
Abstract
Lymphoma is the sixth most common type of cancer worldwide. Under the current treatment standards, patients with lymphoma often fail to respond to treatment or relapse early and require further therapy. Hence, novel therapeutic strategies need to be explored and our understanding of the molecular underpinnings of lymphomas should be expanded. Ferroptosis, a non-apoptotic regulated cell death, is characterized by increased reactive oxygen species and lipid peroxidation due to metabolic dysfunction. Excessive or lack of ferroptosis has been implicated in tumor development. Current preclinical evidences suggest that ferroptosis participates in tumorigenesis, progression, and drug resistance of lymphoma, identifying a potential biomarker and an attractive molecular target. Our review summarizes the core mechanisms and regulatory networks of ferroptosis and discusses existing evidences of ferroptosis induction for the treatment of lymphoma, with intent to provide a framework for understanding the role of ferroptosis in lymphomagenesis and a new perspective of lymphoma treatment.
Similar content being viewed by others
Facts
-
Ferroptosis is an iron-dependent type of regulated cell death characterized by increased reactive oxygen species and excessive lipid peroxidation.
-
The p53, MYC, and PI3K pathways that are often dysregulated in lymphoma also regulate ferroptosis.
-
Crosstalk between ferroptosis and other regulated cell death pathways can compound the role of ferroptosis in lymphoma.
-
Ferroptosis dysregulation is involved in lymphomagenesis of some lymphoma subtypes.
-
Promising preclinical studies show that lymphoma cells are susceptible to ferroptosis induction, yet further investigation is warranted.
Introduction
Ferroptosis is an iron-dependent form of regulated cell death (RCD) first defined in 2012 [1], and differs in morphology, biochemistry, and immune features from other forms of RCD, such as apoptosis, necroptosis, and pyroptosis [2]. The biochemical mechanisms underlying ferroptosis involve lipid peroxidation and intracellular iron accumulation, which in turn result in the production of lethal reactive oxygen species (ROS) and alkyl oxygen radicals, inducing membrane damage and disorganization [3]. Broadly, ferroptosis can be triggered through two pathways [4]: the extrinsic and intrinsic pathways. The extrinsic pathway is a receptor-dependent pathway primarily triggered by the activation of iron transporters or inhibition of cell membrane transporters in the cystine/glutamate (Glu) antiporter system Xc−. The intrinsic pathway is mediated by regulation of intracellular antioxidant enzymes, such as glutathione peroxidase 4 (GPX4) [5] and those in metabolism pathways of amino acids, iron, and polyunsaturated fatty acids (PUFA). Of note, the PUFA pathway is perhaps the most important metabolic pathway in ferroptosis (Fig. 1), as PUFA can interact with peroxides to produce toxic lipid peroxides (PL-PUFA-OOH) under quiescent conditions [6]. Conversely, toxic PL-PUFA-OOH can be reduced by GPX4 into non-toxic products [7].

The core mechanism underlying the induction of ferroptosis is iron-dependent lipid peroxidation, which leads to the accumulation of reactive oxygen species (ROS). The iron metabolism involved in ferroptosis induction is regulated by several proteins, including ferritin components (FTH1, FTL), solute carrier family 40 member 1 (SLC40A1), serotransferrin, lactotransferrin, and transferrin receptor (TFRC). Acetyl-CoA carboxylase (ACAC)-mediated fatty acid synthesis increases accumulation of polyunsaturated fatty acids (PUFA) and promotes the formation of polyunsaturated fatty acid-containing phospholipids (PUFA–PL) via fatty acid–CoA ligase 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3). Monounsaturated fatty acids (MUFA) are resistant to oxidation and inhibit PUFA-PL peroxidation. The glutathione (GSH)/glutathione peroxidase 4 (GPX4) antioxidant system initiated by system Xc− is the main cellular defense against ferroptosis. Other independent defense mechanisms include the CoQH2 system and other antioxidants such as tetrahydrobiopterin/dihydrobiopterin and squalene. FSP1 and mitochondrial DHODH reduce CoQ to CoQH2 and suppress ferroptosis. ACSL3 fatty acid–CoA ligase 3, ALOXs arachidonate lipoxygenases, BECN1 beclin 1, BH4 tetrahydrobiopterin, CoQ10 coenzyme Q10, DDP4 dipeptidyl peptidase-4, DHODH dihydroorotate dehydrogenase, Fer-1 Ferrostatin 1, FSP1 ferroptosis suppressor protein 1, GCH1 GTP cyclohydrolase-1, GCL glutamate-cysteine ligase, Glu glutamic acid, GSR glutathione-disulfide reductase, GSS glutathione synthetase, GSSG glutathione disulfide, oxidized glutathione, GTP guanosine triphosphate, IFN-γ interferon-γ, LIP labile iron pool, NCOA4 nuclear receptor co-activator 4, NOXs NADPH oxidases, PL-PUFA-OOH polyunsaturated fatty acid-containing phospholipids hydroperoxide, TCA tricarboxylic acid cycle, VK vitamin K, VKH2 vitamin K hydroquinone.
Noteworthy progress has been made in our understanding of the role of ferroptosis in the molecular pathogenesis of many cancers. There is crosstalk between ferroptosis and multiple cancer-associated signaling pathways in cancer cells. For example, the tumor suppressor p53, mutated in over 50% of tumors, may promote or suppress ferroptosis in oxidative/metabolic stress conditions (Fig. 2A) [8]. By contrast, the PI3K–AKT–mTOR pathway inhibits ferroptosis via promoting GPX4 protein synthesis [9] or by regulating lipid metabolism (Fig. 2B). On one side, oncogenic signaling-mediated activation of ferroptosis has been implicated in promoting tumor growth, immune evasion, tumor progression, and therapeutic resistance [10]. On the other side, high burden of ROS, metabolic alterations, and specific mutations render cancer cells susceptible to ferroptotic cell death [11, 12]. These findings suggest that ferroptosis plays diverse roles in cancer development and therapeutic responses of tumors [13, 14]. Thus, modulating ferroptosis and targeting cancer vulnerabilities could have great therapeutic efficacy, especially when combined with conventional treatments [15,16,17].

A The role of p53 in ferroptosis. p53 promotes ferroptosis by regulating solute carrier family 7 member 11 (SLC7A11), glutamine synthase 2 (GLS2), and spermidine/spermine N1-acetyltransferase 1 (SAT1)/arachidonate lipoxygenase 15 (ALOX15), and inhibits ferroptosis by targeting p21 and blocking DPP4 activity in a p53-independent and transcription-independent manner. Meanwhile, the p53 pro-ferroptosis role can be mediated by arachidonate lipoxygenase 12 (ALOX12) and NCOA4-mediated ferritinophagy via a p53-dependent pathway. B The regulatory role of Myc in ferroptosis. Myc regulates ferroptosis via TFR1, DMTQ, and PCAT1. Oncogenic MYCN activity sensitizes cells to ferroptosis upon cyst(e)ine depletion, caused by decreased GSH and consequent massive lipid peroxidation. C The PI3K–AKT–mTOR signaling pathway suppresses ferroptosis. PI3K–AKT–mTOR signaling synergizes with the SLC7A11–GSH–GPX4 pathway by upregulating SREBP1 and SCD1 promoting MUFA synthesis, and also augments GPX4 protein synthesis by inhibiting eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4EBP1). Cys cysteine, Cys2 cystine, DMT1 divalent metal transporter 1, PCAT1 peptidase-containing ABC transporter 1, SREBP1 sterol regulatory element-binding protein 1, STEAP3 six-transmembrane epithelial antigen of the prostate 3, SCD1 stearoyl-CoA desaturase-1, TFR1 transferrin receptor 1.
Recent studies have demonstrated that lipids are associated with cancer development and are elevated in lymphoma. Choline metabolism, which is a crucial component of lipid synthesis, is dysregulated in lymphoma [18]. Besides, there is a hypothesis that lymphoma dependent on lipoprotein-mediated cholesterol uptake is susceptible to ferroptotic cell death [19]. However, to date, few studies have examined ferroptosis in lymphoma. As research on ferroptosis increases, it is important to improve our understanding of ferroptosis in lymphoma, especially in the subsets with clinical needs. This review aimed to provide a comprehensive framework for the regulatory networks involved in ferroptosis in lymphoma and targeting ferroptosis as a therapeutic strategy for lymphoma based on the findings of the aforementioned studies.
Oxidative damage inducing ferroptosis is implicated in lymphoma suppression
lipid peroxidation control and ferroptosis regulation
Lipid peroxidation is the primary feature of ferroptosis, and the main mechanism is the peroxidation of PUFA and the fatal accumulation of lipid peroxides (Fig. 1). A PUFA-rich diet has been reported to suppress tumor growth and prolong survival in a mouse tumor model of colorectal carcinoma [20]. Lipid peroxidation has been implicated in cancer progression and is the driving force of ferroptotic cell death in germinal center B cell-like (GCB) diffuse large B-cell lymphoma (DLBCL) [21]. Acyl-CoA synthetase long chain family member 4 (ACSL4) and lysophospholipid acyltransferase 5 encoded by the lysophosphatidylcholine acyltransferase 3 (LPCAT3) gene are essential enzymes that regulate the biosynthesis and remodeling of PUFAs [22, 23]. ACSL4 catalyzes the conversion of free PUFAs to activated coenzyme-A (CoA)-PUFAs [22], and LPCAT3 catalyzes their esterification to form PUFA-PLs [24]. PUFA-PLs are susceptible to oxidation by PL-PUFA-OOH, which induces ferroptosis [25, 26]. Accordingly, knockdown of ACSL4 or LPCAT3 in vitro blocks the peroxidation of PUFA-PLs and prevents or attenuates ferroptosis [22, 27]. In addition, some membrane proteins such as cytochrome P450 oxidoreductase [28], NADPH oxidase [29], and arachidonate lipoxygenase (ALOX) [30] increase lipid peroxidation, which induces production of ROS. In contrast, monounsaturated fatty acids (MUFA) are converted to MUFA-CoA by fatty acid–CoA ligase 3 (ACSL3), which inhibits the peroxidation of PUFA-PLs and in turn inhibits ferroptosis [31]. Ferrostatin-1 exerts an anti-ferroptotic role by inhibiting the activity of lipid hydroperoxides [32]. Moreover, AMPK interferes with the phosphorylation of acetyl-CoA carboxylase (ACAC), which in turn inhibits PUFA production and consequently ferroptosis [33]. These data imply that lipid peroxidation is an essential factor that regulates the development and progression of cancers including lymphoma and maybe a potential new target.
Regulation of iron metabolism in lymphoma
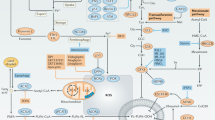
Typically, normal cells regulate iron uptake, utilization, storage, and export to maintain the iron pool balance, whereas malignant cells can be observed as iron unbalanced. Iron, a trace element, is required for the growth of malignant tumors, such as Burkitt lymphoma (BL) [34] and T-cell lymphoma [35]. Consistently, a case report described a patient with follicular lymphoma (FL) characterized by hyperferritinemia and widespread iron deposition [36]. Abnormal iron homeostasis has been implicated in the biogenesis and development of lymphoma, leading to an imbalance in the intracellular labile iron pool (LIP) level (Fig. 1). High LIP levels result in increased peroxidation in lymphoma cells [37], which promotes ferroptosis. Most intracellular iron exists in a free state as Fe2+. Fe2+ reacts with hydrogen peroxide via the Fenton reaction to generate ROS. Ferritin, an iron storage protein, stores iron in the form of Fe3+, thereby inhibits the Fenton reaction. Iron uptake mediated by transferrin receptor (TFRC/TFR) [38], lactotransferrin (LTF) [39] and serotransferrin (STF) [40] enhances ferroptosis, whereas solute carrier family 40 member 1 (SLC40A1) inhibits ferroptotic cell death by facilitating iron export [41]. Furthermore, the interaction of nuclear receptor co-activator 4 (NCOA4) and ferritin heavy chain 1 (FTH1) delivers iron-bound ferritin to autophagosomes for lysosomal degradation, which is termed as ferritinophagy [42]. This process releases iron from ferritin to the LIP [43, 44]. Interestingly, the lymphoma-associated MYC oncogene is involved in iron metabolism and can increase intracellular LIP by activating transferrin receptor 1 (TFR1) (Fig. 3). The modification of iron metabolism by c-Myc can disrupt iron homeostasis and affect lymphoma growth [45]. Consequently, targeting iron homeostasis represents a new therapeutic strategy for patients with lymphoma, especially Myc-driven lymphoma.

(Top) Mitochondrial metabolism plays an indispensable role in ferroptosis regulation by mitochondrial ferritin-mediated iron chelation, cysteine (Cys) deprivation-induced TCA cycle stimulation, and DHODH-mediated ferroptosis defense. (Bottom) Autophagy, a cell survival mechanism supporting cancer cell metabolism and survival under stress, induces autophagy-dependent ferroptosis in cancer cells. This phenomenon is marked by increased intracellular iron concentration and lipid peroxidation driven by hyperactivated NCOA4-mediated autophagy. CoQ10 coenzyme Q10, DHODH dihydroorotate dehydrogenase, GSH glutathione, LIP labile iron pool, NCOA4 nuclear receptor co-activator 4, TCA tricarboxylic acid cycle.
Antioxidant defense systems protect lymphoma cells from ferroptosis
GPX4-dependent pathways
GPX4, an antioxidant enzyme, mitigates lipid peroxidation in lymphoma and hence is a central repressor of ferroptosis [46]. GPX4 plays an essential role in the growth and development of innate-like B cells (such as B1 and marginal zone B cells) and antibody responses of follicular B2 cells [47]. Vitamin E cooperates with GPX4 to maintain the lipid redox balance and prevent ferroptosis in hematopoietic stem and progenitor cells [48]. High GPX4 expression is negatively correlated with clinical outcomes in a study in DLBCL [49]. The activity of GPX4 is regulated by the level of glutathione (GSH) synthesized from cysteine, glycine, and Glu, which alleviates oxidative stress by eliminating ROS [46]. GSH can be transformed into oxidized glutathione (GSSG), which is then reduced to GSH to maintain a steady state. Notably, the system Xc−, which is comprised of solute carrier family 3 member 2 (SLC3A2) and solute carrier family 7 member 11 (SLC7A11), is integral to GSH production. The system Xc−, an antiporter on the cell membrane, transports one cystine (oxidized derivative of cysteine) to the intracellular space for subsequent GSH production and releases one Glu into the extracellular space [50]. The SLC7A11–GSH–GPX4 axis is the primary cellular defense mechanism against ferroptosis (Fig. 1) which is positively regulated by NRF2 [51] and ATF4 [52] and negatively regulated by the suppressor genes p53 [53] and BAP1 [54]. Moreover, the phosphorylation of beclin 1 (BECN1) can suppress the activity of SCL7A11, leading to ferroptotic cell death in an AMPK-mediated manner [55].
Pharmacological inhibitors of system Xc− (erastin, imidazole ketone erastin [IKE], and sulfasalazine) or GPX4 (RSL3 or ML-210) induce ferroptotic cell death, and present attractive therapeutic strategies for clinical application [56]. IKE has been identified as an effective inhibitor of system Xc− in mouse models of lymphoma [57]. On the other hand, several cell lines with functionally inactive GPX4 are resistant to ferroptosis inhibitors, suggesting that the involvement of mechanisms other than the SLC7A11-GSH-GPX4 dependent pathway. Although these inhibitors show high responsiveness, they are not suitable for clinical use due to the toxicity. Gpx4 knockout triggers acute renal failure in mice [58] which can be rescued by ACSL4 inhibitors [22], whereas Lpcat3 knockout is also lethal in vivo, likely because LPCAT3 is essential for membrane phospholipid composition, triglyceride transport, and lipoprotein production [59,60,61].
GPX4-independent pathways
Recent studies have identified that the ubiquinol (CoQH2) system is a GPX4-independent defense mechanism against ferroptosis [62, 63]. The GPX4 and CoQH2 systems are further organized into constituents located in the non-mitochondrial (cytosolic GPX4 in the GPX4 system and ferroptosis suppressor protein 1 [FSP1] in the CoQH2 system [62, 64]) and mitochondrial compartments (mitochondrial GPX4 in the GPX4 system and mitochondrial dihydroorotate dehydrogenase [DHODH] in the CoQH2 system) (Fig. 1). FSP1, previously known as AIFM2, is an oxidoreductase that reduces coenzyme Q10 (CoQ10, also known as ubiquinone-10) to CoQ10H2 using NAD(P)H [62]. The FSP1–CoQ10–NAD(P)H pathway is a self-contained system parallel to and in concert with the SLC7A11–GSH–GPX4 axis to suppress ferroptosis [63]. Vitamin K (more specifically, the reduced form hydroquinone) can protect against detrimental lipid peroxidation via FSP1-mediated reduction [65, 66]. In addition, MDM2 and MDMX can decrease the protein levels of FSP1 and promote ferroptosis by activating PPARα in a p53-independent manner [67]. The DHODH-CoQH2 pathway is a mitochondrial CoQH2 system that acts in parallel to the mitochondrial GPX4 pathway against ferroptosis. When mitochondrial GPX4 is knocked down, DHODH inhibits lipid peroxidation in the mitochondria [68]. Consequently, inactivation of mitochondrial GPX4 and DHODH increases mitochondrial lipid peroxidation and stimulates ferroptosis.
Other GPX4-independent ferroptosis-defense mechanisms include the GTP cyclohydrolase-1–tetrahydrobiopterin-phospholipid pathway [69] and squalene (Fig. 1). Squalene is an antioxidant against lipid peroxidation and ferroptosis, and was identified by Garcia-Bermudez et al. as a metabolite in the cholesterol synthesis pathway abnormally accumulated in cholesterol auxotrophic ALK+ anaplastic large cell lymphoma (ALCL) cells conferring a growth advantage under oxidative stress [27]. As cholesterol is metabolically required by human cells, growth of ALK + ALCL cells is sensitive to cholesterol-depleting therapy or targeting the low-density lipoprotein receptor (LDLR) [27]. Squalene accumulated in the upstream cholesterol synthesis could be a pathogenic mechanism of ALK + ALCL and cause resistance to GPX4-inhibition treatment; inhibition of squalene sensitizes ALK + ALCL cells to GPX4 inhibitors ML162 and RSL3 [27]. Studies on the mechanisms underlying the GPX4-independent pathways will inspire ideas for treating patients who may not respond to GPX4 inhibition-based approaches.
Lymphoma-related pathways are involved in ferroptosis regulation
The p53 signaling pathway
Mutations in the TP53 gene and consequently in p53 proteins result in inactivation of the p53 tumor suppressor pathway in cancers [70, 71]. TP53 encodes p53, a tumor suppressor protein, which plays an important role in lymphoma tumorigenesis [72, 73]. TP53 is commonly mutated in small lymphocytic lymphoma/chronic lymphocytic leukemia (SLL/CLL) [74, 75] and non-Hodgkin lymphoma (NHL) [72] including DLBCL [76], FL [77], mantle cell lymphoma (MCL) [78], and BL [79], and TP53 mutations are associated with poor overall survival (OS) and progression-free survival (PFS). Recent studies have reported that TP53 mutations can modulate the function of p53 in the context of various cellular process, such as antioxidant defense and ferroptosis [8]. For example, the P47S variant of p53 is associated with ferroptosis phenotypes and increased resistance to glutamate-mediated cytotoxicity [80], whereas the 3KR acetylation-defective variant of p53 is associated with defective p53-dependent induction of apoptosis and sensitivity to oxidative stress-dependent ferroptosis [53]. These reports suggest that the p53 pathway contributes to the regulation of ferroptosis.
p53 was first reported to mediate ferroptosis in a reduction/oxidation-dependent manner in 2015 [53]. p53 can efficiently repress the expression of SLC7A11, thereby enhancing the sensitivity of tumor cells to ferroptosis inducers, such as erastin. p53 also promotes ferroptosis by regulating metabolism-related genes, such as GLS2 [81], SAT1/ALOX15 [82], and FDXR [83], and non-coding RNAs, such as LINC00336/miR-6852 [84] and PVT1/miR-214 [85]. Different from the induction of ferroptosis by erastin or GPX4 inhibitors requiring ACSL4, p53-mediated ferroptosis under ROS stress requires ALOX12 and p53 activation [86]. In contrast, p53 inhibits ferroptosis through the target gene p21 [87] and blocking dipeptidyl-peptidase-4 (DPP4) activity [88]. DPP4 binds to the NADPH oxidase 1 (NOX1), and the DPP4-NOX1 complex promotes lipid peroxidation and ferroptosis [88]. Thus, DPP4 inhibitors limit the anti-cancer activity of ferroptosis inducers (such as erastin). Notably, the regulation of ferroptosis by p53 is likely to be dependent on intracellular ROS levels: p53 plays a protective role against ferroptosis when intracellular ROS levels are low, whereas p53 promotes ferroptosis when intracellular ROS levels are abnormally high [89]. Collectively, these reports highlight the dual context-dependent pro- and anti-ferroptosis roles of p53 (Fig. 2A).
In preclinical studies, the natural product kayadiol exhibited a significant anti-tumor effect via induction of the p53-dependent ferroptosis pathway in natural killer/T-cell lymphoma (NKTCL) [90]. Additionally, APR-246, a small molecule that reactivates the activity of p53 mutants, induces DLBCL cell death via ferroptosis, and triggers p53-dependent ferritinophagy in DLBCL cells with TP53 missense mutation on exon 7 [91]. These findings are crucial for understanding the mechanism underlying p53-mediated ferroptosis in lymphoma and provide a new direction for the treatment of p53-dependent lymphoma (Table 1).
MYC signaling pathway
The oncogenic transcription factor MYC plays extensive roles in cancers including lymphoma, and its deregulation is associated with lymphoma progression and poor prognosis [92]. MYC-IGH and MYC-IGL translocations occur in 80% and 10% of BL cases, respectively [93]. Wang et al. speculated that Epstein-Barr virus (EBV)-infected BL is associated with iron deficiency because of its unique dependence on the ferrireductase CYB561A3 pathway [34, 94], suggesting a strong link between tumorigenesis and iron metabolism. The MYC family member MYCN was reported to mediate cysteine addiction, and make the fast proliferating cells vulnerable to cyst(e)ine depletion, inducing MYCN-dependent ferroptosis [95]. Although MYC translocation is uncommon in DLBCL, overexpression of Myc is observed in 30% of DLBCL cases. Recently studies found that MYC regulates downstream target genes involved in iron metabolism and the oxidative stress response (Fig. 2B). MYC can regulate several genes involved in iron metabolism, such as TFRC and the ferrous iron transporter DMT1, which promote ferroptotic cell death by increasing intracellular LIP [34, 45]. Thus, iron chelators or ironomycin treatment can effectively target Myc and TFR1, demonstrating a potential therapeutic strategy for lymphoma patients with Myc overexpression [96]. On the contrary, there are several reports that suggest that Myc can inhibit ferroptosis by interacting with PCAT1 and promoting SLC7A11 expression [97] or by downregulating KMD1A, thereby increasing malondialdehyde and iron levels [98]. Further understanding of the interplay between MYC and ferroptosis may improve the treatment of MYC-driven lymphoproliferative disorders.
PI3K–AKT–mTORC1 signaling pathway
The PI3K–AKT–mTOR pathway is one of the most aberrantly active signaling pathways in various cancers [99]. Fatty acid synthase activity is positively correlated with the PI3K–AKT–mTOR pathway which promotes the oncogenic translation of DLBCL [100, 101]. mTOR complex 1 (mTORC1) maintains protein synthesis and cellular homeostasis [102] and also regulates ferroptosis [103]. Recent studies have elucidated two mechanisms by which mTORC1 suppress ferroptosis (Fig. 2C). First, mTORC1 plays a synergistic role with GPX4 to inhibit ferroptosis. Specifically, mTORC1 augments GPX4 synthesis by inhibiting eukaryotic translation initiation factor 4E-binding protienn1 (4EBP1) phosphorylation [9, 104]. Consistently, Torin1, an ATP-competitive mTOR inhibitor [105], promotes ferroptotic cell death by decreasing GPX4 protein levels [9]. Second, mTORC1 induces cancer cell resistance to ferroptosis by activating sterol regulatory element-binding protein 1 (SREBP1), which regulates the expression of stearoyl-CoA desaturase-1 (SCD1), an enzyme responsible for MUFA synthesis [106]. Accordingly, abnormal PI3K–AKT–mTORC1 signaling allows lymphoma cells to escape ferroptosis, which promotes tumor initiation and progression. Further studies should focus on simultaneously targeting both the mTOR pathway and ferroptosis. Treatment with mTOR inhibitors in combination with ferroptosis inducers could be a promising strategy for patients with lymphoma with intrinsic or acquired resistance.
Mitochondrial metabolism regulating ferroptosis is high in lymphoma subset
Tumors rewire their metabolisms to ensure sufficient nutrients to support their growth. In this regard, cancer cell metabolism is tightly linked with cell death pathways through regulation of mitochondrial functions. OxPhos-DLBCL is a DLBCL subset identified by a gene signature involved in oxidative phosphorylation, mitochondrial metabolism, and the electron transport chain [107]. OxPhos-DLBCLs display enhanced mitochondrial energy transduction but are insensitive to inhibition of the B-cell receptor (BCR) signaling pathway [108]. Recently, mitochondria have also been reported to be involved in ferroptosis, apoptosis, and mitophagy [109, 110]. Mitochondrial metabolism regulates ROS generation and ferroptosis via three pathways (Fig. 3). First, mitochondrial ferritin chelates iron, inhibiting free iron accumulation, thereby blocking lipid peroxidation via the Fenton reaction [111]. DNA damage results in excessive free iron accumulation in the mitochondria, and increased lipid peroxidation of cell membranes, which ultimately induces ferroptosis [112]. Second, cystein deprivation stimulates the tricarboxylic acid cycle, which enhances mitochondrial respiration, increases lipid peroxidation and ROS production in the mitochondria and therefore promotes ferroptosis [109, 113]. Third, mitochondria possess a DHODH-mediated ferroptosis defense mechanism [68]. In the mitochondria, DHODH reduces CoQ10 to CoQ10H2, and this mechanism runs in parallel with mitochondrial GPX4 to inhibit ferroptosis. These findings suggest that the mitochondria play an essential role in ferroptosis regulation and these pathways are potential therapeutic targets for ferroptosis induction in lymphoma cells.
Mitophagy is a mitochondria targeted autophagy with controversial role in ferroptosis [109, 114]. Autophagy, a cellular mechanism that supports cancer cell metabolism and survival under stress [115], is generally thought to induce ferroptosis. Autophagy-dependent ferroptosis has been observed in cancer cells, and is characterized by intracellular iron concentration and lipid peroxidation caused by the hyperactivation of autophagy [116]. In addition, knockdown of autophagy-related genes can deplete intracellular iron and reduce lipid peroxidation, protecting cells from ferroptotic cell death [116]. NCOA4 is a selective cargo receptor for the lysosomal autophagic turnover of ferritin. NCOA4-mediated ferritinophagy promotes ferroptosis by increasing intracellular Fe2+ levels (Fig. 3) [44, 117]. Consistently, inhibition of NCOA4 [44] or proteasomal degradation targeted by HERC2 [42] can decrease ferritin degradation and suppress ferroptotic cell death. In turn, ROS can activate autophagy, which plays a significant role in ferritin degradation during ferroptosis [118]. These data contribute to a more profound understanding of the connection between ferroptosis and autophagy.
Tumor microenvironment may interact with ferroptotic lymphoma cells
The tumor microenvironment (TME) plays complex roles in diverse processes of lymphoma, including pathogenesis, progression, drug resistance, and prognosis, by facilitating sustained tumor proliferation growth and sensitize lymphoma cells to immunotherapy [119, 120]. Cancer immunotherapy can restore and enhance the effector function of cytotoxic CD8+ T cells, thereby improving immune responses to tumor cells [120]. CD8+ T cell mediated-cytotoxicity is primarily mediated by perforin/granzyme- or Fas/Fas ligand signaling-induced apoptosis. Additionally, immunotherapy-activated CD8+ T cells secrete interferon-γ (IFN-γ), which downregulates the expression of SLC7A11 and SLC3A2 by targeting the JAK–STAT pathway. This in turn enhances lipid peroxidation and consequently induces ferroptosis in cancer cells [15]. Consistently, the anti-PD-L1 nivolumab can downregulate the expression of SLC3A2 and upregulate the expression of IFN-γ and CD8 as elucidated by transcriptome analysis. Blocking SLC7A11-mediated cystine uptake in combination with immunotherapy synergistically enhances CD8+ T cell-mediated anti-tumor activity and promotes ferroptotic cell death. Moreover, IFN-γ in combination with PUFA triggers ferroptosis and boosts anti-tumor immune responses dependent of ACSL4. Targeting the ACSL4 pathway may be more effective in increasing the efficacy of immunotherapy. Besides, ferroptotic cancer cells also release HMGB1 in an autophagy-dependent manner, which binds to AGER and promotes inflammatory responses in macrophages (however, the increased inflammatory TNF cytokine production was only confirmed at the transcription level) [121].
However, emerging evidence has also shown that ferroptotic cancer cells negatively affect immune responses. Ferroptotic cancer cells inhibit the maturation of dendritic cells (DCs) and disrupt antigen cross-presentation, thereby hampering DC-mediated anti-tumor activity [122]. In addition, autophagy-dependent ferroptosis of cancer cells release KRASG12D proteins via exosomes, which are uptaken by AGER expressed on macrophages, inducing differentiation of macrophages from a M1-like phenotype to a M2-like tumor-supportive phenotype via STAT3-dependent fatty acid oxidation [123]. Moreover, ferroptotic cancer cells release excess fatty acids into the TME, effectively inhibiting CD8+ T and NK cell efficacy and cytokines secretion, such as IFN-γ, tumor necrosis factor α (TNF-α) and peroxisome activator receptor-γ (PPAR-γ). Fatty acids bind to CD36 on T cells triggering ferroptosis, which limits the anti-tumor activities of T cells by downregulation of the release of cytotoxic cytokines IFN-γ and TNF-α [124, 125]. Decreased production of IFN-γ and increased expression of the PPAR-γ was seen in NK cells of patients with DLBCL and in an Eµ-myc lymphoma model with increased iron metabolism [126]. These findings highlight a dynamic interplay between cancer cells and their TME via regulation of ferroptosis (Fig. 4). Hence, targeting T cell-mediated ferroptosis represents a new approach in immunotherapy, and the combination of ferroptosis modulation and immunotherapy is a potential strategy for cancer treatment. However, the mechanisms of TME and ferroptotic lymphoma cell interaction during lymphoma progression remain to be elucidated.

CD8+ T cells induce ferroptotic cancer cell death by secreting interferon γ, which activates the JAK1–STAT1 signaling pathway, thereby regulating the SLC7A11–GSH–GPX4 axis. Ferroptotic cancer cells can release damage-associated molecular patterns (such as HMGB1) in a ferritinophagy-dependent manner, which binds to AGER to promote inflammatory response of macrophages. Autophagy-dependent ferroptotic cancer death also can release oncogenic KRASG12D protein, which triggers M2 macrophage polarization and hampers dendritic cell-mediated anti-tumor activities. Ferroptotic cancer cells also release fatty acids, which inhibit natural killer (NK) cell cytotoxicity and induce ferroptosis in CD8+ T cells by binding to CD36. Cys cysteine, Cys2 cystine, IFN-γ interferon-γ, GSH glutathione, GPX4 glutathione peroxidase 4, PPAR- γ peroxisome activator receptor γ, PUFA polyunsaturated fatty acids, PUFA–PL polyunsaturated fatty acid-containing phospholipids, TAM tumor associated macrophage, TNF-α tumor necrosis factor-alpha.
Targeting ferroptosis has therapeutic potential in various types of lymphoma
The currently available cancer treatment methods do not effectively resolve the issue of inherent drug resistance in cancer cells. Numerous studies have shown that ferroptosis plays a practical role in eliminating cancer cells and suppressing tumor growth in lung cancer [127], glioblastoma [128], colorectal cancer [129], pancreatic cancer [130], and prostate cancer [131]. Ferroptosis has also been found to play important roles in lymphoma. In particular, various lymphoma-related signaling pathways crosstalk with ferroptosis.
Biological markers associated with clinical outcomes identified through the analysis of samples (e.g., blood, saliva, urine, feces, and/or tumor tissue) guides precision medicine. BODIPYTM 581/591 C11 (Table 2), a fluorescent tracer, monitors lipid peroxidation products in live cells and tissues to measure ferroptosis in vivo [132]. In addition, 3F3 anti−ferroptotic membrane antibody (3F3-FMA), a selective ferroptosis-staining reagent binding to TFR1, has been identified as a histopathological biomarker for ferroptosis [38]. Ferroptosis-related proteins GPX4, HADHB, and SECISBP2 can be prognostic biomarkers of lymphoma [49, 133, 134]. Some ferroptosis-related agents for clinical assessment were listed in Table 1. Moreover, the latest developments in detection technologies, such as small-molecule fluorescent probes [135], enhanced magnetic resonance imaging-guided therapy [136], CRISPR screens and metabolomics [27, 137], may be useful for ferroptosis-directed treatments. Such painstaking efforts will require collaborative and close multidisciplinary cooperation for translation into clinical practice.
Recently, drugs targeting ferroptosis have displayed pre-clinical efficacy through two main approaches: (1) inhibiting antioxidant mechanisms such as the SLC7A11–GSH–GPX4 axis, and (2) inducing ferroptosis by increasing iron uptake, oxidation, and the LIP level modulated by LTF, SLC40A1, TFRC, and NCOA4.
Current research on ferroptosis in lymphoma has focused on targeting ferroptosis-specific molecules to increase the sensitivity of lymphoma cells to ferroptosis, thereby suppressing the growth and progression of lymphoma. In this section, we further discuss the relationships between ferroptosis and various types of lymphoma and summarize the ferroptosis-modulating agents and molecular targets in different types of lymphoma (Table 1 and Fig. 5).

Inducers and inhibitors of ferroptosis are depicted in red and green, respectively. ART Artesunate, ALCL anaplastic large cell lymphoma, BL Burkitt lymphoma, DLBCL diffuse large B cell lymphoma, DMF dimethyl fumarate, HDL-NPs high–density lipoprotein-like nanoparticles, IKE imidazole ketone erastin, MCL mantle cell lymphoma, NKTCL extranodal natural killer/T cell lymphoma, SQLE squalene monooxygenase.
DLBCL
DLBCL is the most common malignant lymphoma accounting for ~30% of NHL, and is characterized by biological heterogeneity [138]. Abnormal iron homeostasis and lipid peroxidation are involved in the carcinogenesis and progression of DLBCL. Serum ferritin and transferrin levels can reflect cellular iron levels, and several anti-TFR1 and malondialdehyde antibodies [38] have been identified as effective reagents to selectively stain ferroptotic cells in tissues and multiple cell culture contexts including DLBCL cells [38].
A Japanese study group found that in a cohort of 93 patients with DLBCL, 35.5% of cases expressed GPX4 and had significantly worse OS and PFS than GPX4-negative cases [49]. However, treatment was heterogeneous in this cohort (47 R-CHOP and 46 CHOP cases). Later, this study group investigated SECISBP2 that regulates GPX4 translation in 165 DLBCL patients treated with R-CHOP, and HADHB involved in fatty acid beta oxidation in 128 DLBCL patients (treatment unknown), and also found significant unfavorable prognostic effects in DLBCL [133, 134] (Table 2). Some other researchers constructed ferroptosis-related risk score models for prognostic prediction (Table 3) based on transcriptional expression of ferroptosis-related genes available in the Gene Expression Omnibus (GEO, including our deposition GSE31312) and The Cancer Genome Atlas (TCGA) databases [96, 139,140,141,142]. In these studies, high-risk scores were consistently associated with significantly shorter survival; and in one study risk scores also correlated with simulated dosing response of DLBCL to doxorubicin [141]. However, there is much disparity in these risk models (Table 3), likely caused by the differences in the bioinformatics methods and cohorts employed for model construction (for example, models were based on clinical results in the overall cohorts of GSE87371 with heterogeneous regimens; GSE11318 and GSE57611 with CHOP-like regimens; and GSE10846 with CHOP-like and R-CHOP-like regimens, respectively). Furthermore, a study estimated enrichment scores of immune cells and immune inhibitory receptors by single-sample gene set enrichment analysis, and found that high ferroptosis-related risk scores were correlated with immunosuppressive cell infiltration (regulatory T cells and macrophages) and expression of immune checkpoint molecules (CTLA-4, PD-L1, LAG-3, and TIM-3) thus immunosuppressive TME [139]. Another study estimated immune infiltrate using CIBERSORT and found that high ferroptosis-based risk score group had higher infiltration levels of activated CD4 memory T cells, resting NK cells, M2 macrophages, and neutrophils, whereas lower levels of follicular helper T cells, activated NK cells and M0 macrophages [141]. Moreover, ferroptosis-related risk score can predict resistance to ibrutinib in DLBCL cells [139].
DLBCL is susceptible to GPX4-regulated ferroptosis. Inhibition of the GPX4 and system Xc− results in accumulation of intracellular ROS, which enhances the sensitivity of DLBCL cells to ferroptosis [46, 143]. However, the clinical usage of GPX4 inhibitors is still limited to laboratory research due to their poor druggability [144]. High–density lipoprotein-like nanoparticles (HDL NPs) are a cholesterol-poor ligand that binds to scavenger receptor type B1 (SCARB1), the receptor for cholesterol-rich HDLs [19]. Treating cholesterol-addicted BL and GCB-DLBCL cells with HDL NPs activated a compensatory response upregulating de novo cholesterol biosynthesis genes which nearly abolished the expression of GPX4, resulting in ferroptotic death of BL and DLBCL cells in vitro and in vivo, as well in primary samples from patients with different types of lymphoma including DLBCL [19]. Similarly, treating previously identified cholesterol auxotrophic ALK + ALCL and histiocytic lymphoma cell lines (which depend on LDLR-mediated cholesterol uptake) [27] with HDL NPs also decreased GPX4 and induced ferroptosis [19].
IKE, an inhibitor of system Xc− that is metabolically stable and suitable for in vivo applications, decreases glutathione and lipid peroxidation and slows tumor growth in mouse DLBCL xenografts with minimal side effects [57]. Schmitt et al. reported that dimethyl fumarate (DMF) induces lipid peroxidation and ferroptosis by inhibiting GSH and GPX4 expression, particularly in GCB-DLBCL [21]. These findings highlight the potential of lipid peroxidation as a therapeutic target for DLBCL.
In addition, targeting cellular iron metabolism has been proven to be an efficient approach. Ironomycin, which regulates iron homeostasis to decrease LIP levels, significantly reduces viability of primary DLBCL cells while presents lower toxicity in normal tissues and hematopoietic progenitors than conventional treatments [96]. Moreover, ironomycin exhibits synergistic effects with doxorubicin, BTK inhibitors, and Syk inhibitors [96]. APR-246, a p53 mutant reactivator, can induce ferroptosis in DLBCL cells by upregulating NCOA4 expression [91]. This in turn results in breakdown of ferritin and cellular LIP level decrease in a TP53-dependent manner. Artesunate, a semisynthetic derivative of artemisinin, can downregulate the expression of GPX4 and induce apoptosis, cell cycle arrest, autophagy and ferroptosis by impairing the STAT3 signaling pathway in DLBCL cells [145].
These studies indicate that ferroptosis is a major mechanism involved in the pathophysiology of DLBCL [46, 49, 57], concomitant with excessive ROS accumulation and lipid peroxidation. These findings added a new molecular mechanism underlying the pathogenesis of DLBCL and may aid the development of new therapies to induce RCD in DLBCL (Table 1 and Fig. 5) and new combination treatment strategies. However, validation studies and the heterogeneity in response to treatment need further investigation.
Mantle cell lymphoma
MCL comprises 5% to 10% of NHL, characterized by genetic alterations involving p53, cyclin-dependent kinase (CDK) 4, CDKN2A, MYC, BCL-2, BCR, and nuclear factor (NF)-κB [146]. MCL cells require large quantities of iron to meet their high proliferation requirements, rendering them more vulnerable to iron deprivation [147]. Thus, iron chelators have been shown to exert cytotoxic effects in MCL cells [148]. Moreover, bortezomib induces mitochondrial depolarization and ROS generation by upregulating the BH3-only protein Noxa in MCL cell lines and primary MCL cells [149], suggesting that ferroptosis-targeted combination strategy could be further explored.
Burkitt lymphoma
BL is a highly aggressive B-cell NHL that is associated with EBV and was the first tumor to be reported to harbor a chromosomal translocation of MYC [150, 151]. A recent study found that different lipid peroxidation byproducts are generated at different stages of EBV transformation. Detoxification of lipid peroxidation productions and ROS by GPX4 and glutathione, respectively, is required for the sustained growth of Burkitt-like B cells. Interference in redox defense mechanisms induces ferroptosis in the earlier stages of EBV transformation or in BL cells (latency I) [152]. This phenomenon highlights the potential of ferroptosis inducers as a therapeutic approach for EBV-associated BL. Wang et al. reported that ART, as a ferroptosis inducer, enhances ferroptosis and suppresses proliferation by activating the ATF4–CHOP–CHAC1 pathway in BL in vitro and in vivo [153]. MYC regulates ferroptosis by facilitating the uptake of iron into the LIP. Thus, iron chelators and ironomycin may be effective treatments for MYC-driven lymphoma [96]. Moreover, deletion of TP53 dramatically accelerates the growth of Myc-induced lymphoma cells; however, blockade of ALOX12 inhibits p53-mediated ferroptosis [86]. Therefore, ALOX12 is a promising molecular target that can inhibit the development and progression of Eμ-Myc lymphoma by regulating the expression and function of p53.
Small lymphocytic lymphoma/chronic lymphocytic lymphoma
SLL/CLL is characterized by the peripheral accumulation of abnormal CD5 + B cells [154] and remains incurable [155]. Ferroptosis-related prognostic score models have been developed to predict the survival of patients with CLL (Table 3) [156, 157]. These models can be used to assess the response of patients with CLL to fludarabine, cyclophosphamide, and ibrutinib, as patients with low risk scores are more likely to benefit from these treatments and exhibit better OS [157]. However, a comprehensive molecular understanding of ferroptosis in CLL is lacking as few studies have reported ferroptosis in CLL.
Extranodal natural killer/T cell lymphoma
NKTCL is a highly aggressive type of NHL with an aggressive clinical course and is closely associated with EBV infection [158], which poses treatment challenges. A study showed that kayadiol, a natural compound extracted from Torreya nucifera, induces accumulation of ROS and ferroptosis by targeting the SLC7A11–GSH–GPX4 axis [90]. Kayadiol effectively killed NKTCL cells but not healthy lymphocytes and hence presents a potential treatment option for NKTCL, particularly in patients who develop chemoresistance.
Conclusions and perspectives
Similar to other types of RCD, ferroptosis plays an essential role in the occurrence and progression of lymphoma. Multiple physiological processes regulate ferroptosis, such as iron metabolism, lipid metabolism, mitochondrial metabolism, and the TME. Although various studies have confirmed the role of ferroptosis in cancer development, the detailed role in lymphoma remains unclear. Ferroptosis is a highly complex process whose role in tumor initiation and progression is context-dependent. Owing to the heterogeneity of lymphoma, ferroptosis signaling pathways and molecular mechanisms are poorly understood. More in-depth insights into the mechanisms of ferroptosis are needed to develop efficient ferroptosis-inducing strategies that can be used for lymphoma treatment.
Furthermore, there is crosstalk between ferroptosis and other RCD pathways as it involves similar gene mutations and protein alterations, making it difficult and all the more necessary to distinguish ferroptosis from non-ferroptotic cell death. To date, no specific biomarker has been identified for ferroptosis. The development of sensitive and reliable specific biomarkers or assays may promote research on ferroptosis in lymphoma and guide the clinical application of ferroptosis-based therapies for lymphoma. In addition, components of the TME are susceptible to ferroptosis, which in turn can influence anti-tumor immune responses. Therefore, balancing the sensitivity of cancer, cytotoxic T cells, and immunosuppressive cells to ferroptosis remains a major hindrance. Exploring the mechanisms underlying these relationships is indispensable to address this issue.
Finally, although many drugs have been reported to induce ferroptosis in cancer and lymphoma cells in preclinical studies, none have entered clinical trials. Ferroptosis-targeted approaches face challenges in clinical application such as the systemic toxicities [159], and future studies need identify novel targets for therapeutic development, such as SCARB1 [19] and LDLR [27]. The effectiveness of HDL NPs observed in both B and T cell lymphomas may suggest common metabolic vulnerability exists in different lymphoma subtypes making targeted therapy appealing. We hope our review on ferroptosis studies provides a comprehensive overview and offer a new research direction for lymphoma therapeutic development for future investigations.
Data availability
Not applicable.
Change history
09 January 2024
A Correction to this paper has been published: https://doi.org/10.1038/s41419-023-06393-9
References
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Stockwell BR, Friedmann Angeli JP, Bayir HBA, Conrad M, Dixon SJ, Fulda S, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–85.
Agmon E, Solon J, Bassereau P, Stockwell BR. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci Rep. 2018;8:5155.
Tang D, Kroemer G. Ferroptosis. Curr Biol. 2020;30:R1292–R1297.
Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22:266–82.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2018;25:486–541.
Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radic Biol Med. 2020;152:175–85.
Liu J, Zhang C, Wang J, Hu W, Feng Z. The regulation of ferroptosis by tumor suppressor p53 and its Pathway. Int J Mol Sci. 2020;21:8387.
Zhang Y, Swanda RV, Nie L, Liu X, Wang C, Lee H, et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat Commun. 2021;12:1589.
Friedmann AJP, Krysko DV, Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer. 2019;19:405–14.
Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature. 2017;551:247–50.
Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453–7.
Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020;30:146–62.
Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022;22:381–96.
Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270–4.
Liang C, Zhang X, Yang M, Dong X. Recent progress in ferroptosis inducers for cancer therapy. Adv Mater. 2019;31:e1904197.
Xu G, Wang H, Li X, Huang R, Luo L. Recent progress on targeting ferroptosis for cancer therapy. Biochem Pharm. 2021;190:114584.
Xiong J, Wang L, Fei XC, Jiang XF, Zheng Z, Zhao Y, et al. MYC is a positive regulator of choline metabolism and impedes mitophagy-dependent necroptosis in diffuse large B-cell lymphoma. Blood Cancer J. 2017;7:e0.
Rink JS, Lin AY, McMahon KM, Calvert AE, Yang S, Taxter T, et al. Targeted reduction of cholesterol uptake in cholesterol-addicted lymphoma cells blocks turnover of oxidized lipids to cause ferroptosis. J Biol Chem. 2021;296:100100.
Excess polyunsaturated fatty acids induce death in acidic cancer cells. Cancer Discov. 2021;11:1872.
Schmitt A, Xu W, Bucher P, Grimm M, Konantz M, Horn H, et al. Dimethyl fumarate induces ferroptosis and impairs NF-kappaB/STAT3 signaling in DLBCL. Blood. 2021;138:871–84.
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–98.
Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10:1604–9.
Jain S, Zhang X, Khandelwal PJ, Saunders AJ, Cummings BS, Oelkers P. Characterization of human lysophospholipid acyltransferase 3. J Lipid Res. 2009;50:1563–70.
Gaschler MM, Stockwell BR. Lipid peroxidation in cell death. Biochem Biophys Res Commun. 2017;482:419–25.
Shah R, Shchepinov MS, Pratt DA. Resolving the role of lipoxygenases in the initiation and execution of ferroptosis. ACS Cent Sci. 2018;4:387–96.
Garcia-Bermudez J, Baudrier L, Bayraktar EC, Shen Y, La K, Guarecuco R, et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature. 2019;567:118–22.
Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16:302–9.
Yao L, Ban F, Peng S, Xu D, Li H, Mo H, et al. Exogenous iron induces NADPH oxidases-dependent ferroptosis in the conidia of Aspergillus flavus. J Agric Food Chem. 2021;69:13608–17.
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA. 2016;113:E4966–75.
Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol. 2019;26:420–32.e429.
Miotto G, Rossetto M, Di Paolo ML, Orian L, Venerando R, Roveri A, et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020;28:101328.
Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 2020;22:225–34.
Wang Z, Guo R, Trudeau SJ, Wolinsky E, Ast T, Liang JH, et al. CYB561A3 is the key lysosomal iron reductase required for Burkitt B-cell growth and survival. Blood. 2021;138:2216–30.
Benadiba J, Rosilio C, Nebout M, Heimeroth V, Neffati Z, Popa A, et al. Iron chelation: an adjuvant therapy to target metabolism, growth and survival of murine PTEN-deficient T lymphoma and human T lymphoblastic leukemia/lymphoma. Leuk Lymphoma. 2017;58:1433–45.
Algahtani H, Absi A, Shirah B, Al-Maghraby H, Algarni H. Hyperferritinemia with iron deposition in the basal ganglia and tremor as the initial manifestation of follicular lymphoma. Int J Neurosci. 2023;133:896–900.
Lipinski P, Drapier JC, Oliveira L, Retmanska H, Sochanowicz B, Kruszewski M. Intracellular iron status as a hallmark of mammalian cell susceptibility to oxidative stress: a study of L5178Y mouse lymphoma cell lines differentially sensitive to H(2)O(2). Blood. 2000;95:2960–6.
Feng H, Schorpp K, Jin J, Yozwiak CE, Hoffstrom BG, Decker AM, et al. Transferrin receptor is a specific ferroptosis marker. Cell Rep. 2020;30:3411–23.e3417.
Wang Y, Liu Y, Liu J, Kang R, Tang D. NEDD4L-mediated LTF protein degradation limits ferroptosis. Biochem Biophys Res Commun. 2020;531:581–7.
Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15:234–45.
Geng N, Shi BJ, Li SL, Zhong ZY, Li YC, Xua WL, et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur Rev Med Pharm Sci. 2018;22:3826–36.
Santana-Codina N, Gikandi A, Mancias JD. The role of NCOA4-mediated ferritinophagy in ferroptosis. Adv Exp Med Biol. 2021;1301:41–57.
Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021–32.
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJR, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–8.
O’Donnell KA, Yu D, Zeller KI, Kim JW, Racke F, Thomas-Tikhonenko A, et al. Activation of transferrin receptor 1 by c-Myc enhances cellular proliferation and tumorigenesis. Mol Cell Biol. 2006;26:2373–86.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31.
Muri J, Thut H, Bornkamm GW, Kopf M. B1 and marginal zone B cells but not follicular B2 cells require Gpx4 to prevent lipid peroxidation and ferroptosis. Cell Rep. 2019;29:2731–44.e2734.
Hu Q, Zhang Y, Lou H, Ou Z, Liu J, Duan W, et al. GPX4 and vitamin E cooperatively protect hematopoietic stem and progenitor cells from lipid peroxidation and ferroptosis. Cell Death Dis. 2021;12:706.
Kinowaki Y, Kurata M, Ishibashi S, Ikeda M, Tatsuzawa A, Yamamoto M, et al. Glutathione peroxidase 4 overexpression inhibits ROS-induced cell death in diffuse large B-cell lymphoma. Lab Investig. 2018;98:609–19.
Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-) : cystine supplier and beyond. Amino Acids. 2012;42:231–46.
Chen D, Tavana O, Chu B, Erber L, Chen Y, Baer R, et al. NRF2 is a major target of ARF in p53-independent tumor suppression. Mol Cell. 2017;68:224–32.e224.
Chen D, Fan Z, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene. 2017;36:5593–608.
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62.
Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. 2018;20:1181–92.
Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc(-) activity. Curr Biol. 2018;28:2388–99.e2385.
Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol. 2019;15:1137–47.
Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, et al. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem Biol. 2019;26:623–33.e629.
Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–91.
Hashidate-Yoshida T, Harayama T, Hishikawa D, Morimoto R, Hamano F, Tokuoka SM, et al. Fatty acid remodeling by LPCAT3 enriches arachidonate in phospholipid membranes and regulates triglyceride transport. Elife. 2015;4:e06328.
Rong X, Wang B, Dunham MM, Hedde PN, Wong JS, Gratton E, et al. Lpcat3-dependent production of arachidonoyl phospholipids is a key determinant of triglyceride secretion. Elife. 2015;4:e06557.
Li Z, Jiang H, Ding T, Lou C, Bui HH, Kuo MS, et al. Deficiency in lysophosphatidylcholine acyltransferase 3 reduces plasma levels of lipids by reducing lipid absorption in mice. Gastroenterology. 2015;149:1519–29.
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–92.
Doll S, Freitas FP, Shah R, Aldrovandi M, Da SMC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–8.
Santoro MM. The antioxidant role of non-mitochondrial CoQ10: mystery solved! Cell Metab. 2020;31:13–15.
Mishima E, Ito J, Wu Z, Nakamura T, Wahida A, Doll S, et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature. 2022;608:778–83.
Hirschhorn T, Stockwell BR. Vitamin K: a new guardian against ferroptosis. Mol Cell. 2022;82:3760–2.
Venkatesh D, O’Brien NA, Zandkarimi F, Tong DR, Stokes ME, Dunn DE, et al. MDM2 and MDMX promote ferroptosis by PPARalpha-mediated lipid remodeling. Genes Dev. 2020;34:526–43.
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586–90.
Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Muller C, Zandkarimi F, et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. 2020;6:41–53.
Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10.
Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. 2017;170:1062–78.
Ichikawa A, Kinoshita T, Watanabe T, Kato H, Nagai H, Tsushita K, et al. Mutations of the p53 gene as a prognostic factor in aggressive B-cell lymphoma. N Engl J Med. 1997;337:529–34.
Koduru PR, Raju K, Vadmal V, Menezes G, Shah S, Susin M, et al. Correlation between mutation in P53, p53 expression, cytogenetics, histologic type, and survival in patients with B-cell non-Hodgkin’s lymphoma. Blood. 1997;90:4078–91.
Gutierrez M, Bhatia K, Diez B, Muriel F, Epelman S, Deandreas M, et al. Prognostic-significance of p53 mutations in small non-cleaved cell lymphomas. Int J Oncol. 1994;4:567–71.
Dohner H, Fischer K, Bentz M, Hansen K, Benner A, Cabot G, et al. p53 gene deletion predicts for poor survival and non-response to therapy with purine analogs in chronic B-cell leukemias. Blood. 1995;85:1580–9.
Karube K, Enjuanes A, Dlouhy I, Jares P, Martin-Garcia D, Nadeu F, et al. Integrating genomic alterations in diffuse large B-cell lymphoma identifies new relevant pathways and potential therapeutic targets. Leukemia. 2018;32:675–84.
Zamò A, Pischimarov J, Schlesner M, Rosenstiel P, Bomben R, Horn H, et al. Differences between BCL2-break positive and negative follicular lymphoma unraveled by whole-exome sequencing. Leukemia. 2018;32:685–93.
Greiner TC, Moynihan MJ, Chan WC, Lytle DM, Pedersen A, Anderson JR, et al. p53 mutations in mantle cell lymphoma are associated with variant cytology and predict a poor prognosis. Blood. 1996;87:4302–10.
Cherney BW, Bhatia KG, Sgadari C, Gutierrez MI, Mostowski H, Pike SE, et al. Role of the p53 tumor suppressor gene in the tumorigenicity of Burkitt’s lymphoma cells. Cancer Res. 1997;57:2508–15.
Leu JI, Murphy ME, George DL. Mechanistic basis for impaired ferroptosis in cells expressing the African-centric S47 variant of p53. Proc Natl Acad Sci USA. 2019;116:8390–6.
Suzuki S, Venkatesh D, Kanda H, Nakayama A, Hosokawa H, Lee E, et al. GLS2 is a tumor suppressor and a regulator of ferroptosis in hepatocellular carcinoma. Cancer Res. 2022;82:3209–22.
Ou Y, Wang SJ, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci USA. 2016;113:E6806–E6812.
Zhang Y, Qian Y, Zhang J, Yan W, Jung YS, Chen M, et al. Ferredoxin reductase is critical for p53-dependent tumor suppression via iron regulatory protein 2. Genes Dev. 2017;31:1243–56.
Wang M, Mao C, Ouyang L, Liu Y, Lai W, Liu N, et al. Correction to: Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death Differ. 2020;27:1447.
Lu J, Xu F, Lu H. LncRNA PVT1 regulates ferroptosis through miR-214-mediated TFR1 and p53. Life Sci. 2020;260:118305.
Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21:579–91.
Venkatesh D, Stockwell BR, Prives C. p21 can be a barrier to ferroptosis independent of p53. Aging. 2020;12:17800–14.
Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017;20:1692–704.
Jiang L, Hickman JH, Wang SJ, Gu W. Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses. Cell Cycle. 2015;14:2881–5.
He C, Wang C, Liu H, Shan B. Kayadiol exerted anticancer effects through p53-mediated ferroptosis in NKTCL cells. BMC Cancer. 2022;22:724.
Hong Y, Ren T, Wang X, Liu X, Fei Y, Meng S, et al. APR-246 triggers ferritinophagy and ferroptosis of diffuse large B-cell lymphoma cells with distinct TP53 mutations. Leukemia. 2022;36:2269–80.
Koh CM, Bezzi M, Low DH, Ang WX, Teo SX, Gay FP, et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature. 2015;523:96–100.
Hummel M, Bentink S, Berger H, Klapper W, Wessendorf S, Barth TF, et al. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N Engl J Med. 2006;354:2419–30.
Farrell PJ. Epstein-Barr virus and cancer. Annu Rev Pathol. 2019;14:29–53.
Alborzinia H, Florez AF, Kreth S, Bruckner LM, Yildiz U, Gartlgruber M, et al. MYCN mediates cysteine addiction and sensitizes neuroblastoma to ferroptosis. Nat Cancer. 2022;3:471–85.
Devin J, Caneque T, Lin YL, Mondoulet L, Veyrune JL, Abouladze M, et al. Targeting cellular iron homeostasis with ironomycin in diffuse large B-cell lymphoma. Cancer Res. 2022;82:998–1012.
Jiang X, Guo S, Xu M, Ma B, Liu R, Xu Y, et al. TFAP2C-mediated lncRNA PCAT1 inhibits ferroptosis in docetaxel-resistant prostate cancer through c-Myc/miR-25-3p/SLC7A11 signaling. Front Oncol. 2022;12:862015.
Lu C, Cai Y, Liu W, Peng B, Liang Q, Yan Y, et al. Aberrant expression of KDM1A inhibits ferroptosis of lung cancer cells through up-regulating c-Myc. Sci Rep. 2022;12:19168.
Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554.
Kapadia B, Nanaji NM, Bhalla K, Bhandary B, Lapidus R, Beheshti A, et al. Fatty acid synthase induced S6kinase facilitates USP11-eIF4B complex formation for sustained oncogenic translation in DLBCL. Nat Commun. 2018;9:829.
Xu W, Berning P, Lenz G. Targeting B-cell receptor and PI3K signaling in diffuse large B-cell lymphoma. Blood. 2021;138:1110–9.
Kim J, Guan KL. mTOR as a central hub of nutrient signalling and cell growth. Nat Cell Biol. 2019;21:63–71.
Lei G, Zhuang L, Gan B. mTORC1 and ferroptosis: regulatory mechanisms and therapeutic potential. Bioessays. 2021;43:e2100093.
Reinke EN, Ekoue DN, Bera S, Mahmud N, Diamond AM. Translational regulation of GPx-1 and GPx-4 by the mTOR pathway. PLoS One. 2014;9:e93472.
Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–32.
Yi J, Zhu J, Wu J, Thompson CB, Jiang X. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci USA. 2020;117:31189–97.
Monti S, Savage KJ, Kutok JL, Feuerhake F, Kurtin P, Mihm M, et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood. 2005;105:1851–61.
Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell. 2012;22:547–60.
Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol cell. 2019;73:354–63.e353.
Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21:85–100.
Wang YQ, Chang SY, Wu Q, Gou YJ, Jia L, Cui YM, et al. The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front Aging Neurosci. 2016;8:308.
Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci USA. 2019;116:2672–80.
Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18:280–96.
Basit F, van Oppen LM, Schöckel L, Bossenbroek HM, van Emst-de Vries SE, Hermeling JC, et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017;8:e2716.
Plantinga TS, Tesselaar MH, Morreau H, Corssmit EP, Willemsen BK, Kusters B, et al. Autophagy activity is associated with membranous sodium iodide symporter expression and clinical response to radioiodine therapy in non-medullary thyroid cancer. Autophagy. 2016;12:1195–205.
Liu J, Kuang F, Kroemer G, Klionsky DJ, Kang R, Tang D. Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem Biol. 2020;27:420–35.
Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. 2020;66:89–100.
Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019;10:822.
Liu Y, Zhou X, Wang X. Targeting the tumor microenvironment in B-cell lymphoma: challenges and opportunities. J Hematol Oncol. 2021;14:125.
Kennedy LB, Salama AKS. A review of cancer immunotherapy toxicity. CA Cancer J Clin. 2020;70:86–104.
Wen Q, Liu J, Kang R, Zhou B, Tang D. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun. 2019;510:278–83.
Wiernicki B, Maschalidi S, Pinney J, Adjemian S, Vanden BT, Ravichandran KS, et al. Cancer cells dying from ferroptosis impede dendritic cell-mediated anti-tumor immunity. Nat Commun. 2022;13:3676.
Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. 2020;16:2069–83.
Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, et al. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab. 2021;33:1001–12.e1005.
CD36 activity causes ferroptosis in tumor-infiltrating CD8(+) T cells. Cancer Discov. 2021;11:OF24.
Kobayashi T, Lam PY, Jiang H, Bednarska K, Gloury R, Murigneux V, et al. Increased lipid metabolism impairs NK cell function and mediates adaptation to the lymphoma environment. Blood. 2020;136:3004–17.
Zhang W, Sun Y, Bai L, Zhi L, Yang Y, Zhao Q, et al. RBMS1 regulates lung cancer ferroptosis through translational control of SLC7A11. J Clin Investig. 2021;131:e152067.
Moujalled D, Southon AG, Saleh E, Brinkmann K, Ke F, Iliopoulos M, et al. BH3 mimetic drugs cooperate with Temozolomide, JQ1 and inducers of ferroptosis in killing glioblastoma multiforme cells. Cell Death Differ. 2022;29:1335–48.
Yan H, Talty R, Johnson CH. Targeting ferroptosis to treat colorectal cancer. Trends Cell Biol. 2023;33:185–8.
Chen X, Kang R, Kroemer G, Tang D. Targeting ferroptosis in pancreatic cancer: a double-edged sword. Trends Cancer. 2021;7:891–901.
Ghoochani A, Hsu EC, Aslan M, Rice MA, Nguyen HM, Brooks JD, et al. Ferroptosis inducers are a novel therapeutic approach for advanced prostate cancer. Cancer Res. 2021;81:1583–94.
Martinez AM, Kim A, Yang WS. Detection of ferroptosis by BODIPY 581/591 C11. Methods Mol Biol. 2020;2108:125–30.
Taguchi T, Kurata M, Onishi I, Kinowaki Y, Sato Y, Shiono S, et al. SECISBP2 is a novel prognostic predictor that regulates selenoproteins in diffuse large B-cell lymphoma. Lab Investig. 2021;101:218–27.
Sekine Y, Yamamoto K, Kurata M, Honda A, Onishi I, Kinowaki Y, et al. HADHB, a fatty acid beta-oxidation enzyme, is a potential prognostic predictor in malignant lymphoma. Pathology. 2022;54:286–93.
Qi YL, Wang HR, Chen LL, Duan YT, Yang SY, Zhu HL. Recent advances in small-molecule fluorescent probes for studying ferroptosis. Chem Soc Rev. 2022;51:7752–78.
Chen Q, Ma X, Xie L, Chen W, Xu Z, Song E, et al. Iron-based nanoparticles for MR imaging-guided ferroptosis in combination with photodynamic therapy to enhance cancer treatment. Nanoscale. 2021;13:4855–70.
Chen Y, Li L, Lan J, Cui Y, Rao X, Zhao J, et al. CRISPR screens uncover protective effect of PSTK as a regulator of chemotherapy-induced ferroptosis in hepatocellular carcinoma. Mol Cancer. 2022;21:11.
Sehn LH, Gascoyne RD. Diffuse large B-cell lymphoma: optimizing outcome in the context of clinical and biologic heterogeneity. Blood. 2015;125:22–32.
Weng J, Chen L, Liu H, Yang XP, Huang L. Ferroptosis markers predict the survival, immune infiltration, and ibrutinib resistance of diffuse large B cell lymphoma. Inflammation. 2022;45:1146–61.
Chen H, He Y, Pan T, Zeng R, Li Y, Chen S, et al. Ferroptosis-related gene signature: a new method for personalized risk assessment in patients with diffuse large B-cell lymphoma. Pharmgenom Pers Med. 2021;14:609–19.
Xiong D, Li M, Zeng C. Construction and validation of a risk scoring model for diffuse large B-cell lymphoma based on ferroptosis-related genes and its association with immune infiltration. Transl Oncol. 2022;16:101314.
Wu H, Zhang J, Fu L, Wu R, Gu Z, Yin C, et al. Identification and development of a 4-gene ferroptosis signature predicting overall survival for diffuse large B-cell lymphoma. Technol Cancer Res Treat. 2023;22:15330338221147772.
Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia. 2001;15:1633–40.
Zheng C, Wang C, Sun D, Wang H, Li B, Liu G, et al. Structure-activity relationship study of RSL3-based GPX4 degraders and its potential noncovalent optimization. Eur J Med Chem. 2023;255:115393.
Chen Y, Wang F, Wu P, Gong S, Gao J, Tao H, et al. Artesunate induces apoptosis, autophagy and ferroptosis in diffuse large B cell lymphoma cells by impairing STAT3 signaling. Cell Signal. 2021;88:110167.
Cheah CY, Seymour JF, Wang ML. Mantle cell lymphoma. J Clin Oncol. 2016;34:1256–69.
Lepelletier Y, Camara-Clayette V, Jin H, Hermant A, Coulon S, Dussiot M, et al. Prevention of mantle lymphoma tumor establishment by routing transferrin receptor toward lysosomal compartments. Cancer Res. 2007;67:1145–54.
Samara A, Shapira S, Lubin I, Shpilberg O, Avigad S, Granot G, et al. Deferasirox induces cyclin D1 degradation and apoptosis in mantle cell lymphoma in a reactive oxygen species- and GSK3beta-dependent mechanism. Br J Haematol. 2021;192:747–60.
Perez-Galan P, Roue G, Villamor N, Montserrat E, Campo E, Colomer D. The proteasome inhibitor bortezomib induces apoptosis in mantle-cell lymphoma through generation of ROS and Noxa activation independent of p53 status. Blood. 2006;107:257–64.
Davis M, Malcolm S, Rabbitts TH. Chromosome translocation can occur on either side of the c-myc oncogene in Burkitt lymphoma cells. Nature. 1984;308:286–8.
Molyneux EM, Rochford R, Griffin B, Newton R, Jackson G, Menon G, et al. Burkitt’s lymphoma. Lancet. 2012;379:1234–44.
Burton EM, Voyer J, Gewurz BE. Epstein-Barr virus latency programs dynamically sensitize B cells to ferroptosis. Proc Natl Acad Sci USA. 2022;119:e2118300119.
Wang N, Zeng GZ, Yin JL, Bian ZX. Artesunate activates the ATF4-CHOP-CHAC1 pathway and affects ferroptosis in Burkitt’s Lymphoma. Biochem Biophys Res Commun. 2019;519:533–9.
Turtle CJ, Hay KA, Hanafi LA, Li D, Cherian S, Chen X, et al. Durable molecular remissions in chronic lymphocytic leukemia treated with CD19-specific chimeric antigen receptor-modified T cells after failure of ibrutinib. J Clin Oncol. 2017;35:3010–20.
Burger JA, O’Brien S. Evolution of CLL treatment - from chemoimmunotherapy to targeted and individualized therapy. Nat Rev Clin Oncol. 2018;15:510–27.
Pan B, Li Y, Xu Z, Miao Y, Yin H, Kong Y, et al. Identifying a novel ferroptosis-related prognostic score for predicting prognosis in chronic lymphocytic leukemia. Front Immunol. 2022;13:962000.
Gong H, Li H, Yang Q, Zhang G, Liu H, Ma Z, et al. A ferroptosis molecular subtype-related signature for predicting prognosis and response to chemotherapy in patients with chronic lymphocytic leukemia. Biomed Res Int. 2022;2022:5646275.
Chi KY, Shen HN. Extranodal natural killer T-cell lymphoma. N Engl J Med. 2020;382:562.
Zou Y, Schreiber SL. Progress in understanding ferroptosis and challenges in its targeting for therapeutic benefit. Cell Chem Biol. 2020;27:463–71.
Chen X, Hu S, Han Y, Cai Y, Lu T, Hu X, et al. Ferroptosis-related STEAP3 acts as predictor and regulator in diffuse large B cell lymphoma through immune infiltration. Clin Exp Med. 2023;23:2601–17.
Acknowledgements
This research was funded by the National Cancer Institute/National Institutes of Health under grants R01CA233490, U54CA272691, and The Duke University startup fund. KHY was also supported by the Hagemeister Lymphoma Foundation.
Author information
Authors and Affiliations
Contributions
Conceptualization: ZYX-M and KHY; Investigation, Methodology, Writing—original draft preparation: TY; validation, ZYX-M; writing—review and editing: ZYX-M, LY, YL, and KHY; resources: KHY; visualization, TY; supervision, LY and KHY; funding acquisition: YL and KHY. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by Professor Boris Zhivotovsky
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yu, T., Xu-Monette, Z.Y., Yu, L. et al. Mechanisms of ferroptosis and targeted therapeutic approaches in lymphoma. Cell Death Dis 14, 771 (2023). https://doi.org/10.1038/s41419-023-06295-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-023-06295-w
