
Abstract
Ischemia/reperfusion (IR) injury, a main reason of mortality and morbidity worldwide, occurs in many organs and tissues. As a result of IR injury, senescent cells can accumulate in multiple organs. Increasing evidence shows that cellular senescence is the underlying mechanism that transforms an acute organ injury into a chronic one. Several recent studies suggest senescent cells can be targeted for the prevention or elimination of acute and chronic organ injury induced by IR. In this review, we concisely introduce the underlying mechanism and the pivotal role of premature senescence in the transition from acute to chronic IR injuries. Special focus is laid on recent advances in the mechanisms as well as on the basic and clinical research, targeting cellular senescence in multi-organ IR injuries. Besides, the potential directions in this field are discussed in the end. Together, the recent advances reviewed here will act as a comprehensive overview of the roles of cellular senescence in IR injury, which could be of great significance for the design of related studies, or as a guide for potential therapeutic target.
Similar content being viewed by others


Facts
-
Ischemia/reperfusion(IR) injury is an inevitable pathological process in many clinical practices, such as transplant surgery, emergency rescue after shock, myocardial infarction and so on. The morbidity and mortality owing to IR injury remain high although many treatments have been developed to prevent this process.
-
Cellular senescence is a state of irreversible cell-cycle arrest that could be caused by many stresses, including IR injury, which is called Stress-induced Cellular Senescence(SIPS).
-
SIPS plays a key role in IR-induced acute and chronic tissue damage/dysfunction of multiple organs, especially in kidney, heart and brain.
Open Questions
-
Currently, the underlying mechanism of IR-induced acute to chronic organ and tissue damage remains complicated and obscure. Therefore, it is of great significance that further study needs to be carried out.
-
Cellular senescence serves as a core mechanism in IR-induced acute to chronic organ and tissue damage. However, the specific mechanism underlying senescence-induced acute and chronic tissue damage remains unclear and needs to be clarified in the future.
-
A large number of reports have confirmed that cellular senescence may serve as a therapeutic target to alleviate IR-induced tissue and organ injury. Hence, the development of treatments targeting cellular senescence will have a wide clinical application in IR-induced acute and chronic injury.
Introduction
Ischemia/reperfusion (IR) injury refers to a condition when the tissues or organs suffer from restricted blood supply, the recovery of blood supply and perfusion do not alleviate ischemic injury, conversely lead to further damage/dysfunction. It is an inevitable pathological process in many clinical practice, such as transplant surgery, emergency rescue after shock, and myocardial infarction. Despite techniques such as thrombolytic therapy, percutaneous coronary angioplasty, and cardiopulmonary bypass have made incredible advancements in reducing tissue ischemia, the morbidity and mortality owing to IR injury after operation still remain high. Extensive studies are focusing on investigating the underline mechanisms of IR injury, involving oxidative stress [1,2,3,4], calcium overload [1, 3], mitochondrial dysfunction [3, 5, 6] and excessive inflammation [1, 3, 7, 8]. These multiple signaling pathways are interrelated and interactive, which eventually contribute to different kinds of cell phenotypes due to different environments and extents of damage: apoptosis [7, 9], necroptosis [9,10,11,12], necrosis [1, 2, 7], pyroptosis [9, 13, 14], ferroptosis [15,16,17] and cellular senescence [18]. However, the full picture of the pathophysiology of IR injury is far from complete and further research is needed.
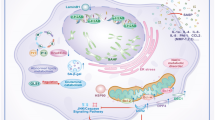
Cellular senescence refers to the state of nonreversible cell-cycle arrest that plays dual roles in different conditions [19, 20] (Fig. 1). In IR-induced acute and chronic tissue damage/dysfunction, cellular senescence is of great significance. In general, senescence can be divided into replicative senescence and premature senescence [21](Fig. 2). Different from replicative senescence induced by telomere shortening after repeated cell division, premature senescence refers to the situation that accelerated by numerous stressful stimuli such as oxidative stress, DNA damage, mitochondrial dysfunction, inflammation, activated oncogenes [22, 23], which is also described as stress-induced premature senescence (SIPS). For the most part, cellular senescence is triggered primarily by the activation of p53/p21CIP1 and/or p16/pRb pathways and characterized as G1/S (or G2/M) cell-cycle arrest [22, 24,25,26]. Moreover, cellular senescence can induce the production of various chemokines, inflammatory cytokines, extracellular matrix remodeling factors, and growth factors by “senescence-associated secretory phenotype” (SASP), including IL-1, IL-6, PAI-1, TGF-β, CTGF, CCN2, and MCP-1 [27,28,29,30,31,32].

Cellular senescence plays dual roles in different conditions. In physiological conditions, cellular senescence contributes to tumor suppression, wound healing, and embryonic development. Cellular senescence is thought to have evolved as an antitumor mechanism where the senescence-associated secretory phenotype(SASP) recruits immune cells to facilitate senescent cells removal. In embryonic development and wound, cell-cycle arrest is induced in damaged cells and results in their elimination by macrophage. Nevertheless, in pathological conditions, senescent cells may result in carcinogenesis if they exist for a long time without clearance. Cellular senescence can also contribute to different kinds of age-associated diseases (such as Alzheimer’s diseases, cardiovascular diseases, osteoporosis, diabetes, renal disease, and liver cirrhosis) and chronic tissue injury progression.

The internal mechanism that leads to cellular senescence varies depending on the triggers and context. Several pathways contribute to the activation of cell-cycle inhibitors, inhibition of retinoblastoma protein (RB) phosphorylation, and cell-cycle arrest which is the main manifestation of cellular senescence. The production of various chemokines, inflammatory cytokines, growth factors, and extracellular matrix remodeling factors which are named “senescence-associated secretory phenotype” (SASP) is also another significant manifestation of cellular senescence. Cellular senescence can be divided into replicative senescence and stress-induced premature senescence(SIPS). (I) In replicative senescence, telomere shortening may trigger activation of ataxia telangiectasia mutated (ATM) or ataxia telangiectasia and RAD3-related protein (ATR) kinases, and result in p53 upregulation, and increased p21. (II) In stress-induced premature senescence, mitochondrial dysfunction and oxidative stress may activate the mitogen-activated protein kinase kinase (MKK3 and MKK6) pathway and their downstream effector p38, leading to the upregulation of p16, p53, and p21 level. DNA damage activates a signaling cascade via ATM/ATR kinases, p53 upregulation, and increased p21. In inflammation response, a component of the senescence-associated secretory phenotype (SASP) pathway named transforming growth factor-β (TGF-β), may upregulate p21 level through SMAD complexes. Lastly, oncogenic signaling or loss of tumor suppressors upregulates p16, p53, and p21 levels, mediated by RAS, MYC, and phosphoinositide 3-kinase (PI3K) and their downstream effectors ATM, ATR, and ARF.
In recent years, studies have confirmed that cellular senescence regulates the development and progression of IR-induced acute and chronic diseases in different organs dynamically and reversibly [33]. More and more evidence revealed that genetic and pharmacological clearance of senescent cells could ameliorate IR-induced acute and chronic disease [34,35,36,37]. However, cellular senescence induced by IR injury is extremely complex and has not yet been fully elucidated. Here, we focus on the recent advances in the role and mechanism of cellular senescence in IR-induced acute and chronic injury in different organs, and clarify the basic and clinical treatment options aiming at treating IR-induced cellular senescence in diverse organs.
Recent advances in ischemia/reperfusion-induced senescence machinery
Consistent with other premature senescence caused by various stressful stimuli, the mechanism underlying IR-induced cellular senescence is complex, including oxidative stress, mitochondrial dysfunction, mitophagy deficiency, inflammation response, and epigenetic modification, which finally contribute to the activation of p53/p21CIP1 and/or p16/pRb senescent pathway (Fig. 3).

IR injury first initiates (I) oxidative stress and mitochondrial dysfunction, followed by (II) inflammation and (III) epigenetic modification, finally activates (IV) p53/p21 and p16 senescence pathway and cause cellular senescence. (I) IR injury may damage the function of mitochondria in parenchymal cells such as renal tubular epithelial cells, neurons, cardiomyocytes, and hepatocytes, lead to ROS generation through downregulation of TREM-1 and klotho and can also mediated by mitophagy defficiency. (II) Inflammation response is characterized by infiltration of immune cells such as macrophages, neutrophils, and lymphocytes in the mesenchyme which are recruited by ROS generated by oxidative stress initially. The infiltrating inflammatory cells will release pro-inflammatory factors (also known as SASP if released by senescent cells) such as IL-6 and IL-8. Besides, senescent parenchymal cells can also release SASP to amplify the inflammation response. (III) Multiple kinds of epigenetic modification including m6A modification, DNA methylation, histone, and p53 acetylation, miRNAs and LncRNAs are involved in IR-induced senescence. (IV) p53/p21 pathway and p16 pathway are the final signaling to induce cellular senescence.
Mitochondrial dysfunction and oxidative stress
IR injury is primarily characterized by mitochondrial dysfunction and burst production of reactive oxygen species (ROS). The excessive production of ROS causes oxidative stress in tissues, leading to cell death and ultimately organ dysfunction [38]. Growing evidence showed that mitochondrial dysfunction and oxidative stress induced by IR might further result in cellular senescence, mediated by p21 and p16 signaling activation [39].
In terms of the internal mechanism, recent studies have shown that mitochondrial autophagy (mitophagy) deficiency after renal IR would alter mitochondrial network and cause the accumulation of dysfunctional mitochondria, which led to excessive ROS-induced senescence [40, 41]. In addition, Miao et al. reported that IR would inhibit Klotho, a widely reported factor associated with anti-senescence, leading to the activation of Wnt1- and Wnt9a-induced mitochondrial injury and cellular senescence in renal [18]. Meanwhile, Tammaro et al. recently indicated that deficiency of the triggering receptor expressed on myeloid cells-1 (TREM-1), an innate immune receptor, would damage mitochondrial metabolism, increase ROS accumulation, drive G2/M arrest and senescence in tubular epithelial cells after renal IR [42]. Moreover, IR can accelerate mitochondrial fission–associated myocardial senescence in mice, following myocardial infarction [43]. These discoveries are consistent with the hypothesis that mitochondrial dysfunction and oxidative stress are involved in IR-induced senescence.
Inflammation
The increased generation of ROS and mtDNA after IR, which are also named damage-associated molecular patterns(DAMPs), would contribute to neutrophil infiltration and a large amount of pro-inflammatory cytokines release, which plays a crucial role in cellular senescence. Increasing studies have shown a complicated interaction between senescence and inflammation in IR injury [44]. On the one hand, the overactive inflammatory response is one of the major predispositions to SIPS. On the other hand, senescent cells may give rise to senescence of their nearby cells through the SASP, amplifying the inflammatory response that follows [45, 46].
As for the internal mechanism, inflammation is considered a complicated interaction between immune cells and parenchymal cells [47] and marked by infiltration of immune cell in the mesenchyme [48]. In the early phases of IR injury, neutrophils are recruited by DAMPs mainly [49]. These DAMPs, interact with pattern recognition receptors(PRRs) on macrophages and contribute to their activation [50], and thus promote cell-cycle arrest through SASP [51]. Another kind of innate immune cells, Dendritic cells(DCs), which serve as a mediator of the recruitment and activation of effector T cells, promoting interstitial immune response [52]. DCs would also aggravate SASP production in immune cells in cisplatin-induced AKI, which serve as crucial amplifiers of local innate immune responses in AKI [53]. These studies emphasize the importance of inflammation microenvironment in cellular senescence. Meanwhile, Qian Li et al. pointed out that renal sympathetic neurotransmitter NE, acting on the α2A-AR of epithelial cells, could promote the crosstalk between inflammation and cellular senescence, contributing to renal fibrosis after IR injury [54]. Weifeng Yao et al. found that aerosol inhalation of a hydrogen-rich solution would attenuate renal macrophage infiltration, the release of pro-inflammatory cytokine, and cellular senescence via TGF-β1 pathway in septic acute kidney injury (AKI) [55]. Besides, inflammation and senescence share a cascade amplification process with each other in cardiac [56] and hepatic IR injury [57]. Recently, Qi et al. found that inhibition of NF-κB pathway would disrupt the reciprocal cycle between inflammation and senescence of TECs [58], and the elimination of senescent myocardiocytes after MI would markedly reduce SASP and induce efferocytosis of macrophage to downregulate inflammation [59]. Putting it all together, further exploring the positive feedback loop between inflammation and cellular senescence might help to alleviate multi-organ injury induced by IR.
Epigenetic modification
In addition to oxidative stress and inflammation, epigenetic modification is critically involved in IR-induced senescence. Epigenetic modification refers to changes of genome that occur without any alteration in DNA sequence, including histone acetylation [60, 61], DNA methylation [62, 63], miRNA [62], LncRNA [64, 65], and m6A [66] modification. Growing evidence indicated that epigenetic modification shared complex interaction with cell senescence in multi-organ IR injury. For instance, renal IR injury enhanced the amount of histone H3 acetylation, triggered G2/M arrest and cellular senescence [62], as well as p53 acetylation in the premature senescence of renal tubular epithelial cell (TEC) [67]. Castellano et al. indicated that after IR, aberrant methylation in DNA regions, which involved in cell-cycle control, would result in cell-cycle arrest and senescence in TEC [68]. Meanwhile, m6A modification was found to be a novel mechanism in IR-induced cellular senescence and organ dysfunction [69, 70]. For example, activation of m6A methyltransferase METTL3 after MI can lead to cell-cycle arrest of cardiomyocyte [66]. Besides, Liu et al. reported that miR-493 targets STMN-1 to promote hypoxia-induced epithelial cell-cycle arrest in G (2)/M, leading to renal fibrosis [71]. Moreover, LncRNAs, including SNHG6, AK028326, and Malat1, were recently reported to regulate the p53-senescent pathway in IR-induced kidney injury [64]. Taken together, epigenetic regulation is closely related to multi-organ IR injury, which may serve as a novel therapeutic target to ameliorate or prevent IR injury through regulating cellular senescence.
The p53/p21 pathway and p16/pR pathway
Under the combined action of oxidative stress [72, 73], inflammation [57], and epigenetic modification [68], the p53/p21CIP1 pathway and p16/pRb pathway are activated to induce cellular senescence in IR injury [74,75,76,77] (Table 1 and Table 2). Specifically, IR induces the persistent DNA damage response (DDR) and triggers cell signaling cascades reaction involved in the cell-cycle arrest process and DNA repair by activating the p53/p21CIP1 [22,23,24] and p16/pRb [73] pathway. As a result, the cyclin-dependent kinases (CDKs) as well as retinoblastoma protein (RB) are inhibited while the checkpoint activity is enhanced, leading to G1/S (or G2/M) cell-cycle arrest.
For instance, oxidative stress happened in IR injury was reported to activate p53-dependent accumulation of p21CIP1 and mediate cardiomyocyte senescence, contributing to cardiac dysfunction as well as pathological remodeling [72]. Meanwhile, Qi et al. found that IR-induced inflammation would consequently cause p16INK4A activation and lead to hepatic cellular senescence [57]. Moreover, a recent study confirmed that IR-induced aberrant methylation involved in cell-cycle control and DNA damage would finally result in p53 upregulation and cell-cycle arrest in specific regions [68].
Cellular senescence is the core mechanism of transition from acute to chronic stage after IR
Besides the acute organ dysfunction, cellular senescence was recently confirmed to make a contribution to the transition from acute to chronic stage in different organs after IR. Here, we will combine the recent research progress and discuss the pivotal role of cellular senescence in acute kidney injury to chronic kidney disease (AKI-to-CKD) transition, cardiac injury progression and in ischemic stroke-induced glial scar and cerebral fibrosis (Fig. 4).

a After kidney IR injury, IR-induced cellular senescence is a major initiative of AKI-to-CKD transition, which is mediated by SASP and chronic inflammation, mitochondrial dysfunction and oxidative stress and myofibroblast activation. Firstly, the existence of senescent TECs will cause persistent inflammation and lead to M1 infiltration and M2 polarization deficiency. Besides, Senescence burden in tubule is aggravated via gap junction and further contributes to chronic inflammation, leading to collagen deposition and vascular rarefaction. Secondly, Mitochondrial dysfunction and ROS generation caused by cellular senescence may result in renal fibrosis. Finally, fibroblast will be activated via Wnt9a-TGF-β1 pathway and intensify renal fibrosis. b After ischemic stroke, IR-induced senescent neurons may lead to reactive gliosis and scar forming. c After heart IR injury, IR-induced senescent cardiomyocytes result in heart remodeling through inflammation and SASP.
Cellular senescence in AKI-to-CKD transition
The kidney receives 20% of cardiac output and consumes 10% of body oxygen, which makes it vulnerable to IR injury [78]. Moreover, although patients suffer from mild AKI can restore normal renal function, more than 70% of them experience renal maladaptive repair, and more than 50% of them will gradually develop into CKD [79], becoming the fifth leading cause of death by 2040 [78, 80]. Hence, research on the underlying mechanism of AKI-to-CKD is becoming increasingly attractive, and one of the recent snapshots is the senescent TECs and senescence-associated fibrosis [9, 74, 81,82,83]. To be specific, cellular senescence participates in AKI-to-CKD through multiple mechanisms, such as SASP, chronic inflammation, mitochondrial dysfunction, oxidative stress, and myofibroblasts activation.
SASP and chronic inflammation
On the one hand, senescent TECs caused by renal IR remain metabolically active and adopt SASP to release inflammatory cytokines and other fibrotic factors that serve as contributors to risk factors in maladaptive repair [84] and renal fibrosis [85, 86], such as collagen deposition, vascular rarefaction and chronic inflammation [86,87,88,89]. Interestingly, recent evidence found that senescent TECs could interrupt the macrophage polarization, increasing M1 infiltration and impaired M2 polarization to induce chronic inflammation in kidney [90]. On the other hand, senescent TECs caused by renal IR might further induce DNA damage response in neighboring cells by cell-cell contact via the gap junction and cause cellular senescence in intact bystander TECs and fibroblasts [91, 92], which might enhance SASP release and lead to renal maladaptive repair and senescence-associated fibrosis [81].
Mitochondrial dysfunction and oxidative stress
On the one hand, Klotho deficiency in IR-induced senescent TECs would further promote mitochondrial injury, ROS generation, and fibrotic lesions [18, 93]. Meanwhile, senescent TECs would downregulate Nrf2 and attenuate anti-oxidative response [39, 88, 94, 95], leading to AKI-CKD transition [35]. On the other hand, senescent cells produce and secrete ROS to induce further DNA damage response in neighboring TECs via gap junction-mediated between adjacent cells [92]. These pieces of evidence suggest that oxidative stress and mitochondrial dysfunction are involved in senescence-induced chronic renal injury.
Myofibroblasts activation
Another key characteristic of maladaptive repair is the activation of numerous myofibroblasts which make contribution to the deposition of collagen and other pro-fibrotic components in kidney [86]. Recently, increasing evidence showed that senescent TECs would enhance myofibroblasts activation via SASP generation in an epithelial-mesenchymal transition (EMT) manner [96]. Meanwhile, senescent TECs would generate TGF-β1, a factor contribute to interstitial fibroblast proliferation and transition to myofibroblasts [97]. Furthermore, a reciprocal activate loop between senescent TECs and myofibroblasts was mediated by Wnt9a-TGF-β1 pathway, which promoted and accelerated the pathogenesis of renal fibrosis [97]. Taken these pieces of evidence together, myofibroblasts activation is substantially relevant to senescence TECs after renal IR.
Cellular senescence in cardiac IR-induced heart remodeling
Consistent with the kidneys, hearts are also prone to suffer from IR injury since they are organs with high energy demand. As a process of cardiac repairment after IR, cardiac remodeling is initially adaptive to the damage induced by IR in the short term, but becomes maladaptive due to the sustained stress, resulting in progressive and irreversible cardiac dysfunction and heart failure [72]. Among all the culprits, cellular senescence induced by IR injury plays an essential role in heart remodeling and transition into chronic heart injury [56, 72].
Recent studies showed that senescent cardiomyocytes induced by cardiac IR would secret SASP [98] and further activate the maladaptive cardiac remodeling, including cellular hypertrophy, inflammation, fibrosis, and attenuation of regeneration, which ultimately contribute to cardiac chronic fibrosis [56, 98]. At the same time, studies revealed that IR-induced senescent cardiomyocyte might provide cardioprotective effects by promoting cellular senescence of fibroblast, which promoted neonatal heart regeneration by accelerating cardiomyocyte proliferation and inhibiting cardiac fibrosis [98]. Together, these results present pieces of evidence of involvement of senescent cells in the acute to the chronic transition of cardiac dysfunction after IR.
Cellular senescence in ischemic stroke-induced glial scar and cerebral fibrosis
In addition to the kidney and heart, brain is also prone to suffer from I/R injury. A recent study also showed that neuron senescence served as a pathogenic mechanism for ischemic stroke-induced brain damage [99, 100], which might result in glial scar and cerebral fibrosis [101].
On the one hand, senescent neurons induced by stroke have a significant increase in the expression of SASP including IL6, TNFα, and CXCL1 [99, 100, 102], which induce cerebral inflammation microenvironment forming and extracellular matrix(ECM) deposition by a pericyte-dependent manner [101]. On the other hand, in the early stages of cerebral IR injury, glial cells especially astrocytes are activated by pro-inflammatory cytokines generated by senescent neurons [101, 103, 104]. Moreover, reactive astrocytes may secrete a myriad of adhesion molecules and pro-inflammatory cytokines, such as VCAM-1, ICAM-1, IL-1β, IL-6, and TNF-α [105], which could involve in a cyclic process of consecutive activation, consequently resulting in cerebral fibrosis, glial scar [106, 107] and regeneration failure in ischemic zones [108].
Therapies that target cellular senescence after IR injury
As mentioned above, premature cell senescence caused by IR injury plays a pivotal role in multiple organs’ acute and chronic dysfunction. Therefore, treatments targeting cellular senescence means a promising prospect in IR injury. Based on the observation, the elimination of senescent cells is mostly beneficial and seems to have few long-term deleterious consequences, researchers have identified various novel agents and strategies to achieve this, which were also called ‘senotherapeutic’ strategies [109]. In summary, it can be classified into four classifications: pharmacological agents named ‘senolytics’ that clear senescent cells, ‘senomorphics’ that prevent harmful effects of senescent cells [110], rejuvenating agents that stimulate SIRT1 to alleviate senescence, and stem cell therapy (Fig. 5).

Several kinds of intervention including senolytics, senomorphics, rejuvenating agents, stem cell therapy, and other intervention, are developed to attenuate the deterioration brought by senescent cells.
Senolytics
Formed by the words “senescence” and “lytic” (destroying), senolytics include pharmacological agents targeting the specific elimination of senescent cells. Increasing pieces of evidence have shown that senolytics might be effective tools to eliminate senescent cells to treat age-related diseases and IR-induced cellular senescence [34]. As for senolytics, the most reported include the combination of dasatinib and quercetin (D + Q), Navitoclax and FOXO4-D-Retro-Inverso peptide (FOXO4-DRI).
A growing body of pieces of evidence suggest that D+Q treatment could disable pro-survival networks and eliminate senescent cells in IR-related organ dysfunctions [111, 112]. For instance, D+Q could reduce senescent cell burden, promote TECs’ proliferation, ameliorate renal fibrosis, and decrease renal inflammation in IR-induced kidney disorders. [111] Moreover, treatment of aged animals with D+Q was reported to eliminate senescent cells and diminish cell-free mitochondrial DNA (cf-mt-DNA) release, attenuating the IR-associated cardiac injury and prolonging the survival of aged cardiac allografts [112]. Meanwhile, D+Q were reported to be beneficial in reducing senescent cell levels and improving renal transplantation outcome [109].
Navitoclax (ABT-263) is a kind of Bcl-2 inhibitors that induces senescent cell apoptosis and death in aged mice [34, 113] and promotes rejuvenation of stem cells for tissue regeneration [109]. A recent study revealed that ABT-263 could reduce senescent cell burdens and restore a regenerative phenotype with improved function, increased tubular proliferation, and reduced fibrosis in the kidneys after renal IR [114]. Furthermore, ABT-263 was shown to eliminate cerebral IR-induced neural cell senescence, reduce the infarct volume and improve neurological function in animal models [102]. Elimination of senescent cells with Navitoclax during cardiac IR injury was proved to be a potential novel therapeutic avenue in improving patients outcomes following cardiac IR [56].
FOXO4-DRI, as a novel cell-penetrating peptide, is designed to interfere with the endogenous p53-FOXO4 interaction [36] and potentially target senescent cells by influencing the p53-dependent apoptosis [36]. Although the clearance of senescent cells with FOXO4-DRI was reported to restore renal function and reduce inflammation markers in the kidney of aged mice [35, 36], whether it is therapeutically feasible in IR injury still needs further exploration. Other approaches rely on immune-system-mediated clearance of senescent cells are emerging consecutively and might become promising methods in mitigating senescent burden after IR-induced senescence in the near future [115].
Senomorphics
The use of senomorphic agents is an alternative to complete the clearance of senescent cells through senolysis against IR injury. Senomorphics is designed to prevent cells occurring growth arrest as well as to disrupt the generation and secretion of SASP while keep the cells alive. This method could interfere with the pro-inflammatory nature of IR-induced cellular senescence and potentially delay the critical effects of IR injury and organ aging [110].
The most commonly reported senomorphics are rapamycin and metformin. Rapamycin is a kind of mTOR inhibitors that have been reported to regulate autophagy and inhibit cellular senescence in renal IR injury [81, 116, 117] via enhancing Wnt signaling [110]. Metformin, an AMPK activator, has been reported to attenuate IR-induced mitochondrial dysfunction [118], decrease the level of p16INK4a and p21CIP1 and inhibit the release of SASP-related cytokines [119]. Increasing evidence also showed that Lipoxin A4 might stimulate inflammation resolution and inhibit cellular senescence in septic AKI [46].
Rejuvenating agents
Rejuvenating agents specifically refer to the interventions that stimulate SIRT1 to alleviate senescence. In mammals, SIRT1 is well-characterized to enhance cell proliferation and inhibit cellular senescence through the suppression and deacetylation of p53, [120,121,122]. Resveratrol (RSV), as the most reported agonist of SIRT1, is potentially to protect organs against IR-induced premature cellular senescence [123]. Further clinical studies are needed to confirm and elaborate the protective effects on the application of RSV in IR injury.
Stem cell therapy
Stem cell therapy is another promising treatment for IR-induced senescence. Multipotent mesenchymal stem cells (MSCs), being a category of adult stem cells springing from the mesoderm, with multi-directional differentiation and self-renewal potential, have recently emerged as a key player in regenerative medicine and clinical translational research [124,125,126]. Typically, MSCs have been extensively studied to inhibit premature senescence by protecting against the IR-induced pro-oxidative state, cell-cycle inhibition [93], and chronic fibrosis [127].
Recently, the increasing underlying mechanism of MSCs against IR-induced senescence has been clarified. On the one hand, MSCs can exert immunomodulatory ability via secreting soluble factors or direct contact with the immune cells, and transform them into an anti‐inflammatory phenotype and further inhibit cellular senescence [128]. On the other hand, MSCs can secret extracellular vesicles (EVs) to inhibit the generation of SASP in senescent cells [129, 130]. For instance, cell-cycle arrest of myocardiocytes after MI can be alleviated by MSC-EVs carrying miR-150-5p via downregulation of TXNIP [131, 132], or by MSC-EVs targeting miR-497/Smad7/TGF-β pathway [133]. Yu et al. also found that EVs carrying mi-202-3p could protect neurons from IR injury via downregulating TLR4-mediated inflammation response [124]. Xiao et al. point that MSC-EVs reduce endothelial cell senescent burden and activate angiogenesis through miR-146a/Src pathway [134].
With the further study of MSCs, more and more novel treatments derived from MSCs have been contrived. For example, prior clearance of senescent cells enhanced the beneficial effects of KIM‐MSC on cellular senescence [135]. Yu et al. suggested that mi-R217 inhibitor may enhance MSCs’ repair of vascular damage and senescence via SIRT1 upregulation [136]. Klotho gene-modified MSCs were recently found to inhibit cellular senescence and show elevated anti-fibrotic effects in kidneys after IR [137]. In general, stem cell therapy provides an innovative approach in IR injury treatment, but the mechanism and clinical application still need further study.
Other interventions
Several other interventions were also found effectively targeting cellular senescence in IR injury. For instance, it was reported that Cilnidipine could prevent hypoxia-induced mitochondrial hyperfission and myocardial senescence [43]. Dexmedetomidine, a highly selective α2 adrenergic receptor (α2-AR) agonist, was proved to be useful in inhibiting cellular senescence and IR-induced renal fibrosis [81]. Moreover, Nicotinamide mononucleotide (NMN) could attenuate renal interstitial fibrosis by suppressing DNA damage and senescence of TECs in AKI [138]. More and more further study may reveal the potential clinical application value of such interventions.
Conclusion and outlook
Increasing pieces of evidence are revealing new insight into the crucial role of cellular senescence in IR injury. Up to now, IR-induced mochondrial dysfunction and oxidative stress, inflammation, epigenetic modification, and activation of p53/p21 and p16/pRb pathways have been reported to ultimately cause cellular senescence. At the same time, IR-induced cellular senescence contributes to the transition from acute organ injury to chronic dysfunction through inflammation, oxidative stress, mitochondrial dysfunction, and myofibroblast activation. However, the currently known function of cellular senescence in IR injury is just the tip of iceberg. For instance, senescence in different types of cells would bring different outcomes. As is mentioned in hepatic and cardiac IR injury, the senescence of hepatic stellate cells [139] and cardiac fibroblast [98] played a protective role in the repair process. Thus, advanced technology such as organoid model [140] and single-cell sequencing should be adopted to explore the precise role of cellular senescence in acute organ injury to chronic disease transition.
With the growing awareness of the importance of cellular senescence in IR-induced acute organ injury and chronic dysfunction comes the potential to target cellular senescence with novel therapeutic strategies. Senolytics, senomorphics, SIRT1 agnoist and stem cell therapy are the most well-reported and promising treatments for cellular senescence in in vivo and in vitro experiments. However, in light of the fact that cellular senescence is instrumental in preventing dangerous DNA mutations, it is important to assess carefully the effects and safety of these drugs to attenuate IR injury in humans.
In a word, more and more researches will certainly shed light on the role of cellular senescence in IR injury and acute to chronic dysfunction transitions. This pervasive disease will certainly be overcome with further research and the novel therapies deserve the higher priority.
Data availability
The data used to support the findings of this study are available from the corresponding author upon request.
References
Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N. Engl. J. Med. 2007;357:1121–35.
Wu MY, Yiang GT, Liao WT, Tsai AP, Cheng YL, Cheng PW, et al. Current mechanistic concepts in ischemia and reperfusion injury. Cell Physiol Biochem. 2018;46:1650–67.
Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Ischemia/reperfusion. Compr Physiol. 2016;7:113–70.
Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–5.
Li YQ, Jiao Y, Liu YN, Fu JY, Sun LK, Su J. PGC-1α protects from myocardial ischaemia-reperfusion injury by regulating mitonuclear communication. J Cell Mol Med. 2021;26:593–600.
Zhao M, Wang Y, Li L, Liu S, Wang C, Yuan Y, et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics. 2021;11:1845–63.
Raghay K, Akki R, Bensaid D, Errami M. Ghrelin as an anti-inflammatory and protective agent in ischemia/reperfusion injury. Peptides. 2020;124:170226.
Smith SF, Hosgood SA, Nicholson ML. Ischemia-reperfusion injury in renal transplantation: 3 key signaling pathways in tubular epithelial cells. Kidney Int. 2019;95:50–56.
Tammaro A, Kers J, Scantlebery AML, Florquin S. Metabolic flexibility and innate immunity in renal ischemia reperfusion injury: the fine balance between adaptive repair and tissue degeneration. Front Immunol. 2020;11:1346.
Yang F, Shang L, Wang S, Liu Y, Ren H, Zhu W, et al. TNFα-mediated necroptosis aggravates ischemia-reperfusion injury in the fatty liver by regulating the inflammatory response. Oxid Med Cell Longev. 2019;2019:2301903.
Qian Y, Guo X, Che L, Guan X, Wu B, Lu R, et al. Klotho reduces necroptosis by targeting oxidative stress involved in renal ischemic-reperfusion injury. Cell Physiol Biochem. 2018;45:2268–82.
Wang X, O’Brien ME, Yu J, Xu C, Zhang Q, Lu S, et al. Prolonged cold ischemia induces necroptotic cell death in ischemia-reperfusion injury and contributes to primary graft dysfunction after lung transplantation. Am J Respir Cell Mol Biol. 2019;61:244–56.
Wu H, Huang T, Ying L, Han C, Li D, Xu Y, et al. MiR-155 is involved in renal ischemia-reperfusion injury via direct targeting of FoxO3a and regulating renal tubular cell pyroptosis. Cell Physiol Biochem. 2016;40:1692–705.
Xiao C, Zhao H, Zhu H, Zhang Y, Su Q, Zhao F, et al. Tisp40 induces tubular epithelial cell GSDMD-mediated pyroptosis in renal ischemia-reperfusion injury via NF-κB signaling. Front Physiol. 2020;11:906.
Li W, Li W, Leng Y, Xiong Y, Xia Z. Ferroptosis is involved in diabetes myocardial ischemia/reperfusion injury through endoplasmic reticulum stress. DNA Cell Biol. 2020;39:210–25.
Lei P, Bai T, Sun Y. Mechanisms of ferroptosis and relations with regulated cell death: a review. Front Physiol. 2019;10:139.
Su L, Jiang X, Yang C, Zhang J, Chen B, Li Y, et al. Pannexin 1 mediates ferroptosis that contributes to renal ischemia/reperfusion injury. J Biol Chem. 2019;294:19395–404.
Miao J, Huang J, Luo C, Ye H, Ling X, Wu Q, et al. Klotho retards renal fibrosis through targeting mitochondrial dysfunction and cellular senescence in renal tubular cells. Physiol Rep. 2021;9:e14696.
Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: defining a path forward. Cell. 2019;179:813–27.
Zhang L, Pitcher LE, Yousefzadeh MJ, Niedernhofer LJ, Robbins PD, Zhu Y. Cellular senescence: a key therapeutic target in aging and diseases. J. Clin. Investig. 2022;132:e158450.
Martínez-Zamudio RI, Robinson L, Roux PF, Bischof O. SnapShot: cellular senescence pathways. Cell. 2017;170:816–816.e811.
van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509:439–46.
Campisi J, d’Adda, di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40.
Childs BG, Durik M, Baker DJ, van Deursen JM. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med. 2015;21:1424–35.
Macip S, Igarashi M, Fang L, Chen A, Pan ZQ, Lee SW, et al. Inhibition of p21-mediated ROS accumulation can rescue p21-induced senescence. EMBO J. 2002;21:2180–8.
He S, Sharpless NE. Senescence in health and disease. Cell. 2017;169:1000–11.
Zhou B, Wan Y, Chen R, Zhang C, Li X, Meng F, et al. The emerging role of cellular senescence in renal diseases. J Cell Mol Med. 2020;24:2087–97.
Paez-Ribes M, Gonzalez-Gualda E, Doherty GJ, Munoz-Espin D. Targeting senescent cells in translational medicine. EMBO Mol Med. 2019;11:e10234.
Matjusaitis M, Chin G, Sarnoski EA, Stolzing A. Biomarkers to identify and isolate senescent cells. Ageing Res Rev. 2016;29:1–12.
Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853–68.
Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118.
Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009;9:81–94.
Tchkonia T, Kirkland JL. Aging, cell senescence, and chronic disease: emerging therapeutic strategies. JAMA. 2018;320:1319–20.
Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–6.
Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530:184–9.
Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell. 2017;169:132–.e116.
Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016;15:973–7.
Yao W, Han X, Ge M, Chen C, Xiao X, Li H, et al. N(6)-methyladenosine (m(6)A) methylation in ischemia-reperfusion injury. Cell Death Dis. 2020;11:478.
Strausser SA, Nakano D, Souma T. Acute kidney injury to chronic kidney disease transition: insufficient cellular stress response. Curr Opin Nephrol Hypertens. 2018;27:314–22.
Korolchuk VI, Miwa S, Carroll B, von Zglinicki T. Mitochondria in cell senescence: is mitophagy the weakest link? EBioMedicine. 2017;21:7–13.
Bakula D, Scheibye-Knudsen M. MitophAging: mitophagy in aging and disease. Front Cell Dev Biol. 2020;8:239.
Tammaro A, Scantlebery AML, Rampanelli E, Borrelli C, Claessen N, Butter LM, et al. TREM1/3 deficiency impairs tissue repair after acute kidney injury and mitochondrial metabolic flexibility in tubular epithelial cells. Front Immunol. 2019;10:1469.
Nishimura A, Shimauchi T, Tanaka T, Shimoda K, Toyama T, Kitajima N, et al. Hypoxia-induced interaction of filamin with Drp1 causes mitochondrial hyperfission-associated myocardial senescence. Sci Signal. 2018;11:eaat5185.
Herranz N, Gil J. Mechanisms and functions of cellular senescence. J Clin Investig. 2018;128:1238–46.
Jin H, Zhang Y, Ding Q, Wang SS, Rastogi P, Dai DF, et al. Epithelial innate immunity mediates tubular cell senescence after kidney injury. JCI Insight 2019;4:e125490.
Chen C, Qiu R, Yang J, Zhang Q, Sun G, Gao X, et al. Lipoxin A4 REstores Septic Renal Function Via Blocking Crosstalk between Inflammation and Premature Senescence. Front Immunol. 2021;12:637753.
Singbartl K, Formeck CL, Kellum JA. Kidney-immune system crosstalk in AKI. Semin Nephrol. 2019;39:96–106.
Meng XM, Nikolic-Paterson DJ, Lan HY. Inflammatory processes in renal fibrosis. Nat Rev Nephrol. 2014;10:493–503.
Zuk A, Bonventre JV. Acute kidney injury. Annu Rev Med. 2016;67:293–307.
Campanholle G, Mittelsteadt K, Nakagawa S, Kobayashi A, Lin SL, Gharib SA, et al. TLR-2/TLR-4 TREM-1 signaling pathway is dispensable in inflammatory myeloid cells during sterile kidney injury. PLoS ONE. 2013;8:e68640.
Djudjaj S, Martin IV, Buhl EM, Nothofer NJ, Leng L, Piecychna M, et al. Macrophage migration inhibitory factor limits renal inflammation and fibrosis by counteracting tubular cell cycle arrest. J Am Soc Nephrol. 2017;28:3590–604.
Kurts C, Ginhoux F, Panzer U. Kidney dendritic cells: fundamental biology and functional roles in health and disease. Nat Rev Nephrol. 2020;16:391–407.
Deng B, Lin Y, Chen Y, Ma S, Cai Q, Wang W, et al. Plasmacytoid dendritic cells promote acute kidney injury by producing interferon-α. Cell Mol Immunol. 2021;18:219–29.
Li Q, Deng Y, Liu L, Zhang C, Cai Y, Zhang T, et al. Sympathetic denervation ameliorates renal fibrosis via inhibition of cellular senescence. Front Immunol. 2021;12:823935.
Yao W, Guo A, Han X, Wu S, Chen C, Luo C, et al. Aerosol inhalation of a hydrogen-rich solution restored septic renal function. Aging. 2019;11:12097–113.
Dookun E, Walaszczyk A, Redgrave R, Palmowski P, Tual-Chalot S, Suwana A, et al. Clearance of senescent cells during cardiac ischemia-reperfusion injury improves recovery. Aging Cell. 2020;19:e13249.
Qi X, Ng KT, Lian Q, Li CX, Geng W, Ling CC, et al. Glutathione peroxidase 3 delivered by hiPSC-MSCs ameliorated hepatic IR injury via inhibition of hepatic senescence. Theranostics. 2018;8:212–22.
Qi R, Wang J, Jiang Y, Qiu Y, Xu M, Rong R, et al. Snai1-induced partial epithelial-mesenchymal transition orchestrates p53-p21-mediated G2/M arrest in the progression of renal fibrosis via NF-κB-mediated inflammation. Cell Death Dis. 2021;12:44.
Lee JR, Park BW, Park JH, Lim S, Kwon SP, Hwang JW, et al. Local delivery of a senolytic drug in ischemia and reperfusion-injured heart attenuates cardiac remodeling and restores impaired cardiac function. Acta Biomaterialia. 2021;135:520–33.
Mercken EM, Hu J, Krzysik-Walker S, Wei M, Li Y, McBurney MW, et al. SIRT1 but not its increased expression is essential for lifespan extension in caloric-restricted mice. Aging cell. 2014;13:193–6.
Cianciolo Cosentino C, Skrypnyk NI, Brilli LL, Chiba T, Novitskaya T, Woods C, et al. Histone deacetylase inhibitor enhances recovery after AKI. J Am Soc Nephrol. 2013;24:943–53.
Tang J, Zhuang S. Histone acetylation and DNA methylation in ischemia/reperfusion injury. Clin Sci (Lond). 2019;133:597–609.
Heylen L, Thienpont B, Naesens M, Lambrechts D, Sprangers B. The emerging role of DNA methylation in kidney transplantation: a perspective. Am J Transpl. 2016;16:1070–8.
Kaucsár T, Róka B, Tod P, Do PT, Hegedűs Z, Szénási G, et al. Divergent regulation of lncRNA expression by ischemia in adult and aging mice. Geroscience. 2021;44:429–45.
Jin L, Song Q, Zhang W, Geng B, Cai J. Roles of long noncoding RNAs in aging and aging complications. Biochim Biophys Acta Mol Basis Dis. 2019;1865:1763–71.
Gong R, Wang X, Li H, Liu S, Jiang Z, Zhao Y, et al. Loss of m(6)A methyltransferase METTL3 promotes heart regeneration and repair after myocardial injury. Pharmacol Res. 2021;174:105845.
Li C, Xie N, Li Y, Liu C, Hou FF, Wang J. N-acetylcysteine ameliorates cisplatin-induced renal senescence and renal interstitial fibrosis through sirtuin1 activation and p53 deacetylation. Free Radic Biol Med. 2019;130:512–27.
Castellano G, Franzin R, Sallustio F, Stasi A, Banelli B, Romani M, et al. Complement component C5a induces aberrant epigenetic modifications in renal tubular epithelial cells accelerating senescence by Wnt4/βcatenin signaling after ischemia/reperfusion injury. Aging. 2019;11:4382–406.
Xiao K, Liu P, Yan P, Liu Y, Song L, Liu Y, et al. N6-methyladenosine reader YTH N6-methyladenosine RNA binding protein 3 or insulin like growth factor 2 mRNA binding protein 2 knockdown protects human bronchial epithelial cells from hypoxia/reoxygenation injury by inactivating p38 MAPK, AKT, ERK1/2, and NF-κB pathways. Bioengineered. 2021;13:11973–86.
Su X, Shen Y, Jin Y, Kim IM, Weintraub NL, Tang Y. Aging-associated differences in epitranscriptomic m6A regulation in response to acute cardiac ischemia/reperfusion injury in female mice. Front Pharmacol. 2021;12:654316.
Liu T, Liu L, Liu M, Du R, Dang Y, Bai M, et al. MicroRNA-493 targets STMN-1 and promotes hypoxia-induced epithelial cell cycle arrest in G(2)/M and renal fibrosis. FASEB J. 2019;33:1565–77.
Luo X, Zhou J, Wang Z, He Y, Yu L, Ma S, et al. An inhibitor role of Nrf2 in the regulation of myocardial senescence and dysfunction after myocardial infarction. Life Sci. 2020;259:118199.
Moonen L, D’Haese PC, Vervaet BA. Epithelial cell cycle behaviour in the injured kidney. Int J Mol Sci. 2018;19:2038.
Sari FT, Sari FT, Sari FT, Arfian N, Sari DCR. Effect of kidney ischemia/reperfusion injury on proliferation, apoptosis, and cellular senescence in acute kidney injury in mice. Med J Malays. 2020;75:20–3.
Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16:393–405.
Franzin R, Stasi A, Fiorentino M, Stallone G, Cantaluppi V, Gesualdo L, et al. Inflammaging and complement system: a link between acute kidney injury and chronic graft damage. Front Immunol. 2020;11:734.
Baisantry A, Berkenkamp B, Rong S, Bhayadia R, Sörensen-Zender I, Schmitt R, et al. Time-dependent p53 inhibition determines senescence attenuation and long-term outcome after renal ischemia-reperfusion. Am J Physiol Ren Physiol. 2019;316:F1124–f1132.
Jiang M, Bai M, Lei J, Xie Y, Xu S, Jia Z, et al. Mitochondrial dysfunction and the AKI-to-CKD transition. Am J Physiol Ren Physiol. 2020;319:F1105–f1116.
Maiwall R, Pasupuleti SSR, Bihari C, Rastogi A, Singh PK, Naik V, et al. Incidence, risk factors, and outcomes of transition of acute kidney injury to chronic kidney disease in cirrhosis: a prospective cohort study. Hepatology. 2020;71:1009–22.
Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N. Engl J Med. 2014;371:58–66.
Li Q, Chen C, Chen X, Han M, Li J. Dexmedetomidine attenuates renal fibrosis via α2-adrenergic receptor-dependent inhibition of cellular senescence after renal ischemia/reperfusion. Life Sci. 2018;207:1–8.
Docherty MH, O’Sullivan ED, Bonventre JV, Ferenbach DA. Cellular senescence in the kidney. J Am Soc Nephrol. 2019;30:726–36.
Sturmlechner I, Durik M, Sieben CJ, Baker DJ, van Deursen JM. Cellular senescence in renal aging and disease. Nat Rev Nephrol. 2017;13:77–89.
Valentijn FA, Falke LL, Nguyen TQ, Goldschmeding R. Cellular senescence in the aging and diseased kidney. J Cell Commun Signal. 2018;12:69–82.
Nastase MV, Zeng-Brouwers J, Wygrecka M, Schaefer L. Targeting renal fibrosis: mechanisms and drug delivery systems. Adv Drug Deliv Rev. 2018;129:295–307.
Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol. 2015;11:264–76.
Zhao L, Han F, Wang J, Chen J. Current understanding of the administration of mesenchymal stem cells in acute kidney injury to chronic kidney disease transition: a review with a focus on preclinical models. Stem Cell Res Ther. 2019;10:385.
Andrade L, Rodrigues CE, Gomes SA, Noronha IL. Acute kidney injury as a condition of renal senescence. Cell Transpl. 2018;27:739–53.
Shimoda H, Doi S, Nakashima A, Sasaki K, Doi T, Masaki T. Inhibition of the H3K4 methyltransferase MLL1/WDR5 complex attenuates renal senescence in ischemia reperfusion mice by reduction of p16(INK4a). Kidney Int. 2019;96:1162–75.
Kim MG, Yang J, Ko YS, Lee HY, Oh SW, Cho WY, et al. Impact of aging on transition of acute kidney injury to chronic kidney disease. Sci Rep. 2019;9:18445.
Schafer MJ, Haak AJ, Tschumperlin DJ, LeBrasseur NK. Targeting senescent cells in fibrosis: pathology, paradox, and practical considerations. Curr Rheumatol Rep. 2018;20:3.
Gu Y, Huang F, Wang Y, Chen C, Wu S, Zhou S, et al. Connexin32 plays a crucial role in ROS-mediated endoplasmic reticulum stress apoptosis signaling pathway in ischemia reperfusion-induced acute kidney injury. J Transl Med. 2018;16:117.
Rodrigues CE, Capcha JM, de Bragança AC, Sanches TR, Gouveia PQ, de Oliveira PA, et al. Human umbilical cord-derived mesenchymal stromal cells protect against premature renal senescence resulting from oxidative stress in rats with acute kidney injury. Stem Cell Res Ther. 2017;8:19.
Rusten TE, Stenmark H. p62, an autophagy hero or culprit? Nat Cell Biol. 2010;12:207–9.
Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–23.
Chen JH, Chao CT, Huang JW, Hung KY, Liu SH, Tarng DC, et al. Early elimination of uremic toxin ameliorates AKI-to-CKD transition. Clin Sci (Lond). 2021;135:2643–58.
Luo C, Zhou S, Zhou Z, Liu Y, Yang L, Liu J, et al. Wnt9a promotes renal fibrosis by accelerating cellular senescence in tubular epithelial cells. J Am Soc Nephrol. 2018;29:1238–56.
Cui S, Xue L, Yang F, Dai S, Han Z, Liu K, et al. Postinfarction hearts are protected by premature senescent cardiomyocytes Via GATA 4-dependent CCN 1 secretion. J Am Heart Assoc. 2018;7:e009111.
Baixauli-Martín J, Aliena-Valero A, Castelló-Ruiz M, Burguete MC, López-Morales MA, Muñoz-Espín D, et al. Brain cell senescence: a new therapeutic target for the acute treatment of ischemic stroke. J Neuropathol Exp Neurol. 2022;81:614–20.
Torres-Querol C, Torres P, Vidal N, Portero-Otín M, Arque G, Purroy F. Acute ischemic stroke triggers a cellular senescence-associated secretory phenotype. Sci Rep. 2021;11:15752.
Amruta N, Rahman AA, Pinteaux E, Bix G. Neuroinflammation and fibrosis in stroke: the good, the bad and the ugly. J Neuroimmunol. 2020;346:577318.
Lim S, Kim TJ, Kim YJ, Kim C, Ko SB, Kim BS. Senolytic therapy for cerebral ischemia-reperfusion injury. Int J Mol Sci. 2021;22:11967.
Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482–96.
Zhang M, Ma Y, Chai L, Mao H, Zhang J, Fan X. Storax protected oxygen-glucose deprivation/reoxygenation induced primary astrocyte injury by inhibiting NF-κB activation in vitro. Front Pharmacol. 2018;9:1527.
Cui M, Huang Y, Tian C, Zhao Y, Zheng J. FOXO3a inhibits TNF-α- and IL-1β-induced astrocyte proliferation:Implication for reactive astrogliosis. Glia. 2011;59:641–54.
Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–47.
Pekny M, Wilhelmsson U, Tatlisumak T, Pekna M. Astrocyte activation and reactive gliosis—a new target in stroke? Neurosci Lett. 2019;689:45–55.
Zhu YM, Lin L, Wei C, Guo Y, Qin Y, Li ZS, et al. The key regulator of necroptosis, RIP1 kinase, contributes to the formation of astrogliosis and glial scar in ischemic stroke. Transl Stroke Res. 2021;12:991–1017.
van Willigenburg H, de Keizer PLJ, de Bruin RWF. Cellular senescence as a therapeutic target to improve renal transplantation outcome. Pharmacol Res. 2018;130:322–30.
Di Micco R, Krizhanovsky V, Baker D, d’Adda, di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. 2021;22:75–95.
Li C, Shen Y, Huang L, Liu C, Wang J. Senolytic therapy ameliorates renal fibrosis postacute kidney injury by alleviating renal senescence. FASEB J. 2021;35:e21229.
Iske J, Seyda M, Heinbokel T, Maenosono R, Minami K, Nian Y, et al. Senolytics prevent mt-DNA-induced inflammation and promote the survival of aged organs following transplantation. Nat Commun. 2020;11:4289.
Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016;22:78–83.
Mylonas KJ, O’Sullivan ED, Humphries D, Baird DP, Docherty MH, Neely SA, et al. Cellular senescence inhibits renal regeneration after injury in mice, with senolytic treatment promoting repair. Sci Transl Med 2021;13:eabb0203.
Prata L, Ovsyannikova IG, Tchkonia T, Kirkland JL. Senescent cell clearance by the immune system: emerging therapeutic opportunities. Semin Immunol. 2018;40:101275.
Goligorsky MS. Chronic kidney disease: a vicarious relation to premature cell senescence. Am J Pathol. 2020;190:1164–71.
Diao C, Wang L, Liu H, Du Y, Liu X. Aged kidneys are refractory to autophagy activation in a rat model of renal ischemia-reperfusion injury. Clin Inter Aging. 2019;14:525–34.
Barreto-Torres G, Parodi-Rullán R, Javadov S. The role of PPARα in metformin-induced attenuation of mitochondrial dysfunction in acute cardiac ischemia/reperfusion in rats. Int J Mol Sci. 2012;13:7694–709.
Kulkarni AS, Gubbi S, Barzilai N. Benefits of metformin in attenuating the hallmarks of aging. Cell Metab. 2020;32:15–30.
Xu C, Wang L, Fozouni P, Evjen G, Chandra V, Jiang J, et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nat Cell Biol. 2020;22:1170–9.
Lodrini AM, Goumans MJ. Cardiomyocytes cellular phenotypes after myocardial infarction. Front Cardiovasc Med. 2021;8:750510.
Engel N, Mahlknecht U. Aging and anti-aging: unexpected side effects of everyday medication through sirtuin1 modulation. Int J Mol Med. 2008;21:223–32.
Feng H, Mou SQ, Li WJ, Zhang N, Zhou ZY, Ding W, et al. Resveratrol inhibits ischemia-induced myocardial senescence signals and NLRP3 inflammasome activation. Oxid Med Cell Longev. 2020;2020:2647807.
Yu G, Sun W, Wang W, Le C, Liang D, Shuai L. Overexpression of microRNA-202-3p in bone marrow mesenchymal stem cells improves cerebral ischemia-reperfusion injury by promoting angiogenesis and inhibiting inflammation. Aging. 2021;13:11877–88.
Galipeau J, Sensébé L. Mesenchymal stromal cells: clinical challenges and therapeutic opportunities. Cell Stem Cell. 2018;22:824–33.
Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–7.
Ishiuchi N, Nakashima A, Doi S, Kanai R, Maeda S, Takahashi S, et al. Serum-free medium and hypoxic preconditioning synergistically enhance the therapeutic effects of mesenchymal stem cells on experimental renal fibrosis. Stem Cell Res Ther. 2021;12:472.
Li W, Chen W, Sun L. An update for mesenchymal stem cell therapy in lupus nephritis. Kidney Dis (Basel, Switz). 2021;7:79–89.
Simon C, Greening DW, Bolumar D, Balaguer N, Salamonsen LA, Vilella F. Extracellular vesicles in human reproduction in health and disease. Endocr Rev. 2018;39:292–332.
van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213–28.
Ou H, Teng H, Qin Y, Luo X, Yang P, Zhang W, et al. Extracellular vesicles derived from microRNA-150-5p-overexpressing mesenchymal stem cells protect rat hearts against ischemia/reperfusion. Aging. 2020;12:12669–83.
Han SH, Jeon JH, Ju HR, Jung U, Kim KY, Yoo HS, et al. VDUP1 upregulated by TGF-beta1 and 1,25-dihydorxyvitamin D3 inhibits tumor cell growth by blocking cell-cycle progression. Oncogene. 2003;22:4035–46.
Chen M, Chen J, Li C, Yu R, Chen W, Chen C. Improvement of cardiac function by mesenchymal stem cells derived extracellular vesicles through targeting miR-497/Smad7 axis. Aging. 2021;13:22276–85.
Xiao X, Xu M, Yu H, Wang L, Li X, Rak J, et al. Mesenchymal stem cell-derived small extracellular vesicles mitigate oxidative stress-induced senescence in endothelial cells via regulation of miR-146a/Src. Signal Transduct Target Ther. 2021;6:354.
Kim SR, Jiang K, Chen X, Puranik AS, Zhu XY, Lerman A, et al. Selective kidney targeting increases the efficacy of mesenchymal stromal/stem cells for alleviation of murine stenotic-kidney senescence and damage. J Tissue Eng Regen Med. 2022;16:550–8.
Yu H, Hua Y, He Y, Wang Y, Hu X, Chen S, et al. Sustained release of MiR-217 inhibitor by nanoparticles facilitates MSC-mediated attenuation of neointimal hyperplasia after vascular injury. Front Cardiovasc Med. 2021;8:739107.
Zhang F, Wan X, Cao YZ, Sun D, Cao CC. Klotho gene-modified BMSCs showed elevated antifibrotic effects by inhibiting the Wnt/β-catenin pathway in kidneys after acute injury. Cell Biol Int. 2018;42:1670–9.
Jia Y, Kang X, Tan L, Ren Y, Qu L, Tang J, et al. Nicotinamide mononucleotide attenuates renal interstitial fibrosis after AKI by suppressing tubular DNA damage and senescence. Front Physiol. 2021;12:649547.
Seki E, Brenner DA. Recent advancement of molecular mechanisms of liver fibrosis. J Hepato-biliary-Pancreat Sci. 2015;22:512–8.
Kishi S, Brooks CR, Taguchi K, Ichimura T, Mori Y, Akinfolarin A, et al. Proximal tubule ATR regulates DNA repair to prevent maladaptive renal injury responses. J Clin Investig. 2019;129:4797–816.
Xie X, Yang X, Wu J, Ma J, Wei W, Fei X, et al. Trib1 contributes to recovery from ischemia/reperfusion-induced acute kidney injury by regulating the polarization of renal macrophages. Front Immunol. 2020;11:473.
Li WF, Yang K, Zhu P, Zhao HQ, Song YH, Liu KC, et al. Genistein Ameliorates Ischemia/Reperfusion-Induced Renal Injury in a SIRT1-Dependent Manner. Nutrients. 2017;9:403.
Shan H, Li T, Zhang L, Yang R, Li Y, Zhang M, et al. Heme oxygenase-1 prevents heart against myocardial infarction by attenuating ischemic injury-induced cardiomyocytes senescence. EBioMedicine. 2019;39:59–68.
Meyer K, Hodwin B, Ramanujam D, Engelhardt S, Sarikas A. Essential role for premature senescence of myofibroblasts in myocardial fibrosis. J Am Coll Cardiol. 2016;67:2018–28.
Acknowledgements
This study was supported partly by the National Natural Science Foundation of China (Grant Nos. 81974296, 82102297), Natural Science Foundation of Guangdong Province (Grant Nos. 2019A1515110020 and 2022A1515012603), the Fundamental Research Funds for the Central Universities of China (Grant No. 22qntd3401) and the Young Talent Support Project of Guangzhou Association for Science and Technology (Grant No. QT20220101257).
Author information
Authors and Affiliations
Contributions
C.C. and M.Z. contributed equally to the manuscript. C.C., M.Z., H.H., and S.F. searched the literature and wrote the manuscript; L.C. and J.Y. designed the table and figures; C.C. and M.Z. conceived the thematic, W.Y., Q.Z., and Z.H. supervised the work. All authors approved the final manuscript and agreed to be responsible for this review.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, C., Zheng, M., Hou, H. et al. Cellular senescence in ischemia/reperfusion injury. Cell Death Discov. 8, 420 (2022). https://doi.org/10.1038/s41420-022-01205-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-022-01205-z
This article is cited by
-
Activation of senescence in critically ill patients: mechanisms, consequences and therapeutic opportunities
Annals of Intensive Care (2024)
-
The mechanism of ferroptosis and its related diseases
Molecular Biomedicine (2023)
