cells integrate the concentration and duration of Shh signal, whereas our assays in MEFs only examine Shh stimulation after 24 h, so we may need to examine JS alleles in in vivo models to fully appreciate their functional consequences.
ARL13B acts as a GEF for ARL3 [5, 43], which plays a role in the release of ciliary cargo from transport proteins, a process necessary for proper cilia function [5]. Interestingly, the c.[223G>A] (p.(Gly75Arg) variant showed marked loss of function in the GEF assay for ARL3, indicating that the variant disrupts this particular ARL13B function. These data argue that the current best in vitro model for disease-causing changes in ARL13B function is to be found in ARL3 GEF activity. In addition, the c.[223G>A] (p.(Gly75Arg) variant was predicted to result in a conformational change in Switch II of ARL13B, which is part of the ARL13B-ARL3 interaction interface [5, 25]. We therefore conclude that an impaired protein–protein interaction underlie the loss of ARL13B-GEF activity toward ARL3.
Taken together, our findings provide further support for the involvement of ARL13B in Joubert syndrome and suggest an extension of the phenotypic spectrum of ARL13B variants. The functional loss of the c.[223G>A] (p.(Gly75Arg) variant is in line with another reported JS-causing ARL13B variant p.(Arg79Gln), which leads to the loss of ARL13B-GEF activity, pointing to ARL13B-GEF activity as the most clinically relevant function [43].
References
Cantagrel V, Silhavy JL, Bielas SL, et al Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am J Hum Genet. 2008;83:170–9.
Larkins CE, Aviles GD, East MP, Kahn RA, Caspary T. Arl13b regulates ciliogenesis and the dynamic localization of Shh signaling proteins. Mol Biol Cell. 2011;22:4694–703.
Thomas S, Cantagrel V, Mariani L, et al Identification of a novel ARL13B variant in a Joubert syndrome-affected patient with retinal impairment and obesity. Eur J Hum Genet. 2015;23:621–7.
Lu H, Toh MT, Narasimhan V, Thamilselvam SK, Choksi SP, Roy S. A function for the Joubert syndrome protein Arl13b in ciliary membrane extension and ciliary length regulation. Dev Biol. 2015;397:225–36.
Gotthardt K, Lokaj M, Koerner C, Falk N, Giessl A, Wittinghofer A. A G-protein activation cascade from Arl13B to Arl3 and implications for ciliary targeting of lipidated proteins. Elife. 2015;4:e11859.
Caspary T, Larkins CE, Anderson KV. The graded response to Sonic Hedgehog depends on cilia architecture. Dev Cell. 2007;12:767–78.
Higginbotham H, Guo J, Yokota Y, et al Arl13b-regulated cilia activities are essential for polarized radial glial scaffold formation. Nat Neurosci. 2013;16:1000–7.
Kasahara K, Miyoshi K, Murakami S, Miyazaki I, Asanuma M. Visualization of astrocytic primary cilia in the mouse brain by immunofluorescent analysis using the cilia marker arl13b. Acta Med Okayama. 2014;68:317–22.
Higginbotham H, Eom TY, Mariani LE, et al Arl13b in primary cilia regulates the migration and placement of interneurons in the developing cerebral cortex. Dev Cell. 2012;23:925–38.
Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: critical elements in the control of small G proteins. Cell. 2007;129:865–77.
Cherfils J, Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev. 2013;93:269–309.
Doherty D. Joubert syndrome: insights into brain development, cilium biology, and complex disease. Semin Pediatr Neurol. 2009;16:143–54.
Romani M, Micalizzi A, Valente EM. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 2013;12:894–905.
Gleeson JG, Keeler LC, Parisi MA, et al. Molar tooth sign of the midbrain-hindbrain junction: occurrence in multiple distinct syndromes. Am J Med Genet A. 2004;125A:125–34
Gorden NT, Arts HH, Parisi MA, et al CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am J Hum Genet. 2008;83:559–71.
Bachmann-Gagescu R, Dempsey JC, Phelps IG, et al Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J Med Genet. 2015;52:514–22.
Kang HG, Lee HK, Ahn YH, et al Targeted exome sequencing resolves allelic and the genetic heterogeneity in the genetic diagnosis of nephronophthisis-related ciliopathy. Exp Mol Med. 2016;48:e251.
Shaheen R, Szymanska K, Basu B, et al Characterizing the morbid genome of ciliopathies. Genome Biol. 2016;17:242.
Rafiullah R, Aslamkhan M, Paramasivam N, et al Homozygous missense mutation in the LMAN2L gene segregates with intellectual disability in a large consanguineous Pakistani family. J Med Genet. 2016;53:138–44.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, Handsaker B, Wysoker A, et al The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Rimmer A, Phan H, Mathieson I, et al Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet. 2014;46:912–8.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat. 2011;32:894–9.
Miertzschke M, Koerner C, Spoerner M, Wittinghofer A. Structural insights into the small G-protein Arl13B and implications for Joubert syndrome. Biochem J. 2014;457:301–11.
Eswar N, Webb B, Marti-Renom MA et al. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2007;Chapter 2:Unit 2 9.
Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815.
John B, Sali A. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Res. 2003;31:3982–92.
Mariani LE, Bijlsma MF, Ivanova AI, Suciu SK, Kahn RA, Caspary T. Arl13b regulates Shh signaling from both inside and outside the cilium. Mol Biol Cell. 2016;27:3780–90.
Schindelin J, Arganda-Carreras I, Frise E, et al Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82.
Cooper MK, Porter JA, Young KE, Beachy PA. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science. 1998;280:1603–7.
Taipale J, Chen JK, Cooper MK, et al Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406:1005–9.
Bowzard JB, Sharer JD, Kahn RA. Assays used in the analysis of Arl2 and its binding partners. Methods Enzymol. 2005;404:453–67.
Bowzard JB, Cheng D, Peng J, Kahn RA. ELMOD2 is an Arl2 GTPase-activating protein that also acts on Arfs. J Biol Chem. 2007;282:17568–80.
Cavenagh MM, Breiner M, Schurmann A, et al ADP-ribosylation factor (ARF)-like 3, a new member of the ARF family of GTP-binding proteins cloned from human and rat tissues. J Biol Chem. 1994;269:18937–42.
Saleheen D, Natarajan P, Armean IM, et al Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature. 2017;544:235–9.
Lek M, Karczewski KJ, Minikel EV, et al Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Ivanova AA, East MP, Yi SL, Kahn RA. Characterization of recombinant ELMOD (cell engulfment and motility domain) proteins as GTPase-activating proteins (GAPs) for ARF family GTPases. J Biol Chem. 2014;289:11111–21.
Hori Y, Kobayashi T, Kikko Y, Kontani K, Katada T. Domain architecture of the atypical Arf-family GTPase Arl13b involved in cilia formation. Biochem Biophys Res Commun. 2008;373:119–24.
Randazzo PA, Terui T, Sturch S, Fales HM, Ferrige AG, Kahn RA. The myristoylated amino terminus of ADP-ribosylation factor 1 is a phospholipid- and GTP-sensitive switch. J Biol Chem. 1995;270:14809–15.
Amor JC, Horton JR, Zhu X, et al Structures of yeast ARF2 and ARL1: distinct roles for the N terminus in the structure and function of ARF family GTPases. J Biol Chem. 2001;276:42477–84.
Seidel RD 3rd, Amor JC, Kahn RA, Prestegard JH. Conformational changes in human Arf1 on nucleotide exchange and deletion of membrane-binding elements. J Biol Chem. 2004;279:48307–18.
Ivanova AA, Caspary T, Seyfried NT et al. Biochemical characterization of purified mammalian ARL13B indicate that it is an atypical GTPase and ARL3 guanine nucleotide exchange factor (GEF). J Biol Chem. 2017;292:11091–108.
Baron-Cohen S, Knickmeyer RC, Belmonte MK. Sex differences in the brain: implications for explaining autism. Science. 2005;310:819–23.
Acknowledgments
We thank the families for participating and supporting this study. We also thank Dr. Ute Hehr (Center for Human Genetics, Regensburg, Germany) for her opinion on the CT scans. We thank Cheryl Timms Strauss for editing of the manuscript. We also thank Christine Fischer for statistical advice.
Funding
This study was supported by the Medical Faculty of Heidelberg (R.R., S.B., and G.A.R.), the National Institutes of Health (GM110663 to T.C., A.B.L., A.A.I., and R.A.K.), and the Emory Integrated Genomics Core (EIGC), which is subsidized by the Emory University School of Medicine. R.R. was supported by a scholarship from the German Academic Exchange Service (DAAD; 91541533) and G.M. and R.C.W. gratefully acknowledge the support of the Klaus Tschira Foundation.
Author contributions
R.R. performed the genetic analysis, expression analysis in mice and SH-SY5Y cells, and data analysis. A.B.L. and T.C. performed and analyzed the data in Arl13b hnn MEFs. A.A.I. and R.A.K. performed and analyzed the GAP and GEF assays. H.A. evaluated the patients, clinical data, and CT scans. S.B. contributed to the data interpretation. N.P., M.S., and S.W. performed and analyzed WES data. G.M. and R.C.W. performed and analyzed the 3D structure modeling. E.B. interpreted the CT scans of the patients. G.A.R. initiated, supervised, and supported the project. R.R., S.B., R.K., T.C., and G.A.R. wrote the manuscript. All authors commented on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Patient consent
Obtained.
Ethical approval
This study was approved by the Institutional Ethical Review Committee, University of Health Sciences Lahore, Pakistan, and the Ethikkommission, Medical Faculty Heidelberg, Germany (S-035/2014).
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Rafiullah, R., Long, A.B., Ivanova, A.A. et al. A novel homozygous ARL13B variant in patients with Joubert syndrome impairs its guanine nucleotide-exchange factor activity. Eur J Hum Genet 25, 1324–1334 (2017). https://doi.org/10.1038/s41431-017-0031-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-017-0031-0
