
Abstract
The phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway plays an essential role in glucose metabolism, promoting glycolysis and resisting gluconeogenesis. PI3K/AKT signaling can directly alter glucose metabolism by phosphorylating several metabolic enzymes or regulators of nutrient transport. It can indirectly promote sustained aerobic glycolysis by increasing glucose transporters and glycolytic enzymes, which are mediated by downstream transcription factors. E3 ubiquitin ligase RING-finger proteins are mediators of protein post-translational modifications and include the cullin-RING ligase complexes, the tumor necrosis factor receptor-associated family, the tripartite motif family and etc. Some members of the RING family play critical roles in regulating cell signaling and are involved in the development and progression of various metabolic diseases, such as cancer, diabetes, and dyslipidemia. And with the progression of modern research, as a negative or active regulator, the RING-finger adaptor has been found to play an indispensable role in PI3K/AKT signaling. However, no reviews have comprehensively clarified the role of RING-finger E3 ligases in PI3K/AKT-mediated glucose metabolism. Therefore, in this review, we focus on the regulation and function of RING ligases in PI3K/AKT-mediated glucose metabolism to establish new insights into the prevention and treatment of metabolic diseases.
Similar content being viewed by others

Facts
-
1.
PI3K/AKT signaling can directly or indirectly alter glucose metabolism, which is a crucial component of mammalian organ function.
-
2.
The mechanisms of RING-finger E3 ligases in PI3K/AKT-mediated glucose metabolism are widely researched.
-
3.
Zinc-binding RING-finger adaptor proteins such as CRLs, TRAF, TRIM, and MARCH play a significant role in the maintenance of glucose homeostasis.
Open questions
-
1.
Which RING-finger proteins are active regulators in PI3K/AKT-mediated glucose metabolism?
-
2.
Which RING-finger proteins are negative regulators in PI3K/AKT-mediated glucose metabolism?
-
3.
What is the exact mechanism of RING-finger ligase function in PI3K/AKT-mediated glucose metabolism?
Introduction
Glucose is an important nutrient that provides most of the energy required by various organs to achieve homeostasis. Therefore, stability of glucose metabolism is crucial for maintaining organ function in humans [1]. However, the prevalence of high-glucose diets has increased attention on glucose metabolism-related disorders [2]. Abnormalities in glucose metabolism contribute to multiple health conditions, including obesity [3], diabetes mellitus [4], hypertension [5], cancer [6], and polycystic ovary syndrome [7]. Under aerobic conditions, tumor tissues in human cancers are more likely to utilize glucose to produce lactate instead of entering the mitochondria and tricarboxylic acid cycle, a phenomenon termed the Warburg effect [8]. Moreover, disorders related to glucose metabolism are associated with several complications. Extreme hyperglycemia might cause ketoacidosis, which alters blood vessels and the nervous system [9]. Phosphatidylinositol 3-kinase (PI3K)/AKT pathway-mediated glucose metabolism signaling, which promotes glycolysis and resists gluconeogenesis, plays a key role in cell biology. The PI3K/AKT pathway alters glucose metabolism, both directly and indirectly [10].
PI3K/AKT signaling, as a well-known pathway, is activated among many diseases, especially cancer [11] and diabetes mellitus [12]. It directly alters glucose metabolism via phosphorylation-associated regulation of metabolic enzymes as well as glucose transporters 4 (GLUT4) trafficking [13]. Such regulatory events rely substantially on PI3K/AKT-associated phosphorylation and suppression of AS160 [14]. In addition to glucose uptake, PI3K/AKT regulates critical glycolytic events by phosphorylating and activating various enzymes. Following entry into the cells, glucose is activated for metabolic use by hexokinases, which carry out glucose phosphorylation to generate glucose-6-phosphate, whose transport out of the cell by GLUT1 is impossible [15]. PI3K/AKT signaling also influences glycolysis by indirectly stimulating the activity of phosphofructokinase-1. Fructose-2,6- biphosphate is phosphorylated and activated by PI3K/AKT. This enzyme catalyzes the formation of fructose-2,6-biphosphate and activates phosphofructokinase-1, a rate-limiting glycolytic enzyme, thereby committing glucose carbons to glycolysis [16] (Fig. 1a).

a AKT directly stimulates glucose metabolic changes by phosphorylating key glycolysis enzymes and influencing glucose transporters 4 translocation. b AKT indirectly changes glucose metabolism by impacting key downstream effectors through a combination of transcriptional, translational, and post-translational mechanisms. HK2: hexokinase 2; PFK1: phosphofructokinase-1; PFKFB2: fructose-2,6-biphosphatase; TSC2: tuberous sclerosis complex 2; RHEB: Ras homolog enriched in brain; mTORC1: mechanistic target of rapamycin complex 1; FOXO: forkhead box O; GSK3: glycogen synthase kinase 3; HIF1α: hypoxia-inducible factor 1α.
PI3K/AKT directly phosphorylates multiple enzymes or modulators involved in metabolism or nutrient transport. It further induces major downstream effectors with significant roles in cell metabolic reprogramming, such as the mechanistic target of rapamycin complex 1 (mTORC1) and glycogen synthase kinase 3 (GSK3) [17]. PI3K/AKT signaling also sustains aerobic glycolysis by upregulating glucose transporters and glycolytic enzymes through the regulation of downstream transcription factors such as forkhead box O (FOXO)-related proteins and MYC [10]. This pathway primarily induces mTORC1 by phosphorylating and inhibiting tuberous sclerosis complex 2, an important part of the TSC complex. It does so by alleviating TSC complex-related suppression of a Ras homolog enriched in the brain [18]. GSK3, which blocks glycogen production by phosphorylating and suppressing its eponym substrate, is suppressed by growth factors and insulin through PI3K/AKT-related phosphorylation [19]. GSK3-associated phosphorylation of the transcription factors MYC and hypoxia-inducible factor 1α (HIF1α) promotes their proteasomal degradation [20, 21] (Fig. 1b). Previous studies have demonstrated that protein post-translational modifications play an essential role in PI3K/AKT-mediated glucose metabolism. Here, we summarize the underlying molecular mechanisms of RING E3 ligases in the regulation of PI3K/AKT-mediated glucose metabolism.
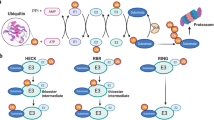
Protein ubiquitination is an important type of post-translational modification for ubiquitin-related protein degradation via the ubiquitin-proteasome system. The system comprises three enzymes: ubiquitin-activating enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3). Under the condition of providing energy by ATP, ubiquitin-activating enzyme E1 catalyzes the activation of ubiquitin. The active ubiquitin is then transferred to ubiquitin-conjugating enzyme E2 and ubiquitin ligase E3, which covalently binds ubiquitin to a target protein [22]. Depending on the presence of characteristic domains, E3 ligases comprise three major families: RING (Really Interesting New Gene) family [23], HECT (homologous to the EA6P carboxyl terminus) domain family [24], and the U-box families [25]. RING-finger proteins are broadly found in eukaryotic cells [26] and are the most abundant of E3 ubiquitin ligases [27]. They perform functions by zinc-binding RING-finger adaptor or a U-box domain, which are responsible for binding the ubiquitin-conjugating enzyme E2 and promoting ubiquitin transfer. RING-finger E3 proteins control cellular events such as DNA damage response [28], cell signaling, and glucose homeostasis [29]. Therefore, abnormal expression of these proteins may result in multiple metabolic disorders. Cullin, tumor necrosis factor receptor-associated factor (TRAF), tripartite motif (TRIM), and Membrane-associated RING-CH-type finger (MARCH) proteins regulate PI3K/AKT-mediated glucose homeostasis. As such, comprehensive analysis of these versatile RING-finger E3s might help to develop new management tools for metabolic dysfunction. Previous studies have reported the role of ubiquitination in PI3K/AKT-mediated glucose metabolism. However, a review that highlights the relationship between RING-finger E3 ubiquitin ligase and PI3K/AKT-mediated glucose metabolism is still lacking. Here, we summarize the roles of RING-finger E3 ubiquitinated ligases, such as Cullin, TRAF and TRIM families in PI3K/AKT-mediated glucose metabolism.
The Cullin Family and PI3K/AKT-mediated glucose metabolism
Cullins, as molecular scaffolds, ubiquitinate via simultaneous interaction with a substrate and anchorage of an E2 ubiquitin-conjugating enzyme via the RING domain. They are also known as cullin-RING ligase complexes (CRLs) and constitutes the most abundant RING E3 ligases [30]. CRLs act via cognate substrate-recognition molecules, including F-box [31], suppressors of cytokine signaling (SOCS) [32], Bric-a-brac, Tramtrack, Broad-complex [33], and von Hippel-Lindau (VHL) proteins. These protein families comprise characteristic motifs recognized by specific adaptors conjugated to the respective cognate cullins. Therefore, a CRL brings a substrate near the E2 that transfers ubiquitin to the substrate [34] (Fig. 2a). Mammals have eight Cullin members, including CUL1, CUL2, CUL3, CUL4a, CUL4b, CUL5, CUL7 and PARC. CRL1, CRL2, CRL4, and CRL7 play important roles in PI3K/AKT-mediated glucose metabolism [29, 35,36,37].

a The components of a CRL. b–e Specific protein factors for each CRL family. Skp1 S phase kinase-associated protein-1, FOXO1 forkhead box O 1, E2 ubiquitin conjugating enzyme, Rbx1 regulator of cullins 1, VHL von Hippel-Lindau, HIF1α hypoxia-inducible factor 1α, DDB1 damage-specific DNA binding protein 1, COP1 constitutive photomorphogenesis protein 1, Fbw8 F-box and WD-repeat-domain-containing protein 8, IRS1 insulin receptor substrate 1.
The CRL1 complex
The multi-subunit CRL1 is a member of the RING E3 ligase family. It has broad substrate specificity and regulates multiple cellular events [38]. Most F-box proteins are involved in the formation of the SCF E3 ubiquitin ligase complex via Skp1, which interacts with CUL1 (Fig. 2b). Additionally, reports assessing F-box family members, such as β-TrCP, Skp2, F-box, and WD-repeat-domain-containing protein 7 (Fbw7), have demonstrated that a given substrate recognition protein can interact with many substrates, thus increasing the functional scope of CRLs and mediating ubiquitination and degradation of various signaling proteins [34]. The Skp2-SCF complex is an essential E3 ligase for PI3K/AKT ubiquitination and membrane recruitment. Skp2 suppression impairs PI3K/AKT activation, GLUT1 expression, glucose uptake, and glycolysis [35]. Moreover, the substrate-binding F-box protein Skp2 of the SCF-Skp2 E3 ligase binds equally to ubiquitinates and promotes the degradation of FOXO1 through PI3K/AKT-phosphorylated FOXO1 at Ser256 [39]. FOXO family members, including FOXO1, FOXO3A, and FOXO4, are canonical substrates of PI3K/AKT. They are phosphorylated and sequestered from the nuclear compartment, subsequently downregulating their respective target genes. FOXO1, the major hepatic FOXO protein that drives PEPCK and G6Pase expression, determines the ability of insulin to maintain glucose homeostasis and regulate glucose biosynthesis in the liver [40] (Fig. 3). By antagonizing the function of MYC, FOXO transcription factors inhibit the expression of glycolytic genes. The activated Skp2-SCF complex degrades FOXO1, thereby relieving the inhibitory effects of FOXO transcription factors on glycolytic enzymes and inhibiting gluconeogenesis [41] (Table 1).

The upstream and major downstream effectors of AKT signaling in glycogen synthesis, gluconeogenesis, glucose transport, and protein synthesis. IRS insulin receptor substrate, PI3K phosphatidylinositol 3-kinase, PTP1B protein-tyrosine phosphatase 1B, PIP3 phosphatidylinositol 3,4,5-trisphosphate, PDK1 phosphoinositide-dependent protein kinase 1, mTORC mechanistic target of rapamycin complex, FOXO forkhead box O, GSK3 glycogen synthase kinase 3, TSC2 tuberous sclerosis complex 2, RHEB Ras homolog enriched in brain.
Fbw7, an F-box protein, is another substrate recognition protein of SCF ubiquitin ligase [42]. It promotes proteasome-dependent c-Myc ubiquitination in vivo and in vitro [43]. The MYC oncoprotein is an important transcription factor that regulates many genes involved in cell growth, proliferation, and metabolic pathways [44]. MYC, a transcription factor, induces the expression of GLUT1 and pyruvate kinase and hexokinase. In addition, MYC activity is generally decided by its abundance and is regulated via the way of transcriptional, translational mechanisms and PTM [45]. MTORC1 signaling regulates the abundance of MYC by enhancing the translation of MYC [46, 47], whereas PI3K/AKT promotes MYC stabilization by inhibiting proteasomal degradation. GSK3, a serine-threonine kinase, can phosphorylate MYC and facilitate its degradation [43, 48, 49] (Fig. 1b). Recent studies have suggested that downregulation of the PI3K/AKT-GSK3β-FBW7 signaling axis promotes destabilization of c-Myc, which downregulates hexokinase 2 [50] (Table 1).
The CRL2 complex
The Elongin B/C-Cul2/Cul5-SOCS-box protein complex belongs to the RING E3 ubiquitin ligase family, which shares a Cullin-Rbx (RING box protein) module. SOCS-box proteins perform substrate recruitment to the complex and are associated with Cullin-Rbx through Elongin B/C (Fig. 2c). VHL is considered a SOCS-box protein, although it lacks the C-terminus of the SOCS box. It binds to endogenous Cul2-Rbx1 in mammalians [51]. CRL2pVHL is essential for controlling oxygen homeostasis via the regulation of hypoxia-inducing transcription factor for degradation [52]. HIF1α, whose degradation depends on oxygen levels, is stabilized under hypoxia. Furthermore, HIF1α upregulates GLUT1 and almost all glycolytic enzymes [53]. To adapt to hypoxic conditions, this protein also upregulates lactate dehydrogenase 1 and pyruvate dehydrogenase kinase 1, which promotes anaerobic glycolysis and inhibits oxidative phosphorylation [54]. Several reports have shown that PI3K/AKT signaling activates mTORC1, and further upregulates the expression of HIF1α, which upregulates gene targets such as phosphofructokinase, and enhances glucose uptake and glucose transformation into lactate, even in normoxia [55,56,57,58]. Thus, degradation of VHL enhances the function of HIF1α, which reprograms glucose and energy metabolism, including enhanced glucose uptake, glycolytic processes, and lactate synthesis [59, 60] (Table 1).
Many proinflammatory cytokines upregulate SOCS protein binding through the Src homology 2 domains to activate cytokine receptors or related Janus kinases, contributing to a negative feedback loop that attenuates cytokine signaling. The SOCS protein also interacts with the Elongin BC-containing E3 ubiquitin ligase complex via the SOCS box. SOCS1 and SOCS3 have been suggested to inhibit heterogeneous pathways by ubiquitinating and degrading insulin receptor substrate 1 (IRS1) and IRS2 [61], thereby suppressing insulin-associated phosphorylation of the p85 subunit of PI3K and AKT [62] (Table 1).
Phosphorylation of IRS by the insulation receptor at a tyrosine residue enables its interaction with Src homology 2 domain proteins, such as the regulatory subunits of class 1 A PI3K. PI3K activation at the cytoplasmic membrane induces phosphorylation of its phospholipid substrate, phosphatidylinositol 4,5-bisphosphate, to generate the second messenger, phosphatidylinositol 3,4,5-trisphosphate (PIP3) [63]. Threonine kinase AKT interacts with PIP3 and undergoes phosphorylation by phosphoinositide-dependent protein kinase 1 (PDPK1 or PDK1) at Thr308 or by mTORC2 at Ser473, which further induces the activation of AKT [64, 65] (Fig. 3). Further, PI3K/AKT signaling partly promotes cell proliferation by altering metabolic pathways in cells. In addition, ubiquitination of IRS1 mediated by the Cul2-Rbx1-SOCS complex suppresses the activation of PI3K/AKT, which causes changes in glucose metabolism.
The CRL4 complex
Constitutive photomorphogenesis protein 1 (COP1), part of the Cul4A-RING E3 ubiquitin ligase complex, is an evolutionarily conserved protein [66]. COP1 can facilitate substrate degradation through its function of adaptor protein of other factors, including damage-specific DNA binding protein 1 (DDB1), CUL4 and Rbx1, to form a large complex [67] (Fig. 2d). Plant studies have deeply assessed COP1, which is also expressed in mammalian organisms, controlling gluconeogenesis, lipid metabolism and tumorigenesis [68]. This protein affects glucose homeostasis by downregulating gluconeogenic genes, thereby suppressing glucose production in the liver [37]. In the presence of insulin, COP1 can induce IRS phosphorylation by inhibiting the activity of protein-tyrosine phosphatase 1B (PTP1B), which is a negative regulator of insulin receptor and IRS [69] (Fig. 3). The regulator mechanism activates multiple protein kinases that reduce hepatic glucose biosynthesis, thereby maintaining glucose homeostasis [70]. Moreover, insulin upregulates COP1, which interacts with FOXO1 and induces its degradation through the ubiquitin-proteasome system [37, 71]. In this manner, the CUL4A-DDB1-COP1 complex inhibits gluconeogenesis and maintains glucose homeostasis (Table 1).
The CRL7 complex
CUL7 belongs to the cullin family of RING E3 ubiquitin ligases and interacts with the Rbx1 RING-finger protein to generate a complex. In this complex, Fbxw8 and Fbxw11 are the only F-box proteins that interact with CUL7 [72]. Several studies have shown that SCF-Fbxw8 E3 ligase targets IRS-1 for degradation. However, the mechanism employed by CUL7 to selectively interact with Fbw8 is unclear. CUL7 has been proven to resemble CUL1 in utilizing the Skp1 adaptor [73] (Fig. 2e). In addition, CUL7 suppression in the C2C12 cell line and heterozygous knockout of mouse CUL7 or Fbxw8 elevated IRS-1 protein levels, PI3K/AKT activity, and glucose uptake after insulin stimulation [29, 74]. These findings confirm that CUL7 modulates the insulin pathway and glucose homeostasis. The function of Fbw8 is regulated by mTORC2, upstream of AKT, which leads to the phosphorylation and activation of AKT [75]. MTORC2 induces Fbw8 stabilization via phosphorylation at Ser86, promoting insulin-associated cytoplasmic translocation of Fbw8 with subsequent IRS1 degradation [76]. The CD36 receptor functions via ubiquitination-dependent interaction with IRS1, suppressing binding to CUL7. Furthermore, Fyn (a Src family kinase) dissociation from CD36 using free fatty acids or Fyn silencing/suppression accelerates insulin-associated IRS1 degradation. This is possibly due to the inhibition of IRS1 binding to CD36, which enhances the interaction with CUL7. This process is critical for the negative feedback modulation of insulin pathway induction and glucose metabolism [77] (Table 1).
The TRAF Family and PI3K/AKT-mediated glucose metabolism
TRAF domain, which is subdivided into two regions: the TRAF-N domain and TRAF-C domain, is a characteristic feature of TRAF family. At the N-terminal, most TRAFs possess a RING domain, followed by many zinc finger domains [78], and various intracellular signaling molecules bind to the TRAF-N domain [79]. The TRAF family serves as adaptor proteins that mediate cytokine signaling and overtly regulate cell survival, proliferation, and stress response [80]. TRAF6 and TRAF4, which are members of the TRAF family, regulate multiple protein-to-protein interactions through the TRAF and RING-finger domains with nonconventional E3 ubiquitin ligase activity [81]. Ubiquitin binds via a lysine residue (K63, K48, K33, K29, K27, K11, or K6) or N-terminal methionine, allowing the assembly of specific polymers [82], although K48 and K63 are the greatest players. K48, K11, and K29-linked poly-Ub directly interact with substrates for proteasomal degradation [83, 84], whereas mono-ubiquitination and K63-associated poly-Ub chains play nonproteolytic roles [85]. Interestingly, TRAF4 and TRAF6-mediated ubiquitination of AKT act via K63-induced ubiquitination, but not K48-dependent ubiquitination, which has no effect on AKT stability but controls PI3K/AKT pathway activation by promoting the AKT membrane recruitment and phosphorylation by serine/threonine kinase [86,87,88].
TRAF6-mediated membrane recruitment and AKT induction may occur via K63-linked ubiquitination of APPL1. APPL1, an adaptor protein, binds to AKT and suppresses AKT binding to its endogenous suppressor tribble 3 via direct competition, thereby inducing AKT translocation to the cytoplasmic membrane and endosomes for subsequent activation [89,90,91]. K63-linked ubiquitination of APPL1, which is induced by TRAF6, enhances membrane recruitment and activation of Akt. A recent study showed that TRAF6 downregulation alleviates insulin-induced ubiquitination and membrane targeting of APPL1, impairing insulin-induced PI3K/AKT activation [92]. In addition, histone lysine demethylase 4B promotes glucose uptake by regulating GLUT1, which is ubiquitously expressed and controls cellular glucose uptake. Histone lysine demethylase 4B interacts with TRAF6 and promotes PI3K/AKT ubiquitination and activation [93]. Furthermore, thioredoxin-interacting protein (TXNIP), a downstream substrate of PI3K/AKT, contributes to the control of GLUT1 trafficking [94]. The protein enhances GLUT1 endocytosis and suppresses glucose uptake [95]. PI3K/AKT signaling phosphorylates and suppresses TXNIP, rapidly increasing GLUT1 and GLUT4 levels in the cytoplasmic membrane and enhancing cellular glucose uptake [96] (Fig. 3). Furthermore, studies have indicated that TRAF4 was overexpressed in human lung cancer cells and tissues and is necessary for activating the critical cell survival kinase PI3K/AKT via ubiquitination [97]. These studies demonstrated that PI3K/AKT undergoes K63-associated ubiquitination by TRAF6 and TRAF4, which is critical for PI3K/AKT membrane recruitment and facilitates glycolysis and glucose uptake [86, 88, 97] (Table 2).
The TRIM family and PI3K/AKT-mediated glucose metabolism
TRIM-related proteins are E3 ubiquitin ligases containing a RING-finger domain. However, not all proteins with a RING-finger domain may act as E3 ubiquitin ligases [98]. In addition to the RING-finger domain, TRIM proteins have one or two zinc-binding motifs termed B-boxes and a coiled-coil region [99]. Multiple substrate specificities of TRIM proteins may be determined by shuffling the respective ligands [100]. Most TRIM proteins, which act as E3 ubiquitin ligases can contribute to many oncogenic events, including transcriptional regulation, cell proliferation, apoptosis, and tumorigenesis [101].
Several studies have demonstrated that TRIM proteins regulate glucose homeostasis by increasing PI3K/AKT induction and glucose transportation [102, 103]. TRIM31-knockout (KO) mice developed glucose intolerance and insulin resistance. Furthermore, decreased AKT Thr308 phosphorylation has been detected in TRIM31-KO mice [102]. TRIM31 also exerted its function by directly interacting with the tuberous sclerosis complex and promoting the degradation of this complex, the upstream suppressor of mTORC1, which facilitates the glycolysis and glucose uptake [104] (Table 2). TRIM32, another TRIM protein, represents an E3 ubiquitin ligase [105]. TRIM32 levels are elevated in many cancers, promoting malignant cell proliferation [106, 107]. Furthermore, TRIM32 is a proliferation and anti-apoptotic protein involved in PI3K/AKT signaling in gastric cancer. This protein potentially controls glycolysis by upregulating GLUT1 and hexokinase 2 in gastric cancer cells [103] (Table 2).
However, some TRIM proteins are considered as negative regulators that suppress the phosphorylation of PI3K/AKT [108, 109]. TRIM63, also known as muscle-specific RING-finger protein-1 (MuRF1), plays an important role in skeletal and cardiac muscle atrophy. This protein is a negative regulator of PI3K/AKT [110]. MuRF1-KO mice showed higher serum glucose and triglyceride levels, and decreased glucose tolerance. MuRF1-KO skeletal muscle had an altered PI3K/AKT pathway, enhanced AKT-Ser-473 induction, and decreased oxidative mitochondrial function, suggesting possible mechanisms underlying MuRF1-associated modulation of glucose and fat metabolism (Table 2). Meanwhile, MyoMed-205, the MuRF1 inhibitor in mice with experimental diabetes significantly affects serum glucose [108]. TRIM26 acts as a tumor suppressor in multiple forms of cancer [111]. A recent study investigated the effect of this protein on proliferative, metastatic, and glycolytic processes in papillary thyroid carcinoma (PTC). TRIM26 overexpression was found to inhibit the malignant potential of PTC cells and substantially reduce glucose uptake and lactate synthesis. Further studies demonstrated that TRIM26 overexpression inhibited PI3K/AKT signaling (Table 2). Administration of an inducer (740Y-P) of PI3K/AKT signaling reversed the anticancer effects of TRIM26 in PTC cells. These data provide evidence that TRIM26 acts as a negative regulator of PI3K/AKT in PTC cells [109]. TRIM proteins exhibit both the biological effects of upregulation and downregulation of PI3K/AKT signaling, thus playing different roles in cell metabolism.
The MARCH Family and PI3K/AKT-mediated glucose metabolism
MARCH proteins, as a subfamily of the RING-finger E3 ligases, contains a C4HC3-type RING domain. K3 and K5, MARCH E3 ubiquitin ligases that are expressed by Kaposi’s sarcoma-associated herpesvirus, are the initial membrane-associated ubiquitin E3 ligases [112] and with further researches, more other members which have RING-CH domains were identified [113]. On the mitochondrial outer membrane, MARCH5 is ubiquitously expressed in human organs such as the heart, brain, liver, and lungs. MARCH5 has four transmembrane domains and an N-terminal C4HC3-type RING-finger domain, which is important for ubiquitin ligase activity [114]. In an earlier study, the authors found that besides a potential effect on antiviral immune responses [115], MARCH5 equally enhances proliferation and alters metabolism in cells [116]. This ubiquitin ligase enhances aerobic glycolysis and lactate biosynthesis by regulating cell growth factor-binding receptor tyrosine kinase endocytosis, thereby enhancing cell sensitivity to autocrine and paracrine factors, altering the pattern of cell phosphorylation, and increasing PI3K/AKT activation [117] (Table 2).
Conclusion
By organizing RING-finger proteins that direct many substrates for ubiquitin-associated degradation or K63-mediated nonproteolytic functions, RING family proteins generate an important regulatory network that controls glucose homeostasis via the PI3K/AKT pathway, thereby regulating multiple biological processes. This review summarized the roles and regulation of RING-finger proteins in association with PI3K/AKT-mediated glucose metabolism. Among the zinc-binding RING-finger adaptor proteins, CRLs, TRAF, TRIM, and MARCH are involved in maintaining glucose homeostasis.
In recent years, the association of RING family members, especially CRLs and TRAF, with metabolic disorders have received extensive attention. The PI3K/AKT pathway is a classical signaling transduction pathway that is related to cell metabolism. Exploring the relationship between RING-finger proteins and PI3K/AKT-mediated glucose metabolism would provide new insights into the treatment of metabolic diseases.
Data availability
The data used to support the findings of this study are available from the corresponding author upon request.
References
Du H, Zhao Y, Yin Z, Wang DW, Chen C. The role of miR-320 in glucose and lipid metabolism disorder-associated diseases. Int J Biol Sci. 2021;17:402–16.
Hirode G, Wong RJ. Trends in the prevalence of metabolic syndrome in the United States, 2011–2016. JAMA. 2020;323:2526–8.
Lu Q, Guo P, Liu A, Ares I, Martínez-Larrañaga M-R, Wang X, et al. The role of long noncoding RNA in lipid, cholesterol, and glucose metabolism and treatment of obesity syndrome. Med Res Rev. 2021;41:1751–74.
DeFronzo RA, Ferrannini E, Groop L, Henry RR, Herman WH, Holst JJ, et al. Type 2 diabetes mellitus. Nat Rev Dis Prim. 2015;1:15019.
Xu W, Janocha AJ, Erzurum SC. Metabolism in pulmonary hypertension. Annu Rev Physiol. 2021;83:551–76.
Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 2015;17:351–9.
van Helden J, Evliyaoglu O, Küberl A, Weiskirchen R. Disorders of the glucose metabolism correlate with the phenotype and the severity in women with polycystic ovary syndrome. Clin Endocrinol (Oxf). 2020;93:44–51.
Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41:211–8.
Dhatariya KK, Glaser NS, Codner E, Umpierrez GE. Diabetic ketoacidosis. Nat Rev Dis Prim. 2020;6:40.
Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. 2020;20:74–88.
Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501.
Huang X, Liu G, Guo J, Su Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int J Biol Sci. 2018;14:1483–96.
Ng Y, Ramm G, Lopez JA, James DE. Rapid activation of Akt2 is sufficient to stimulate GLUT4 translocation in 3T3-L1 adipocytes. Cell Metab. 2008;7:348–56.
Eguez L, Lee A, Chavez JA, Miinea CP, Kane S, Lienhard GE, et al. Full intracellular retention of GLUT4 requires AS160 Rab GTPase activating protein. Cell Metab. 2005;2:263–72.
Roberts DJ, Tan-Sah VP, Smith JM, Miyamoto S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J Biol Chem. 2013;288:23798–806.
Houddane A, Bultot L, Novellasdemunt L, Johanns M, Gueuning M-A, Vertommen D, et al. Role of Akt/PKB and PFKFB isoenzymes in the control of glycolysis, cell proliferation and protein synthesis in mitogen-stimulated thymocytes. Cell Signal. 2017;34:23–37.
Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169:381–405.
Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H, et al. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell. 2014;156:771–85.
Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–9.
Xie L, Yi J, Song Y, Zhao M, Liqiang F, Liming Z. Suppression of GOLM1 by EGCG through HGF/HGFR/AKT/GSK-3β/β-catenin/c-Myc signaling pathway inhibits cell migration of MDA-MB-231. Food Chem Toxicol. 2021;157:112574.
Dai T, Li L, Qi W, Liu B, Jiang Z, Song J, et al. Balanophorin B inhibited glycolysis with the involvement of HIF-1α. Life Sci. 2021;267:118910.
Francesca EM, Helen W. Types of ubiquitin ligases. Cell. 2016;165:248.
Wang D, Ma LN, Wang B, Liu J, Wei WY. E3 ubiquitin ligases in cancer and implications for therapies. Cancer Metastasis Rev. 2017;36:683–702.
Li ZY, Chen SY, Jhong JH, Pang YX, Huang KY, Li SF, et al. UbiNet 2.0: a verified, classified, annotated and updated database of E3 ubiquitin ligase-substrate interactions. Database (Oxf). 2021;2021:baab010.
Yang Q, Zhao JY, Chen D, Wang Y. E3 ubiquitin ligases: styles, structures and functions. Mol Biomed. 2021;2:23.
Sun J, Sun Y, Ahmed RI, Ren A, Xie AM. Research progress on plant RING-finger proteins. Genes. 2019;10:E973.
Joazeiro CA, Weissman AM. RING finger proteins: mediators of ubiquitin ligase activity. Cell. 2000;102:549–52.
Moore LE, Nickerson ML, Brennan P, Toro JR, Jaeger E, Rinsky J, et al. Von Hippel-Lindau (VHL) inactivation in sporadic clear cell renal cancer: associations with germline VHL polymorphisms and etiologic risk factors. PLoS Genet. 2011;7:e1002312.
Scheufele F, Wolf B, Kruse M, Hartmann T, Lempart J, Mühlich S, et al. Evidence for a regulatory role of Cullin-RING E3 ubiquitin ligase 7 in insulin signaling. Cell Signal. 2014;26:233–9.
Harper JW, Schulman BA. Cullin-RING ubiquitin ligase regulatory circuits: a quarter century beyond the F-Box hypothesis. Annu Rev Biochem. 2021;90:403–29.
Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416:703–9.
Bullock AN, Debreczeni JE, Edwards AM, Sundström M, Knapp S. Crystal structure of the SOCS2-elongin C-elongin B complex defines a prototypical SOCS box ubiquitin ligase. Proc Natl Acad Sci USA. 2006;103:7637–42.
Zhuang M, Calabrese MF, Liu J, Waddell MB, Nourse A, Hammel M, et al. Structures of SPOP-substrate complexes: insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol Cell. 2009;36:39–50.
Sarikas A, Hartmann T, Pan Z-Q. The cullin protein family. Genome Biol. 2011;12:220.
Chan C-H, Li C-F, Yang W-L, Gao Y, Lee S-W, Feng Z, et al. The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptin sensitivity, and tumorigenesis. Cell. 2012;149:1098–111.
Sudhagar S, Sathya S, Lakshmi BS. Rapid non-genomic signalling by 17β-oestradiol through c-Src involves mTOR-dependent expression of HIF-1α in breast cancer cells. Br J Cancer. 2011;105:953–60.
Sanchez-Barcelo EJ, Mediavilla MD, Vriend J, Reiter RJ. Constitutive photomorphogenesis protein 1 (COP1) and COP9 signalosome, evolutionarily conserved photomorphogenic proteins as possible targets of melatonin. J Pineal Res. 2016;61:41–51.
Asmamaw MD, Liu Y, Zheng Y-C, Shi X-J, Liu H-M. Skp2 in the ubiquitin-proteasome system: A comprehensive review. Med Res Rev. 2020;40:1920–49.
Huang H, Tindall DJ. Dynamic FoxO transcription factors. J Cell Sci. 2007;120:2479–87.
Wang L, Scott I, Zhu L, Wu K, Han K, Chen Y, et al. GCN5L1 modulates cross-talk between mitochondria and cell signaling to regulate FoxO1 stability and gluconeogenesis. Nat Commun. 2017;8:523.
Daitoku H, Fukamizu A. FOXO transcription factors in the regulatory networks of longevity. J Biochem (Tokyo). 2007;141:769–74.
Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83–93.
Welcker M, Orian A, Jin J, Grim JE, Grim JA, Harper JW, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci USA. 2004;101:9085–90.
Farrell AS, Sears RC. MYC degradation. Cold Spring Harb Perspect Med. 2014;4:a014365.
Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, metabolism, and cancer. Cancer Disco. 2015;5:1024–39.
West MJ, Stoneley M, Willis AE. Translational induction of the c-myc oncogene via activation of the FRAP/TOR signalling pathway. Oncogene. 1998;17:769–80.
Csibi A, Lee G, Yoon S-O, Tong H, Ilter D, Elia I, et al. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr Biol. 2014;24:2274–80.
Gregory MA, Qi Y, Hann SR. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J Biol Chem. 2003;278:51606–12.
Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2501–14.
Yuan J, Peng G, Xiao G, Yang Z, Huang J, Liu Q, et al. Xanthohumol suppresses glioblastoma via modulation of Hexokinase 2 -mediated glycolysis. J Cancer. 2020;11:4047–58.
Kamura T, Maenaka K, Kotoshiba S, Matsumoto M, Kohda D, Conaway RC, et al. VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 2004;18:3055–65.
Kim JA, Choi DK, Min JS, Kang I, Kim JC, Kim S, et al. VBP1 represses cancer metastasis by enhancing HIF-1α degradation induced by pVHL. FEBS J. 2018;285:115–26.
Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32.
Semba H, Takeda N, Isagawa T, Sugiura Y, Honda K, Wake M, et al. HIF-1α-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat Commun. 2016;7:11635.
Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–83.
Zhong H, Chiles K, Feldser D, Laughner E, Hanrahan C, Georgescu MM, et al. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 2000;60:1541–5.
Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, et al. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol. 2002;22:7004–14.
Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med. 2004;10:594–601.
Semenza GL. HIF-1 mediates the Warburg effect in clear cell renal carcinoma. J Bioenerg Biomembr. 2007;39:231–4.
Haque M, Kendal JK, MacIsaac RM, Demetrick DJ. WSB1: from homeostasis to hypoxia. J Biomed Sci. 2016;23:61.
Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem. 2002;277:42394–8.
Bose SK, Shrivastava S, Meyer K, Ray RB, Ray R. Hepatitis C virus activates the mTOR/S6K1 signaling pathway in inhibiting IRS-1 function for insulin resistance. J Virol. 2012;86:6315–22.
Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–51.
Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7:261–9.
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101.
Xu D. COP1 and BBXs-HY5-mediated light signal transduction in plants. N. Phytol. 2020;228:1748–53.
Wertz IE, O’Rourke KM, Zhang Z, Dornan D, Arnott D, Deshaies RJ, et al. Human De-etiolated-1 regulates c-Jun by assembling a CUL4A ubiquitin ligase. Science. 2004;303:1371–4.
Song Y, Liu Y, Pan S, Xie S, Wang Z-W, Zhu X. Role of the COP1 protein in cancer development and therapy. Semin Cancer Biol. 2020;67:43–52.
Li D, Zhang S, Yang C, Li Q, Wang S, Xu X, et al. A novel PTP1B inhibitor-phosphate of polymannuronic acid ameliorates insulin resistance by regulating IRS-1/Akt signaling. Int J Mol Sci. 2021;22:12693.
Ren W, Sun Y, Cheema S, Du K. Interaction of constitutive photomorphogenesis 1 protein with protein-tyrosine phosphatase 1B suppresses protein-tyrosine phosphatase 1B activity and enhances insulin signaling. J Biol Chem. 2013;288:10902–13.
Kato S, Ding J, Pisck E, Jhala US, Du K. COP1 functions as a FoxO1 ubiquitin E3 ligase to regulate FoxO1-mediated gene expression. J Biol Chem. 2008;283:35464–73.
Shi L, Du D, Peng Y, Liu J, Long J. The functional analysis of Cullin 7 E3 ubiquitin ligases in cancer. Oncogenesis. 2020;9:98.
Dias DC, Dolios G, Wang R, Pan Z-Q. CUL7: A DOC domain-containing cullin selectively binds Skp1.Fbx29 to form an SCF-like complex. Proc Natl Acad Sci USA. 2002;99:16601–6.
Xu X, Sarikas A, Dias-Santagata DC, Dolios G, Lafontant PJ, Tsai S-C, et al. The CUL7 E3 ubiquitin ligase targets insulin receptor substrate 1 for ubiquitin-dependent degradation. Mol Cell. 2008;30:403–14.
Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–76.
Kim SJ, DeStefano MA, Oh WJ, Wu C, Vega-Cotto NM, Finlan M, et al. mTOR complex 2 regulates proper turnover of insulin receptor substrate-1 via the ubiquitin ligase subunit Fbw8. Mol Cell. 2012;48:875–87.
Sun S, Tan P, Huang X, Zhang W, Kong C, Ren F, et al. Ubiquitinated CD36 sustains insulin-stimulated Akt activation by stabilizing insulin receptor substrate 1 in myotubes. J Biol Chem. 2018;293:2383–94.
Das A, Middleton AJ, Padala P, Ledgerwood EC, Mace PD, Day CL. The structure and ubiquitin binding properties of TRAF RING heterodimers. J Mol Biol. 2021;433:166844.
Hyun HP. Structure of TRAF family: current understanding of receptor recognition. Front Immunol. 2018;9:1999.
Inoue JI, Ishida T, Tsukamoto N, Kobayashi N, Naito A, Azuma S, et al. Tumor necrosis factor receptor-associated factor (TRAF) family: adapter proteins that mediate cytokine signaling. Exp Cell Res. 2000;254:14–24.
Walsh MC, Lee J, Choi Y. Tumor necrosis factor receptor- associated factor 6 (TRAF6) regulation of development, function, and homeostasis of the immune system. Immunol Rev. 2015;266:72–92.
F I, I D. Atypical ubiquitin chains: new molecular signals. ‘Protein Modifications: beyond the Usual Suspects’ review series. EMBO Rep. 2008;9:536–42.
Jin L, Williamson A, Banerjee S, Philipp I, Rape M. Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell. 2008;133:653–65.
Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–83.
Tanno H, Komada M. The ubiquitin code and its decoding machinery in the endocytic pathway. J Biochem (Tokyo). 2013;153:497–504.
Chan C-H, Jo U, Kohrman A, Rezaeian AH, Chou P-C, Logothetis C, et al. Posttranslational regulation of Akt in human cancer. Cell Biosci. 2014;4:59.
Yang W-L, Wang J, Chan C-H, Lee S-W, Campos AD, Lamothe B, et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science. 2009;325:1134–8.
Yang W-L, Wu C-Y, Wu J, Lin H-K. Regulation of Akt signaling activation by ubiquitination. Cell Cycle Georget Tex. 2010;9:487–97.
Cheng KKY, Iglesias MA, Lam KSL, Wang Y, Sweeney G, Zhu W, et al. APPL1 potentiates insulin-mediated inhibition of hepatic glucose production and alleviates diabetes via Akt activation in mice. Cell Metab. 2009;9:417–27.
Wang Y, Cheng KKY, Lam KSL, Wu D, Wang Y, Huang Y, et al. APPL1 counteracts obesity-induced vascular insulin resistance and endothelial dysfunction by modulating the endothelial production of nitric oxide and endothelin-1 in mice. Diabetes. 2011;60:3044–54.
Cheng KKY, Lam KSL, Wu D, Wang Y, Sweeney G, Hoo RLC, et al. APPL1 potentiates insulin secretion in pancreatic β cells by enhancing protein kinase Akt-dependent expression of SNARE proteins in mice. Proc Natl Acad Sci USA. 2012;109:8919–24.
Cheng KKY, Lam KSL, Wang Y, Wu D, Zhang M, Wang B, et al. TRAF6-mediated ubiquitination of APPL1 enhances hepatic actions of insulin by promoting the membrane translocation of Akt. Biochem J. 2013;455:207–16.
Li H, Lan J, Wang G, Guo K, Han C, Li X, et al. KDM4B facilitates colorectal cancer growth and glucose metabolism by stimulating TRAF6-mediated AKT activation. J Exp Clin Cancer Res. 2020;39:12.
Waldhart AN, Dykstra H, Peck AS, Boguslawski EA, Madaj ZB, Wen J, et al. Phosphorylation of TXNIP by AKT mediates acute influx of glucose in response to insulin. Cell Rep. 2017;19:2005–13.
Wu N, Zheng B, Shaywitz A, Dagon Y, Tower C, Bellinger G, et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell. 2013;49:1167–75.
Hong SY, Yu F-X, Luo Y, Hagen T. Oncogenic activation of the PI3K/Akt pathway promotes cellular glucose uptake by downregulating the expression of thioredoxin-interacting protein. Cell Signal. 2016;28:377–83.
Li W, Peng C, Lee M-H, Lim D, Zhu F, Fu Y, et al. TRAF4 is a critical molecule for Akt activation in lung cancer. Cancer Res. 2013;73:6938–50.
Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, et al. The tripartite motif family identifies cell compartments. EMBO J. 2001;20:2140–51.
Meroni G, Diez-Roux G. TRIM/RBCC, a novel class of ‘single protein RING finger’ E3 ubiquitin ligases. BioEssays N. Rev Mol Cell Dev Biol. 2005;27:1147–57.
Herquel B, Ouararhni K, Khetchoumian K, Ignat M, Teletin M, Mark M, et al. Transcription cofactors TRIM24, TRIM28, and TRIM33 associate to form regulatory complexes that suppress murine hepatocellular carcinoma. Proc Natl Acad Sci USA. 2011;108:8212–7.
Hatakeyama S. TRIM proteins and cancer. Nat Rev Cancer. 2011;11:792–804.
Cheng J, Xue F, Zhang M, Cheng C, Qiao L, Ma J, et al. TRIM31 deficiency is associated with impaired glucose metabolism and disrupted gut microbiota in mice. Front Physiol. 2018;9:24.
Wang J, Fang Y, Liu T. TRIM32 promotes the growth of gastric cancer cells through enhancing AKT activity and glucose transportation. BioMed Res Int. 2020;2020:4027627.
Guo P, Ma X, Zhao W, Huai W, Li T, Qiu Y, et al. TRIM31 is upregulated in hepatocellular carcinoma and promotes disease progression by inducing ubiquitination of TSC1-TSC2 complex. Oncogene. 2018;37:478–88.
Locke M, Tinsley CL, Benson MA, Blake DJ. TRIM32 is an E3 ubiquitin ligase for dysbindin. Hum Mol Genet. 2009;18:2344–58.
Cui X, Lin Z, Chen Y, Mao X, Ni W, Liu J, et al. Upregulated TRIM32 correlates with enhanced cell proliferation and poor prognosis in hepatocellular carcinoma. Mol Cell Biochem. 2016;421:127–37.
Zhao T-T, Jin F, Li J-G, Xu Y-Y, Dong H-T, Liu Q, et al. TRIM32 promotes proliferation and confers chemoresistance to breast cancer cells through activation of the NF-κB pathway. J Cancer. 2018;9:1349–56.
Labeit S, Hirner S, Bogomolovas J, Cruz A, Myrzabekova M, Moriscot A, et al. Regulation of glucose metabolism by MuRF1 and treatment of myopathy in diabetic mice with small molecules targeting MuRF1. Int J Mol Sci. 2021;22:2225.
Wang K, Chai L, Qiu Z, Zhang Y, Gao H, Zhang X. Overexpression of TRIM26 suppresses the proliferation, metastasis, and glycolysis in papillary thyroid carcinoma cells. J Cell Physiol. 2019;234:19019–27.
Peris-Moreno D, Taillandier D, Polge C. MuRF1/TRIM63, master regulator of muscle mass. Int J Mol Sci. 2020;21:E6663.
Wang Y, He D, Yang L, Wen B, Dai J, Zhang Q, et al. TRIM26 functions as a novel tumor suppressor of hepatocellular carcinoma and its downregulation contributes to worse prognosis. Biochem Biophys Res Commun. 2015;463:458–65.
Lehner PJ, Hoer S, Dodd R, Duncan LM. Downregulation of cell surface receptors by the K3 family of viral and cellular ubiquitin E3 ligases. Immunol Rev. 2005;207:112–25.
Lin H, Li S, Shu HB. The membrane-associated MARCH E3 ligase family: emerging roles in immune regulation. Front Immunol. 2019;10:1751.
Nobuhiro N, Kimura Y, Tokuda M, Honda S, Hirose S. MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 2006;7:1019–22.
Pardieu C, Vigan R, Wilson SJ, Calvi A, Zang T, Bieniasz P, et al. The RING-CH ligase K5 antagonizes restriction of KSHV and HIV-1 particle release by mediating ubiquitin-dependent endosomal degradation of tetherin. PLoS Pathog. 2010;6:e1000843.
Bond ST, Moody SC, Liu Y, Civelek M, Villanueva CJ, Gregorevic P, et al. The E3 ligase MARCH5 is a PPARγ target gene that regulates mitochondria and metabolism in adipocytes. Am J Physiol Endocrinol Metab. 2019;316:E293–E304.
Karki R, Lang SM, Means RE. The MARCH family E3 ubiquitin ligase K5 alters monocyte metabolism and proliferation through receptor tyrosine kinase modulation. PLoS Pathog. 2011;7:e1001331.
Acknowledgements
We thank Chang Gao and Mohan Li of China Medical University for their guidance on this manuscript. This study was supported by the National Natural Science Foundation of China (Nos. 82071607, 82171571, and 81900355); LiaoNing Revitalization Talents Program (No. XLYC1907071); National Postdoctoral Innovative Talents Support Program (BX2021376).
Author information
Authors and Affiliations
Contributions
WW contributed to the manuscript and figures. BS and RC and MH modified the grammar. YP and HY and JS and DF summarized the molecular mechanisms of PI3K/AKT and glucose metabolism. NZ and DL contributed to the conception and design. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, W., Shi, B., Cong, R. et al. RING-finger E3 ligases regulatory network in PI3K/AKT-mediated glucose metabolism. Cell Death Discov. 8, 372 (2022). https://doi.org/10.1038/s41420-022-01162-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-022-01162-7
This article is cited by
-
PECAM-1 drives β-catenin-mediated EndMT via internalization in colon cancer with diabetes mellitus
Cell Communication and Signaling (2023)
-
Tripartite motif containing 26 prevents steatohepatitis progression by suppressing C/EBPδ signalling activation
Nature Communications (2023)
