
Abstract
The transcription factor EB (TFEB) regulates the expression of target genes bearing the Coordinated Lysosomal Expression and Regulation (CLEAR) motif, thereby modulating autophagy and lysosomal biogenesis. Furthermore, TFEB can bind to the promoter of autophagy-associated genes and induce the formation of autophagosomes, autophagosome–lysosome fusion, and lysosomal cargo degradation. An increasing number of studies have shown that TFEB stimulates the intracellular clearance of pathogenic factors by enhancing autophagy and lysosomal function in multiple kidney diseases, such as cystinosis, acute kidney injury, and diabetic nephropathy. Taken together, this highlights the importance of developing novel therapeutic strategies against kidney diseases based on TFEB regulation. In this review, we present an overview of the current data on TFEB and its implication in kidney disease.
Similar content being viewed by others


Facts
-
The transcription factor EB regulates the expression of CLEAR motif-containing target genes involved in autophagy and lysosomal biogenesis.
-
Aberrant autophagy and impaired lysosomal function are important in kidney diseases.
-
TFEB deficiency is involved in the development of kidney diseases.
-
Thus, targeting TFEB activity, autophagy, and mitophagy could be a novel therapeutic strategy for patients with kidney diseases.
Introduction
Autophagy is an evolutionarily conserved intracellular homoeostatic process wherein cytoplasmic cargo-containing autophagosomes fuse with lysosomes to degrade the cargo1. Macroautophagy and selective autophagy (e.g., mitophagy, aggrephagy) influence cellular processes, such as cell death, inflammation, and immune responses, thereby exerting adaptive and maladaptive roles in the pathogenesis of multiple human diseases, such as skeletal muscle diseases, cancer, neurodegenerative diseases, systemic lupus erythematosus, chronic kidney disease etc.2,3.
Transcription factor EB (TFEB), a basic helix–loop–helix-leucine-zipper (bHLH-Zip) protein in the microphthalmia/transcription factor E (MiT/TFE) family, primarily controls the expression of genes in the autophagy–lysosomal pathway4,5,6. TFEB regulates autophagic flux by promoting the biogenesis of lysosomes, formation of autophagosomes, and fusion with lysosomes, thereby facilitating substrate clearance. TFEB also functions in selective autophagy and lysosomal exocytosis7. Overexpression of TFEB enhances the degradation of bulk amounts of substrates, lipid droplets, and damaged mitochondria and alleviates the phenotypes associated with various diseases, such as Parkinson’s and Alzheimer’s disease, in murine models by promoting autophagy and lysosomal biogenesis8. In particular, current researches declared that TFEB is associated with kidney disease pathogenesis in diverse conditions, such as diabetic nephropathy (DN)9 and acute kidney disease10. In this review, we have described the mechanisms involved in the regulation of TFEB activation and, subsequently, coordinating lysosomal function and autophagy. We have emphasised the role of TFEB in kidney diseases and its potential as a therapeutic in rescuing renal function.
MIT/TFE family of transcription factors
Four members of the microphthalmia family of bHLH-Zip transcription factors have been identified: MITF/TFEF, TFEB, TFE3, and TFEC4,8 (Fig. 1). The common features of the MIT/TFE proteins include a DNA-binding motif, HLH, and Zip region necessary for dimerisation4. MITF/TFEF, TFEB, and TFE3 also possess an activation domain required for its transcriptional activation function8,11. TFEC does not contain this activation domain and seems to play a role of inhibition to its downstream gene’s transcription rather than activation12 (Fig. 2 shows the domain structure and homology model of TFEB.)

a Comparison of the amino acid sequences of the bHLH and leucine zipper regions of TFEB, TFE3, TFEC, and MITF. b The various domains found in TFEB, TFE3, TFEC, and MITF. Gln glutamine-rich region, AD acidic domain, bHLH basic helix–loop–helix, LZ leucine zipper domain, Pro proline-rich segment, Pro+Arg proline- and arginine-rich region, Ser serine-rich region.

The homology model was adopted from the SWISS-MODEL (https://swissmodel.expasy.org/; a fully automated protein structure homology-modelling server).
MIT/TFE proteins are conservative in vertebrates13 and primarily expressed in the retinal pigment epithelia, macrophages, osteoclasts, mast cells, melanocytes, and natural killer cells14, while TFEC expression is limited to myeloid cells15. TFE3 and TFEB are expressed in multiple cell types16. Researches of MIT/TFE proteins have shown that they play a critical role in the maintenance of physiological and pathological processes17. Aberrant expression of these proteins stimulate the development of various human cancers, including renal carcinomas18,19, melanomas20, and alveolar sarcomas21.
Transcriptional control in lysosomes by TFEB
The lysosome was discovered in the early 1950s as a membrane-bound organelle containing more than 50 types of acid hydrolases for a wide variety of substrates, including proteins, carbohydrates, lipids, and nucleic acids22. Lysosomes are primary sites of intracellular degradation and molecular recycling system and maintenance of cellular homoeostasis23. Lysosomes are crucial for endocytosis, autophagy, and lysosomal exocytosis24. Lysosomal genes share a 10-base E-box-like palindromic sequence (GTCACGTGAC) typically found within 200 base pairs of the transcription initiation site. This motif, named the Coordinated Lysosomal Expression and Regulation (CLEAR) element, comprises an E-box (CANNTG) that was recognised by the MIT/TFE family transcription factors5.
TFEB enhances the expression of its target genes by specifically binding to the CLEAR motif present in the target promoters5,7. Thus, TFEB overexpression increases the biogenesis of lysosomes and improves their capacity for degrading lysosomal substrates, such as glycosaminoglycans and substrates for autophagy5,8. In addition, TFEB can promote lysosomal exocytosis which is a process that lysosomes can secrete content out of cell through fusing to cell membrane25. This highlights the importance of transcriptional control of gene expression in lysosomal function.
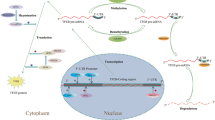
TFEB is involved in intracellular clearance by enhancing lysosomal biogenesis and function, autophagy, and lysosomal exocytosis (Fig. 3a)7. TFEB overexpression improves the rate of degradation of the cargo, e.g., long-lived proteins6 and enhances the elimination of lipid droplets26 and damaged mitochondria27. Hence, TFEB is the central modulator of the autophagy–lysosomal pathway and organelle-specific autophagy, including lipophagy and mitophagy.

a TFEB regulates several biological processes, such as autophagy, lysosomal biogenesis, lysosomal exocytosis, lipophagy, mitophagy, etc. b Under nutrient-rich conditions, TFEB is predominantly found in its inactive state that is characterised by mTORC1 and ERK2-mediated phosphorylation. When cells experience stress or disease, TFEB is activated by dephosphorylation.
Mechanism involved in regulating TFEB activity
TFEB regulates biological processes by modulating the expression of downstream genes. TFEB activity is tightly controlled by post-translational modifications and protein–protein interactions8. During nutrient-rich conditions, TFEB is predominantly in its inactive phosphorylated form in the cytosol5,6. However, under conditions of starvation, oxidative stress, or lysosomal disorder, TFEB translocates to the nucleus to bind its target genes and stimulate autophagy and lysosomal function28 (Fig. 3b).
The phosphorylation state of TFEB depends on two serine residues (Ser142 (refs. 6,29) and Ser211 (refs. 30,31)) that regulates its activity and cellular localisation. When Ser142 and Ser211 are phosphorylated, TFEB is inactive and localises to the cytoplasm. Mutating either of the serine residues to alanine enables TFEB to be constitutively active and localise to the nucleus6,29,30,31.
Mechanistic target of rapamycin complex 1 (mTORC1) is an atypical Ser/Thr kinase that is stimulated by the nutrient content and provides balance between anabolism and catabolism32. Ser211 is phosphorylated by mTORC1, which phosphorylates TFEB at Ser211 and induces the binding between TFEB and 14-3-3 proteins, thereby inhibiting TFEB from shuttling into the nucleus. After mTORC1 is inhibited, TFEB gets dephosphorylated and translocates into the nucleus30,31,33. It has been recently reported that mTORC1 can phosphorylate Ser211 and Ser122 to regulate TFEB activity32. Ser142, another serine involved in the subcellular localisation of TFEB, is phosphorylated by mTORC1 and ERK2 (refs. 6,29). Studies have shown that TFEB phosphorylation at Ser142 competes with that at Ser211, but the mechanism is yet to be elucidated34,35. Therefore, understanding the phosphorylation status of these amino acid residues is critical for TFEB activity and localisation.
ERK2 is another important Ser/Thr kinase that regulates cell proliferation, differentiation, transformation, apoptosis, and autophagy6,36. Under normal cellular conditions, ERK2 phosphorylates Ser142 in TFEB and holds its cytosolic localisation. During cell starvation, ERK2 no longer maintains the phosphorylation of TFEB, thereby activating TFEB and enabling its translocation to the nucleus6. ERK2 localises to multiple subcellular compartments, including the lysosome37. Further research is required to understand whether ERK2-mediated TFEB phosphorylation occurs on the lysosomal surface or in the other subcellular compartments.
Calcineurin is a serine/threonine phosphatase that regulates TFEB activity38. During nutrient deficiency or lysosomal stress, Ca2+ is released from the lysosome via mucolipin 1, thereby stimulating calcineurin, TFEB dephosphorylation, and nuclear translocation33. Upon nutrient deprivation, the depletion of mucolipin 1 decreases the activation of TFEB and inhibits autophagy by hindering lysosomal Ca2+ release and calcineurin activation33.
The protein kinase C (PKC) family of proteins comprise Ser/Thr kinases that are involved in cell growth, differentiation, apoptosis, transformation, tumorigenicity, synaptic function, behaviour, and cognition39. Activated isoforms of PKC, primarily PKCα, PKCβ, and PKCδ, stimulate the biogenesis of lysosomes by coupling two parallel signalling pathways without affecting mTORC1. Activated PKCα and PKCδ inactivate GSK3β, a ubiquitous Ser/Thr kinase that is involved in translational regulation, thereby activating TFEB via reduced phosphorylation and increased nuclear localisation35. PKCδ phosphorylates JNK and p38 MAPK, thereby inactivating ZKSCAN3 and translocating it out of the nucleus35,40. Therefore, PKC activates TFEB and inactivates the transcriptional repressor ZKSCAN3 by targeting two parallel signalling cascades.
Interestingly, some regulators of TFEB are in turn regulated by TFEB. PGC-1α (peroxisome proliferator-activated receptor g coactivator 1 α) is the main modulator of mitochondrial biogenesis and respiration41,42. Several reports show that TFEB regulates mitochondrial biogenesis and glucose through controlling cellular uptake of PGC-1α26,42. Moreover, PGC-1α upregulates and activates TFEB26. These phenotypes demonstrate that the regulation of and by TFEB dictates survival in response to environmental stresses that is balanced by numerous feedback loops8. Furthermore, TFEB upregulation induces its own transcriptional activation26, revealing the presence of another feedback loop that controls lysosomal biogenesis and function.
Research status and application of regulated TFEB activity as therapy in kidney diseases
Cystinosis
Cystinosis is a lysosomal storage disorder due to mutations in CTNS gene (encoding cystinosin) that is a proton-cystine symporter responsible for the secretion of cystine dimers and protons from the lysosomal lumen43,44,45. Cystinosis is a rare autosomal recessive disease that is found in 1 out of 100,000 individuals46. Cystinosis manifests with the accumulation of cystine in lysosomes, thereby resulting in cellular dysfunction and affecting multiple organs, such as kidneys, eyes, liver, and brain47. Fanconi syndrome occurs during the first year of life and is characterised by kidney (primarily proximal tubular) dysfunction with polyuria and loss of glucose, proteins, and amino acids in the urine47,48. Without treatment, patients develop progressive glomerular damage and end-stage renal disease (ESRD) in ~10 years49. ESRD, dehydration, and electrolyte imbalance can be attributed to the rate of mortality in patients with Fanconi syndrome46.
It has been recently reported that proximal tubular injury results in increased incidence of apoptosis50, anomalous autophagy51,52, endoplasmic reticulum stress53, endo-lysosomal dysfunction54, and cell dedifferentiation55. Cysteamine has been the only treatment available for cystinosis since the late 1970s48,56; it reduces lysosomal cystine content and delays glomerular aggravation while improving growth of children with cystinosis. However, even treated with cysteamine, patients require a kidney transplantation owing to cellular dysfunction, progressive renal injury, and tissue failure49,57,58. Thus, there is an urgent need for developing novel therapeutic strategies against cystinosis that improves the quality of life and prognosis of patients with nephropathic cystinosis.
TFEB is the primary transcription factor involved in regulating the expression of lysosomal elements, including cystinosin, and lysosomal biogenesis and clearance by coordinating autophagy and anabolism24. Similar to the lysosome-related genes, TFEB regulates the expression of CTNS by binding to the E-box in its promoter7. Furthermore, Rega et al.48 reported that cystinotic cells, derived from a patient with cystinosis or CTNS-depleted (short interfering RNA-mediated) healthy cells, possess decreased levels of TFEB that cannot be rescued by cysteamine. Moreover, depleting cells of CTNS results in the decrease of TFEB without affecting the accumulation of intracellular cystine, indicating that decreased levels of TFEB lead to the deficiency in cystinosin, but not cystine accumulation. Similarly, Zhang et al.58 demonstrated that CTNS−/− mouse fibroblasts exhibited downregulated endogenous TFEB that, upon upregulation, promoted the expression of cystinosin and rescued cystinosis. Andrzejewska et al.59 showed that clearance of lysosomal cystine using cysteamine cannot reactivate mTORC1 (upstream effector of TFEB) in cystinotic cells, further suggesting that mTORC1 inactivation is a result of cystinosin deficiency rather than cystine accumulation and cystinosin directly regulates mTORC1 activity. Thus, cystinosin functions directly, and indirectly, in regulating the intracellular expression and activity of TFEB, thereby limiting cysteamine from fully curing patients with Fanconi syndrome.
Cystinosis is also characterised by the presence of abnormal lysosomes that result in aberrant autophagy. Ivanova et al.54 showed that cells deficient for cystinosin have atypical shape, disordered endo-lysosomal compartments, and damaged endocytosis. Cysteamine treatment promotes surface expression of receptors (megalin and cubilin) and elimination of lysosomal substrates but failed to recover the aberrant morphology of the endo-lysosomal compartments. Activation of TFEB increases its expression, nuclear translocation, and induction of lysosome biogenesis by genistein, thereby effectively promoting the clearance of aggregates60,61,62,63. Impaired autophagy has been recently shown to be associated with the pathogenesis of cystinosis52,64. Genetic and chemical manipulation stimulates TFEB activity that promotes the clearance of accumulated cystine, degradation of delayed cargo, and recovery of the aberrant morphology of the lysosomal compartments in cystinotic cells48. Taken together, cystinosis results from the reduction in expression and activity of TFEB and regulating TFEB may be a potential therapeutic strategy against cystinosis.
Acute kidney injury
Acute kidney injury (AKI) is a commonly occurring disease of the kidney that affects ∼13.3 million patients and causes 1.7 million deaths per year65 and characterised by sudden kidney failure or injury, thereby resulting in the deposition of metabolic end-products and/or decreased urine output66. Patients with AKI can develop chronic kidney disease and are at a higher risk of ESRD in the future67. The pathogenesis of AKI involves inflammation, damage to the tubular epithelia, endothelial and vascular disorder, reperfusion, sepsis, and nephrotoxins68,69,70,71,72,73. The renal tubules are severely injured in patients with AKI that results in tubular malfunction and necrosis and apoptosis; proximal tubules are particularly vulnerable to injury70,71,72,73,74,75. Tubular cells that survive restore the integrity of the remnant tubule by increased proliferation; inaccurate and suboptimal repairs lead to renal fibrosis and, ultimately, chronic kidney disease76.
Autophagosomes were discovered in tubular epithelial cells from a rodent model for renal ischaemia–reperfusion injury (IRI)77. Subsequently, the induction of autophagy has been observed in in vivo and in vitro models of IRI. Inhibiting autophagy aggravates IRI as indicated by renal function, histology, and tubular cell apoptosis, suggesting that autophagy plays a renoprotective role in cell survival in IRI78. Cisplatin-induced AKI is similar to IRI: inhibiting autophagy using pharmacological inhibitors (3-methyladenine or bafilomycin A1) or genetic manipulation (knockdown of Beclin-1 or ATG5) increases apoptosis in cisplatin-treated RPTC cells, suggesting autophagy is important in protecting renal function in cisplatin-induced tubular cell injury79. The group of Yang et al.80 showed that cisplatin-induced autophagy in LLC-PK1 cells play a protective role against cell apoptosis. Similarly, cisplatin-treated AKI model revealed that the inhibition in autophagic flux remarkably enhances kidney injury, while the activation of autophagy protects proximal tubules from injury81. Wang et al.10 demonstrated that TFEB, activated by urolithin A, promotes autophagy by regulating CLEAR motif-containing genes and attenuates IRI by reducing inflammation and kidney injury. Thus, the induction of autophagy by TFEB can be used as a novel therapeutic strategy for AKI.
Mitochondria are dynamic organelles that undergo continuous biogenesis, fusion, and fission and are selectively removed by mitophagy; all of these processes are crucial for its function and have been proven to be associated with AKI82. Whitaker et al.83 demonstrated disrupted mitochondrial homoeostasis and biogenesis after ischaemia–reperfusion, rhabdomyolysis-induced AKI, and folic acid-induced AKI. Tran et al.84 indicated inhibiting PGC-1α and mitochondrial biogenesis may take part in disease progression in sepsis-induced AKI. Persistent stimulation of mitochondrial biogenesis continually replenishes the cellular mitochondrial content and ameliorates tubular damage induced by ischaemia/reperfusion, toxins, and oxidants85,86. Disrupted mitochondrial dynamics result in mitochondrial dysfunction, tubular cell damage, and death in ischaemia–reperfusion and cisplatin-induced AKI86. Gonzalez et al.87 demonstrated that the imbalance between mitochondrial fusion and fission determines the differential progression of experimental sepsis. Liu et al.88 found that the balance between mitochondrial fusion/fission is tilted towards fission in the later stages of sepsis-induced AKI. They also reported impaired mitophagy during this stage although mitophagy is stimulated in the earlier stages of disease. Mitophagy maintains mitochondrial biogenesis and energy metabolism by removing dysfunctional or excrescent mitochondria and preventing the accumulation of reactive oxygen species, inflammation, and apoptosis89,90,91,92. Therefore, moderating mitochondrial biogenesis, mitochondrial dynamics, and mitophagy may be beneficial in preventing AKI. As Scott et al.93 have shown that TFEB (downstream effector of PGC-1α) controls the cellular mitochondrial content. Lynch et al.94 demonstrated that TFEB is required for PGC-1α-dependent tubular protection because PGC-1α promotes lysosomal biogenesis via TFEB as AKI progressing. Tan et al.95 reported that pomegranate extract potentiates mitophagy to eliminate the accumulation of dysfunctional mitochondria and reactive oxygen species in a TFEB-dependent manner. Liu et al.96 demonstrated that TFEB overexpression induces mitophagy by promoting the expression of autophagy-related genes. Moreover, Prajapati et al.97 found that TFEB expression induces the biogenesis of lysosomes and promotes the recovery of mitochondrial function. Taken together, TFEB activation promotes mitophagy and rescues mitochondrial function and can be a promising candidate to target AKI.
As mentioned, TFEB plays a critical role in promoting lysosomal function and autophagy by regulating the CLEAR motif-containing genes. TFEB and the CLEAR network are associated with molecular interactions in AKI. TFEB binds to the promoters of its targets and regulates various degradation pathways, such as glycosaminoglycan, haemoglobin, chitin, and sphingolipid7, that are critical in AKI98,99. TFEB also regulates other processes associated with AKI100,101,102, such as protein degradation, energy metabolism, steroid biosynthesis, antigen processing and presentation, signalling pathways, and DNA metabolism7. TFEB is involved in eliminating dead and injured cells (critical for the recovery of AKI7). Taken together, TFEB is a promising therapeutic target for AKI.
Diabetic nephropathy
DN is a serious complication associated with diabetes and one of the most important factors in the development of ESRD worldwide103. Approximately 35–40% diabetic patients (type 1 or 2) ultimately develop DN; this has led to the poor clinical outcome and increase in mortality in diabetic patients104. The International Diabetes Federation has predicted a global increase in the number of patients with DN from 382 million to 592 million between 2013 and 2035 (ref. 105). Unfortunately, this will be associated with increased economic burden for patients and the society.
Diabetic patients manifest with disrupted multi-factor interactions between metabolic processes and haemodynamics, thereby enabling the development of DN104. Clinical studies have shown that hyperglycaemia is the primary risk factor for complications associated with diabetes, including DN106. DN can be attributed to the alterations in cellular metabolism mediated by hyperglycaemia, including the accumulation of advanced glycation end-products (AGEs), activation of PKC, and oxidative stress107,108. Furthermore, chronic hyperglycaemia activates the diacylglycerol–PKC pathway that regulates vascular permeability, vasoconstriction, and the synthesis and turnover of the extracellular matrix109. Moreover, the pathogenesis of DN is influenced by the haemodynamic changes that lead to systemic and glomerular hypertension and renin–angiotensin system110,111,112,113.
Impaired autophagy has also been reported to be involved in the pathogenesis of DN. Renal cortical tubules of streptozotocin (STZ)-induced early diabetic rats exhibit inhibited autophagy that can be reversed upon treatment with insulin or islet transplantation114,115. Impaired autophagy results in the renal deposition of p62/Sequestosome 1 (SQSTM1) and degradation products from autophagy in mouse models of type 1 and type 2 diabetes, respectively116,117. Liu et al.118 showed that autophagosomes and SQSTM1-positive amass in tubular epithelial cells in patients with DN or HK-2 cells treated with AGEs, indicating that AGEs disturb autophagy–lysosomal pathway of renal tubular epithelial cell. Moreover, biopsies from animal models and human kidney showed that aberrant autophagy is involved in renal cell injuries and the pathogenesis of DN119.
Recent studies have revealed that impaired autophagy contributes to the development of DN via nutrient-sensing pathways. Excess nutrition leads to the activation of mTOR and reduction of AMPK (AMP-activated protein kinase) and Sirt1 (Sirtuin 1), thereby leading to the inhibition of autophagy in human and experimental type 1 and 2 DN120,121,122,123. Autophagy can be inhibited in podocytes upon prolonged treatment with high concentrations of glucose that can be recovered by the addition of rapamycin, indicating that mTOR inhibits autophagy during hyperglycaemia, leading to podocyte injury124. Similarly, impaired autophagy can be reversed by rapamycin-mediated inhibition of mTOR in STZ-induced diabetic mice125. Zhao et al.126 demonstrated that mTOR-induced autophagic dysfunction can be attributed to podocyte injury stimulated by high content of AGEs; pharmacologic downregulation of mTOR reverses impaired autophagy. This reinforces the potential for targeting autophagic flux in patients with DN. Thus, TFEB regulates autophagy via the mTOR pathways and, as compared to mTOR, is a more direct target to activate autophagy.
DN is also characterised by TFEB dysfunction. Brijmohan et al.9 found that in humans with DN, the accumulation of misfolded proteins coincided with a decrease in TFEB expression. Takahashi et al.127 demonstrated that lysosomal biogenesis and function were induced in vitro by increasing amounts of AGEs, but was inhibited in autophagy-deficient renal proximal tubular epithelial cells due to the cytoplasmic retention of TFEB. Similarly, STZ-treated mice with defective autophagy do not induce lysosomal biogenesis and exhibit amassed AGEs in the glomeruli, renal vasculature, and proximal tubular epithelial cells. These findings indicate that autophagy promotes the clearance of AGEs by increasing lysosomal biogenesis and function; this can be augmented by activating TFEB. Zhao et al.126 also showed that AGEs inhibit the formation and turnover of autophagosomes in podocytes by disrupting the binding of TFEB to the promoters of autophagy-related genes and activating mTOR to prevent the transcriptional activation of TFEB. Conversely, TFEB overexpression restores autophagic flux, confirming that downregulation of TFEB leads to impaired autophagy stimulated by AGEs in podocytes. Taken together, these studies reveal a pivotal role of TFEB in the pathogenesis of DN that can be used as a therapeutic strategy to protect autophagy.
Conclusion
TFEB is an important transcriptional modulator that regulates the expression of genes involved in the autophagy–lysosomal pathways, thereby controlling intracellular clearance and energy metabolism. Understanding the role of TFEB has helped provide insights into the mechanisms involved in cellular response to environmental conditions, such as nutrient deficiency. The advancements in research on TFEB and MIT/TFE proteins have led to a better understanding of the function, regulatory mechanisms, and pathways affected by these transcription factors. However, how cells integrate multiple extra- and intracellular signals to modulate an appropriate response remains to be investigated. Numerous studies have shown that improving intracellular clearance may alleviate the symptoms associated with a variety of diseases, especially neurodegenerative diseases. Regulating TFEB activity may be a promising therapeutic strategy against kidney diseases (Table 1). However, the function and mechanism of TFEB involved in the treatment process of kidney disease are still notemployed by TFEB in these processes remain to be fully understood. Therefore, further studies need to be performed to develop TFEB as a therapeutic target for kidney diseases.
References
Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 20, 521–527 (2018).
Choi, M. E. Autophagy in kidney disease. Annu. Rev. Physiol. 82, 297–322 (2020).
Jiang, P. & Mizushima, N. Autophagy and human diseases. Cell Res. 24, 69–79 (2014).
Steingrímsson, E., Copeland, N. G. & Jenkins, N. A. Melanocytes and the Microphthalmia Transcription Factor Network. Annu. Rev. Genet. 38, 365–411 (2004).
Sardiello, M. et al. A gene network regulating lysosomal biogenesis and function. Science 325, 473–477 (2009).
Settembre, C. et al. TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433 (2011).
Palmieri, M. et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 20, 3852–3866 (2011).
Napolitano, G. & Ballabio, A. TFEB at a glance. J. Cell Sci. 129, 2475–2481 (2016).
Brijmohan, A. S. et al. HDAC6 inhibition promotes transcription factor EB activation and is protective in experimental kidney disease. Front. Pharm. 9, 1–12 (2018).
Wang, Y. et al. Role of TFEB in autophagic modulation of ischemia reperfusion injury in mice kidney and protection by urolithin A. Food Chem. Toxicol. 131, 110591 (2019).
Beckmann, H., Su, L. K. & Kadesch, T. TFE3: a helix-loop-helix protein that activates transcription through the immunoglobulin enhancer μE3 motif. Genes Dev. 4, 167–179 (1990).
Zhao, G. Q., Zhao, Q., Zhou, X., Mattei, M. G. & de Crombrugghe, B. TFEC, a basic helix-loop-helix protein, forms heterodimers with TFE3 and inhibits TFE3-dependent transcription activation. Mol. Cell. Biol. 13, 4505–4512 (1993).
Bouché, V. et al. Drosophila Mitf regulates the V-ATPase and the lysosomal-autophagic pathway. Autophagy 12, 484–498 (2016).
Martina, J. A. et al. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci. Signal. 7, ra9–ra9 (2014).
Martina, J. A., Diab, H. I., Li, H. & Puertollano, R. Novel roles for the MiTF/TFE family of transcription factors in organelle biogenesis, nutrient sensing, and energy homeostasis. Cell. Mol. Life Sci. 71, 2483–2497 (2014).
Jebbink, J. M. et al. Increased glucocerebrosidase expression and activity in preeclamptic placenta. Placenta 36, 160–169 (2015).
Yang, M. et al. Emerging roles and regulation of MiT/TFE transcriptional factors. Cell Commun. Signal. 16, 1–11 (2018).
Kauffman, E. C. et al. Molecular genetics and cellular features of TFE3 and TFEB fusion kidney cancers. Nat. Rev. Urol. 11, 465–475 (2014).
Inamura, K. Translocation renal cell carcinoma: an update on clinicopathological and molecular features. Cancers (Basel). 9, 111 (2017).
Garraway, L. A. et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 436, 117–122 (2005).
Tanaka, M. et al. Modeling alveolar soft part sarcoma unveils novel mechanisms of metastasis. Cancer Res. 77, 897–907 (2017).
De Duve, C., Pressman, B. C., Gianetto, R., Wattiaux, R. & Appelmans, F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 60, 604–617 (1955).
Ballabio, A. The awesome lysosome. EMBO Mol. Med. 8, 73–76 (2016).
Settembre, C., Fraldi, A., Medina, D. L. & Ballabio, A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 14, 283–296 (2013).
Medina, D. L. et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell 21, 421–430 (2011).
Settembre, C. et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 15, 647–658 (2013).
Nezich, C. L., Wang, C., Fogel, A. I. & Youle, R. J. MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. J. Cell Biol. 210, 435–450 (2015).
Raben, N. & Puertollano, R. TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu. Rev. Cell Dev. Biol. 32, 255–278 (2016).
Settembre, C. et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 31, 1095–1108 (2012).
Martina, J. A., Chen, Y., Gucek, M. & Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8, 903–914 (2012).
Roczniak-Ferguson, A. et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 5, ra42 (2012).
Vega-Rubin-de-Celis, S., Peña-Llopis, S., Konda, M. & Brugarolas, J. Multistep regulation of TFEB by MTORC1. Autophagy 13, 464–472 (2017).
Medina, D. L. et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 17, 288–299 (2015).
Nabar, N. R. & Kehrl, J. H. The transcription factor EB links cellular stress to the immune response. Yale J. Biol. Med. 90, 301–315 (2017).
Li, Y. et al. Protein kinase C controls lysosome biogenesis independently of mTORC1. Nat. Cell Biol. 18, 1065–1077 (2016).
Kaneko, H. et al. Taurine is an amino acid with the ability to activate autophagy in adipocytes. Amino Acids 50, 527–535 (2018).
Nada, S. et al. The novel lipid raft adaptor p18 controls endosome dynamics by anchoring the MEK-ERK pathway to late endosomes. EMBO J. 28, 477–489 (2009).
Chen, D. et al. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat. Commun. 9, 873 (2018).
Sun, M. K. & Alkon, D. L. The “memory kinases”: roles of PKC isoforms in signal processing and memory formation. Prog Mol Biol Transl Sci. 122, 31–59 (2014).
Saftig, P. & Haas, A. Turn up the lysosome. Nat. Cell Biol. 18, 1025–1027 (2016).
St-Pierre, J. et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 127, 397–408 (2006).
Tsunemi, T. et al. PGC-1a rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci. Transl. Med. 4, 142ra97 (2012).
Town, M. et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat. Genet. 18, 319–324 (1998).
Kalatzis, V., Cherqui, S., Antignac, C. & Gasnier, B. Cystinosin, the protein defective in cystinosis, is a H+-driven lysosomal cystine transporter. EMBO J. 20, 5940–5949 (2001).
Zhang, J. et al. Chaperone-mediated autophagy upregulation rescues megalin expression and localization in cystinotic proximal tubule cells. Front. Endocrinol. (Lausanne) 10, 1–11 (2019).
Nesterova, G. & Gahl, W. Nephropathic cystinosis: late complications of a multisystemic disease. Pediatr. Nephrol. 23, 863–878 (2008).
Gahl, W. A., Thoene, J. G. & Schneider, J. A. Medical progress: cystinosis. N. Engl. J. Med. 347, 111–121 (2002).
Rega, L. R. et al. Activation of the transcription factor EB rescues lysosomal abnormalities in cystinotic kidney cells. Kidney Int. 89, 862–873 (2016).
Elmonem, M. A. et al. Cystinosis: a review. Orphanet J. Rare Dis. 11, 1–17 (2016).
Laube, G. F. et al. Glutathione depletion and increased apoptosis rate in human cystinotic proximal tubular cells. Pediatr. Nephrol. 21, 503–509 (2006).
Sansanwal, P. & Sarwal, M. M. Abnormal mitochondrial autophagy in nephropathic cystinosis. Autophagy 6, 971–973 (2010).
Napolitano, G. et al. Impairment of chaperone-mediated autophagy leads to selective lysosomal degradation defects in the lysosomal storage disease cystinosis. EMBO Mol. Med. 7, 158–174 (2015).
Johnson, J. L. et al. Upregulation of the Rab27a-dependent trafficking and secretory mechanisms improves lysosomal transport, alleviates endoplasmic reticulum stress, and reduces lysosome overload in cystinosis. Mol. Cell. Biol. 33, 2950–2962 (2013).
Ivanova, E. A. et al. Endo-lysosomal dysfunction in human proximal tubular epithelial cells deficient for lysosomal cystine transporter cystinosin. PLoS ONE 10, 1–18 (2015).
Raggi, C. et al. Dedifferentiation and aberrations of the endolysosomal compartment characterize the early stage of nephropathic cystinosis. Hum. Mol. Genet. 23, 2266–2278 (2014).
Thoene, J. G., Oshima, R. G. & Crawhall, J. C. Intracellular cystine depletion by aminothiols in vitro and in vivo. J. Clin. Invest. 58, 180–189 (1976).
Cherqui, S. Cysteamine therapy: a treatment for cystinosis, not a cure. Kidney Int. 81, 127–129 (2012).
Zhang, J. et al. Cystinosin, the small GTPase Rab11, and the Rab7 effector RILP regulate intracellular trafficking of the chaperone-mediated autophagy receptor LAMP2A. J. Biol. Chem. 292, 10328–10346 (2017).
Andrzejewska, Z. et al. Cystinosin is a component of the vacuolar H+-ATPase-ragulator-rag complex controlling mammalian target of rapamycin complex 1 signaling. J. Am. Soc. Nephrol. 27, 1678–1688 (2016).
Moskot, M. et al. The phytoestrogen genistein modulates lysosomal metabolism and Transcription Factor EB (TFEB) activation. J. Biol. Chem. 289, 17054–17069 (2014).
Kilpatrick, K., Zeng, Y., Hancock, T. & Segatori, L. Genetic and chemical activation of TFEB mediates clearance of aggregated α-synuclein. PLoS ONE 10, e0120819 (2015).
Pastore, N. et al. Gene transfer of master autophagy regulator TFEB results in clearance of toxic protein and correction of hepatic disease in alpha-1-anti-trypsin deficiency. EMBO Mol. Med. 5, 397–412 (2013).
Spampanato, C. et al. Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol. Med. 5, 691–706 (2013).
Sansanwal, P., Li, L. & Sarwal, M. M. Inhibition of intracellular clusterin attenuates cell death in nephropathic cystinosis. J. Am. Soc. Nephrol. 26, 612–625 (2015).
Mehta, R. L. et al. International Society of Nephrology’s 0by25 initiative for acute kidney injury (zero preventable deaths by 2025): a human rights case for nephrology. Lancet 385, 2616–2643 (2015).
Khwaja, A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin. Pract. 120, c179–c184 (2012).
He, L., Livingston, M. J. & Dong, Z. Autophagy in acute kidney injury and repair. Nephron Clin. Pract. 127, 56–60 (2014).
Zarjou, A. & Agarwal, A. Sepsis and acute kidney injury. J. Am. Soc. Nephrol. 22, 999–1006 (2011).
Farrar, A. Acute kidney injury. Nurs. Clin. North Am. 53, 499–510 (2018).
Kanagasundaram, N. S. Pathophysiology of ischaemic acute kidney injury. Ann. Clin. Biochem. 52, 193–205 (2015).
Pabla, N. & Dong, Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int. 73, 994–1007 (2008).
Price, P. M., Safirstein, R. L. & Megyesi, J. The cell cycle and acute kidney injury. Kidney Int. 76, 604–613 (2009).
Bonventre, J. V. & Yang, L. Cellular pathophysiology of ischemic acute kidney injury. Sci. Med. 121, 4210–4221 (2011).
Padanilam, B. J. Cell death induced by acute renal injury: a perspective on the contributions of apoptosis and necrosis. Am. J. Physiol. Ren. Physiol. 284, 608–627 (2003).
Havasi, A. & Borkan, S. C. Apoptosis and acute kidney injury. Kidney Int. 80, 29–40 (2011).
Tang, C. et al. P53 in kidney injury and repair: mechanism and therapeutic potentials. Pharmacol. Ther. 195, 5–12 (2019).
Suzuki, C. et al. Participation of autophagy in renal ischemia/reperfusion injury. Biochem. Biophys. Res. Commun. 368, 100–106 (2008).
Jiang, M., Liu, K., Luo, J. & Dong, Z. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am. J. Pathol. 176, 1181–1192 (2010).
Periyasamy-Thandavan, S. et al. Autophagy is cytoprotective during cisplatin injury of renal proximal tubular cells. Kidney Int. 74, 631–640 (2008).
Yang, C., Kaushal, V., Shah, S. V. & Kaushal, G. P. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am. J. Physiol. Ren. Physiol. 294, F777–F787 (2008).
Jiang, M. et al. Autophagy in proximal tubules protects against acute kidney injury. Kidney Int. 82, 1271–1283 (2012).
Martin, J. L., Gruszczyk, A. V., Beach, T. E., Murphy, M. P. & Saeb-Parsy, K. Mitochondrial mechanisms and therapeutics in ischaemia reperfusion injury. Pediatr. Nephrol. 34, 1167–1174 (2019).
Whitaker, R. M., Wills, L. P., Stallons, L. J. & Schnellmann, R. G. CGMP-selective phosphodiesterase inhibitors stimulate mitochondrial biogenesis and promote recovery from acute kidney injury. J. Pharmacol. Exp. Ther. 347, 626–634 (2013).
Tran, M. et al. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Invest. 121, 4003–4014 (2011).
Rasbach, K. A. & Schnellmann, R. G. PGC-1α over-expression promotes recovery from mitochondrial dysfunction and cell injury. Biochem. Biophys. Res. Commun. 355, 734–739 (2007).
Brooks, C., Wei, Q., Cho, S. G. & Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Invest. 119, 1275–1285 (2009).
Gonzalez, A. S. et al. Abnormal mitochondrial fusion-fission balance contributes to the progression of experimental sepsis. Free Radic. Res. 48, 769–783 (2014).
Liu, J. X. et al. Disturbance of mitochondrial dynamics and mitophagy in sepsis-induced acute kidney injury. Life Sci. 235, 116828 (2019).
Quoilin, C., Mouithys-Mickalad, A., Lécart, S., Fontaine-Aupart, M. P. & Hoebeke, M. Evidence of oxidative stress and mitochondrial respiratory chain dysfunction in an in vitro model of sepsis-induced kidney injury. Biochim. Biophys. Acta 1837, 1790–1800 (2014).
Picca, A. et al. Fueling inflamm-aging through mitochondrial dysfunction: mechanisms and molecular targets. Int. J. Mol. Sci. 18, 933 (2017).
Patil, N. K., Parajuli, N., Macmillan-Crow, L. A. & Mayeux, P. R. Inactivation of renal mitochondrial respiratory complexes and manganese superoxide dismutase during sepsis: mitochondria-targeted antioxidant mitigates injury. Am. J. Physiol. Ren. Physiol. 306, 734–743 (2014).
Hyttinen, J. M. T., Viiri, J., Kaarniranta, K. & Błasiak, J. Mitochondrial quality control in AMD: does mitophagy play a pivotal role? Cell. Mol. Life Sci. 75, 2991–3008 (2018).
Scott, I. et al. GCN5-like protein 1 (GCN5L1) controls mitochondrial content through coordinated regulation of mitochondrial biogenesis and mitophagy. J. Biol. Chem. 289, 2864–2872 (2014).
Lynch, M. R. et al. TFEB-driven lysosomal biogenesis is pivotal for PGC1α-dependent renal stress resistance. JCI Insight. 4, e126749 (2019).
Tan, S. et al. Pomegranate activates TFEB to promote autophagy-lysosomal fitness and mitophagy. Sci. Rep. 9, 1–18 (2019).
Liu, W., Li, C. C., Lu, X., Bo, L. Y. & Jin, F. G. Overexpression of transcription factor EB regulates mitochondrial autophagy to protect lipopolysaccharide-induced acute lung injury. Chin. Med. J. (Engl.) 132, 1298–1304 (2019).
Prajapati, P. et al. Enforced lysosomal biogenesis rescues erythromycin- and clindamycin-induced mitochondria-mediated cell death in human cells. Mol. Cell. Biochem. 461, 23–36 (2019).
Gyebi, L., Soltani, Z. & Reisin, E. Lipid nephrotoxicity: new concept for an old disease. Curr Hypertens Rep. 14, 177–181 (2012).
Guebre-Egziabher, F. et al. Ectopic lipid accumulation: a potential cause for metabolic disturbances and a contributor to the alteration of kidney function. Biochimie 95, 1971–1979 (2013).
Tang, J. & Zhuang, S. Epigenetics in acute kidney injury. Curr. Opin. Nephrol. Hypertens. 24, 351–358 (2015).
Kinsey, G. R., Li, L. & Okusa, M. D. Inflammation in acute kidney injury. Nephron Exp. Nephrol. 109, e102–e107 (2008).
Kezic, A., Spasojevic, I., Lezaic, V. & Bajcetic, M. Mitochondria-Targeted antioxidants: future perspectives in kidney ischemia reperfusion injury. Oxid. Med. Cell. Longev. 2016, 2950503 (2016).
Levin, A. et al. Global kidney health 2017 and beyond: a roadmap for closing gaps in care, research, and policy. Lancet 390, 1888–1917 (2017).
Cao, Z. & Cooper, M. E. Pathogenesis of diabetic nephropathy. J. Diabetes Investig. 2, 243–247 (2011).
Shi, Y. & Hu, F. B. The global implications of diabetes and cancer. Lancet 383, 1947–1948 (2014).
Kitada, M. Clinical therapeutic strategies for early stage of diabetic kidney disease. World J. Diabetes 5, 342 (2014).
Giacco, F. & Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 107, 1058–1070 (2010).
Calcutt, N. A., Cooper, M. E., Kern, T. S. & Schmidt, A. M. Therapies for hyperglycaemia-induced diabetic complications: from animal models to clinical trials. Nat. Rev. Drug Discov. 8, 417–429 (2009).
Noh, H. & King, G. L. The role of protein kinase C activation in diabetic nephropathy. Kidney Int. 72, 49–53 (2007).
Brenner, B. M. et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N. Engl. J. Med. 345, 861–869 (2001).
Lewis, E. J. et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N. Engl. J. Med. 345, 851–860 (2001).
Har, R. et al. The effect of renal hyperfiltration on urinary inflammatory cytokines/chemokines in patients with uncomplicated type 1 diabetes mellitus. Diabetologia 56, 1166–1173 (2013).
Ruggenenti, P., Cravedi, P. & Remuzzi, G. The RAAS in the pathogenesis and treatment of diabetic nephropathy. Nat. Rev. Nephrol. 6, 319–330 (2010).
Han, K., Zhou, H. & Pfeifer, U. Inhibition and restimulation by insulin of cellular autophagy in distal tubular cells of the kidney in early diabetic rats? Kidney Blood Press. Res. 20, 258–263 (1997).
Barbosa, A. & de, A. et al. Inhibition of cellular autophagy in proximal tubular cells of the kidney in streptozotocin-diabetic and uninephrectomized rats. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 61, 359–366 (1992).
Kitada, M. et al. Dietary restriction ameliorates diabetic nephropathy through anti-inflammatory effects and regulation of the autophagy via restoration of sirt1 in diabetic wistar fatty (fa/fa) rats: a model of type 2 diabetes. Exp. Diabetes Res. 2011, 1–11 (2011).
Vallon, V. et al. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am. J. Physiol. - Ren. Physiol. 304, F156–F167 (2013).
Liu, W. J. et al. Autophagy-lysosome pathway in renal tubular epithelial cells is disrupted by advanced glycation end products in diabetic nephropathy. J. Biol. Chem. 290, 20499–20510 (2015).
Tagawa, A. et al. Impaired podocyte autophagy exacerbates proteinuria in diabetic nephropathy. Diabetes 65, 755–767 (2016).
Mori, H. et al. The mTOR pathway is highly activated in diabetic nephropathy and rapamycin has a strong therapeutic potential. Biochem. Biophys. Res. Commun. 384, 471–475 (2009).
Gödel, M. et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J. Clin. Invest. 121, 2197–2209 (2011).
Mori, H. et al. The mTOR pathway is highly activated in diabetic nephropathy and rapamycin has a strong therapeutic potential. Biochem. Biophys. Res. Commun. 384, 471–475 (2009).
Kitada, M., Kume, S., Imaizumi, N. & Koya, D. Resveratrol improves oxidative stress and protects against diabetic nephropathy through normalization of Mn-SOD dysfunction in AMPK/SIRT1- independent pathway. Diabetes 60, 634–643 (2011).
Fang, L. et al. Autophagy attenuates diabetic glomerular damage through protection of hyperglycemia-induced podocyte injury. PLoS ONE 8, e60546 (2013).
Xiao, T. et al. Rapamycin promotes podocyte autophagy and ameliorates renal injury in diabetic mice. Mol. Cell. Biochem. 394, 145–154 (2014).
Zhao, X. et al. Advanced glycation end-products suppress autophagic flux in podocytes by activating mammalian target of rapamycin and inhibiting nuclear translocation of transcription factor EB. J. Pathol. 245, 235–248 (2018).
Takahashi, A. et al. Autophagy inhibits the accumulation of advanced glycation end products by promoting lysosomal biogenesis and function in the kidney proximal tubules. Diabetes 66, 1359–1372 (2017).
Acknowledgements
This research was funded by Natural Science Foundation of China, grant number 81974095 and 81670654.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by: M. V. Niklison Chirou
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, W., Li, X., Wang, S. et al. Regulation of TFEB activity and its potential as a therapeutic target against kidney diseases. Cell Death Discov. 6, 32 (2020). https://doi.org/10.1038/s41420-020-0265-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-020-0265-4
This article is cited by
-
From the regulatory mechanism of TFEB to its therapeutic implications
Cell Death Discovery (2024)
-
Chronic dehydration induces injury pathways in rats, but does not mimic histopathology of chronic interstitial nephritis in agricultural communities
Scientific Reports (2023)
-
TFEB: a double-edged sword for tumor metastasis
Journal of Molecular Medicine (2023)
-
FGF21 via mitochondrial lipid oxidation promotes physiological vascularization in a mouse model of Phase I ROP
Angiogenesis (2023)
-
RagD auto-activating mutations impair MiT/TFE activity in kidney tubulopathy and cardiomyopathy syndrome
Nature Communications (2023)
