
Abstract
Lipid droplets (also known as lipid bodies) are lipid-rich, cytoplasmic organelles that play important roles in cell signaling, lipid metabolism, membrane trafficking, and the production of inflammatory mediators. Lipid droplet biogenesis is a regulated process, and accumulation of these organelles within leukocytes, epithelial cells, hepatocytes, and other nonadipocyte cells is a frequently observed phenotype in several physiologic or pathogenic situations and is thoroughly described during inflammatory conditions. Moreover, in recent years, several studies have described an increase in intracellular lipid accumulation in different neoplastic processes, although it is not clear whether lipid droplet accumulation is directly involved in the establishment of these different types of malignancies. This review discusses current evidence related to the biogenesis, composition and functions of lipid droplets related to the hallmarks of cancer: inflammation, cell metabolism, increased proliferation, escape from cell death, and hypoxia. Moreover, the potential of lipid droplets as markers of disease and targets for novel anti-inflammatory and antineoplastic therapies will be discussed.
Similar content being viewed by others

Facts
-
Lipid droplets are dynamic and multifunctional organelles involved in energy metabolism, signaling, and inflammatory mediator production.
-
Lipid droplets accumulate in a variety of cancer cells.
-
Lipid droplets modulate the cross-talk between tumors and other cell types in tumor microenvironment.
-
Lipid droplet accumulation and catabolism are tightly coupled to energetic metabolism, cell signaling, and are critical to cancer cell proliferation, resistance to death, and aggressiveness.
Open questions
-
Does LD play a causal role or is a consequence of tumorigenesis?
-
How important is LD accumulation during the distinct phases of tumor development?
-
Can lipid droplet be a cytoplasmic hub for protumorigenic cellular signaling?
-
Are there roles for inhibitors of lipid droplet biogenesis as target for cancer therapy?
Introduction
Lipid droplets (LDs) are cytoplasmic lipid-enriched organelles delimited by a monolayer of phospholipid, which covers a hydrophobic core composed of neutral lipids, mainly triacylglycerol (TAG) and cholesteryl esters (CEs), with a diverse content of proteins that may vary according to the cell and stimulatory conditions. In addition, LDs are coated by structural proteins of the PAT family that include perilipin-1, perilipin-2, and perilipin-31,2,3,4. LDs are organelles that originate and are intimately related to the endoplasmic reticulum (ER), formed by the transfer of lipids and proteins from the ER to newly formed LDs5. Although LD biogenesis involves specific and well-regulated mechanisms, the cellular and molecular mechanisms involved are still not completely understood.
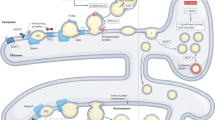
The first reports of LDs in human tumors date from the 1960s6,7; nevertheless, for many years, the study of LDs in cancer was restricted to descriptions in different tumors. A major breakthrough in LD research in cancer, came from the demonstration that LDs are major sites for prostaglandin E2 (PGE2) synthesis in colon cancer cells and have roles in tumor cell proliferation8. Over the last decade, there was a considerable expansion of the mechanisms that regulate the formation and functions of LDs in cancer. LDs have been identified in all the processes involved in cancer development, including initiation, promotion, and progression. Hanahan and Weinberg9 outlined hallmarks that characterized the capabilities acquired during tumor development. Among these are sustaining proliferation, resisting death, evading growth suppressors, promoting replicative immortality, activating invasion and metastasis, as well as promoting angiogenesis, genome instability, inflammation, energy metabolism deregulation, and evasion of immune destruction10. Here, we review the current knowledge of LD functions according to categories comprising the hallmarks of cancer, and two additional topics comprise LD roles in biomarkers and cancer stem cells (Fig. 1).

Based on the increasingly information about the role of lipid droplets in cancer, emerging from several different models, we suggest the association of lipid droplets with some of the currently established Hallmarks of Cancer—biological traits acquired by cells during cancer multistep development, a concept originally conceived by Hanahan and Weinberg in 2000. Although there are many unanswered questions of fundamental importance to better understand the relationship between these organelles and tumorigenesis, these associations may be explored for future anticancer therapies.
Implications of lipid droplets in cancer
Lipid droplets and metabolism in cancer
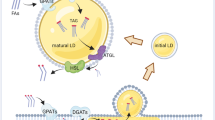
Lipid droplet accumulation in transformed cancer cells involves complex and mediated mechanisms, including increased lipid uptake, de novo lipid synthesis and remodeling, as well as regulations in lipolysis (Fig. 2). Lipogenesis is paramount among the anabolic processes in cancer, where fatty acid (FA) synthesis is critical for generating building blocks for more complex lipids11. Signaling pathways that trigger tumor lipogenesis culminate in the accumulation of newly formed LDs8,12,13,14,15,16,17,18,19,20,21,22. Indeed, accumulating evidence support a relationship between tumor development and both lipolytic or lipogenic enzymes, implying that lipid mobilization from LDs may be an appropriate target for cancer therapy. The main factors in this phenomenon are sterol regulatory element-binding proteins (SREBPs), a key regulator of lipid homeostasis23, and mammalian target of rapamycin (mTOR), a crucial sensor that connects cellular growth to the availability of extracellular nutrients24. SREBP1 upregulation triggers tumor growth and LD accumulation together with lipogenesis enzyme overexpression12,14,15,18,20,22,25,26. Moreover, both mTOR catalytic complexes, namely, mTORC1 and mTORC2, were implicated in LD biogenesis in cancer by SREBP1-dependent14,26 and SREBP1-independent20 mechanisms.

Different stimuli and cellular pathways contribute for lipid droplets formation, depending on environmental conditions, such as hypoxia, obesity, infection, or extracellular signaling molecules. These processes invariably involve changes in gene expression that regulates de novo lipid synthesis, induction of extracellular lipid uptake and LD biogenesis. Lipid droplets formed by these different stimuli harbor specific lipid content; and a set of enzymes directly related to lipogenesis, such as FAS and DGAT, promote its increase. Lipolytic enzymes could also be located in lipid droplets for fatty acid mobilization upon activation. ABHD5 α-β hydrolase domain containing 5 (also known as CGI-58—Comparative Gene Identification-58), ACC acetyl-CoA carboxylase, ATGL adipose triglyceride lipase, DGAT diacylglycerol O-acyltransferase, DAG diacylglycerol, FA fatty acid, FABP fatty acid-binding protein, FAS fatty acid synthase, G0S2 G0/G1 switch 2, HIF hypoxia-inducible factors, mTOR mammalian target of rapamycin, PI3K phosphoinositide 3-kinase, PLIN2 perilipin-2, PPARγ peroxisome proliferator-activated receptor gamma, RXR retinoid X receptor, SatFA saturated fatty acid, SCD stearoyl-CoA desaturase, SREBP sterol regulatory element-binding protein, TAG triacylglycerol, TLR4 Toll-like receptor, TRIF TIR-domain-containing adapter-inducing interferon-β, UnsatFA unsaturated fatty acids, CD36 fatty acid translocase, CE cholesteryl ester, FC free cholesterol, LDL low-density lipoprotein, LDL-R low-density lipoprotein receptor, SRB1 scavenger receptor class B type, ACAT acyl-CoA:cholesterol acyltransferase.
Obesity and its associated metabolic dysregulation are established risk factors for many cancers. Obesity-drive ectopic LD accumulation in non-adipose tissues have been associated with insulin-resistance, cardiovascular disease, and cancer1. Different mechanisms may contribute to obesity-induced increased LDs in epithelial cells and other in non-adipose tissues. In addition to excess of free FAs and other lipids, adipose-derived leptin through mTOR activation may contribute to the obesity-related enhanced susceptibility to colon carcinoma by altering the intracellular lipid metabolism and inflammatory environment27.
TAG is one of the major neutral lipids in LDs. TAG is formed by a glycerol joined to three FAs and shares many steps with glycerophospholipid synthesis until the generation of DAG that is esterified by acyl-CoA diacylglycerol acyltransferase (DGAT) to TAG. The role of TAG metabolism remains poorly understood in cancer. Ackerman et al.28 showed that TAG acts as a buffer for lipid remodeling, especially under O2 and nutrient limitation. LD TAGs were enriched with unsaturated FAs, particularly oleate; however, during O2 and serum deprivation, oleate was released from LDs into phospholipid pools, which prevents cellular stress by avoiding the use of fully saturated, potentially toxic lipids28. They also showed that inhibition of both DGAT isoforms increased cancer cell death in vivo, impairing tumor growth28. In addition, DGAT1 was overexpressed in prostate cancer cells, and its inhibition decreased LD density, microtubule-organizing center numbers and microtubule stability, which affects cell migration and growth29.
TAG is sequentially hydrolyzed by three different lipases: adipose triglyceride lipase (ATGL), hormone sensitive lipase (HSL), and monoacylglycerol lipase (MAGL). Consecutively, they hydrolyze TAG, DAG, and MAG into a glycerol backbone and free FAs. ATGL function could be regulated by CGI-58/ABHD5, a coactivator, and G0S2 (G0/G1 switch gene2), an inhibitor30. The following data indicate that the role of lipases in cancer seems to be dependent on the cell type or protein studied. Loss of ATGL is a common feature in many human tumors and induces spontaneous lung cancer in animal models31. In addition, ATGL deletion can induce a more aggressive phenotype in lung cancer cells through lipid accumulation32. Moreover, CGI-58/ABHD5 deletion was correlated with cancer development and progression33 and epithelial–mesenchymal transition33,34. In contrast, ATGL inhibition by G0S2 decreased proliferation in tumor cell lines, suggesting that ATGL activity is common in some cancer types35. MAGL regulates the network of free FAs in tumors, such as colorectal cancer, neuroblastoma, hepatocarcinoma, and nasopharyngeal carcinoma, by enabling tumor cells to mobilize and utilize FAs from stored TAGs36,37,38,39,40. These released FAs, including lysophosphatidic acid, PGE2, and endocannabinoids, were involved in signal cascades that induce carcinogenesis, tumor progression, and metastasis39,41. Interestingly, lipolysis also plays an important role in cancer-associated cachexia, a multifactorial metabolic syndrome associated with loss of muscle and adipose mass42. Increased ATGL and, to a less extent, HSL activities were observed in white adipose tissue from cancer-associated cachexia patients, whereas ATGL-deficient mice with tumors maintained adipose tissue and gastrocnemious muscle mass43. Therefore, further studies are needed to elucidate the role of TAG lipases in cancer.
Another LD component is cholesteryl ester (CE), the storage form of cholesterol synthesized by acyl coenzyme A: cholesterol acyltransferase (ACAT)44. Free cholesterol maintains membrane fluidity and can be either de novo synthesized via the mevalonate pathway or taken up from exogenous lipoproteins45. Aberrant CE accumulation in LDs is an important target of tumor metabolism remodeling. Increased CE is a metabolic signature in renal cell carcinoma, glioblastoma, breast, prostate and pancreatic cancer17,21,46,47,48,49. In addition, CE accumulation was positively correlated with advanced clinical staging, metastasis, and poor survival21,46,47,48. Inhibition of ACAT significantly suppressed cancer proliferation, migration, invasion, and tumor growth in vitro and in vivo17,21,46. CE accumulation is driven by PTEN loss and PI3k/Akt/mTOR upregulation21 and modulates signaling pathways, such as SREBP by blocking the SREBP-negative feedback loop caused by excess free cholesterol, maintaining tumor growth17,21,46, and also Wnt/β-catenin by regulating FA availability for Wnt3a acylation50. These studies suggest that cholesterol esterification may be a major target in cancer therapy.
LDs are directed to autophagy pathways as an alternative route for lipid storage mobilization51,52. Lipid droplet inclusion in autophagossomes and subsequent degradation in lysosomes (known as macrolipophagy) regulates cellular lipid content, as inhibition of lipophagy decreases TAG breakdown53. Upon engulfment, LD content are broken by lysosomal acid lipases (LAL), mostly known for its deficiency in Wolman disease and CE storage disease54,55. Another mechanism for LD hydrolysis in lysosomes is through chaperone-mediated autophagy (CMA), a lysosomal proteolysis carried by heat shock protein 70 (HSP70)56 and lysosome-associated membrane protein 2A (LAMP-2A)57. Degradation of perilipin-2 and perilipin-3 by CMA act as a prerequisite to stimulate both ATGL lipolysis and macrolypophagy58. Several CMA-targeted proteins are relevant to cancer biology59, though the relationship between CMA-dependent lipolysis and cancer is currently unknown. For more in-depth information, we recommend recently published reviews where lipophagy regulatory mechanisms and functions in other physiopathological conditions are comprehensively discussed52,60.
Though poorly studied, lipophagy appears to play a dual role in cancer. LDs degradation by lipophagy increases viability of hepatocarcimoma cells during starvation61, and protects androgen-sensitive prostate cancer cells during androgen-deprivation in vitro62. Also, pharmacological inhibition of lipolysis in colorectal cancer cell line induced both LDs accumulation and cell death, while altering the profile of cancer stem cells toward a more invasive mesenchymal phenotype63. On the other hand, lipophagy can display an antitumoral role in some cancer models. Overexpression of autophagy regulatory protein ATG14 increased LD lipophagy while sensitized HeLa cells to apoptosis64.
Lipid droplets in inflammation and avoiding immune destruction
In inflammation and cancer, LDs are linked to the regulation of immune and inflammatory responses by acting as specialized hubs of signaling with major roles in eicosanoid and other lipid mediator formation65. Eicosanoids are signaling molecules derived from the enzymatic oxygenation of arachidonic acid (AA), which control key cellular processes, including cell activation, metabolism, proliferation, and death66. It is well established that LDs are sites of esterified AA as well as of several enzymes involved in eicosanoid synthesis including cPLA2, cyclooxygenases and prostaglandin synthases8,67,68,69,70,71. By means of eicosacell, a technique to localize newly formed eicosanoids at their sites of synthesis72, it was established that LDs are major intracellular locales for the activation-elicited formation of PGE2 in cancer cells8. Moreover, leukocytes, endothelial and epithelial cells involved in pathological conditions, such as in cancer, hypoxia, and during infections were shown to contain increased numbers of eicosanoid-synthesizing LDs leading to amplified eicosanoid production8,73,74,75,76.
The amplification of eicosanoid formation through the compartmentalization of eicosanoid-synthetic machinery at LDs in tumor cells may have implications to promote tumor growth by paracrinally regulating cancer cells, as well as by orchestrating the complex interactions to establish the tumor microenvironment (Fig. 3). Indeed, LD and LD-derived PGE2 were shown to promote tumor epithelial cell proliferation8,27,73. Also, several reports indicate that PGE2 has a protumoral role by stimulating tumor cell proliferation and suppressing host immune surveillance of tumor77,78,79,80,81.

Lipid droplets (LDs) were associated with distinct roles in heterogeneous cell population of tumor microenvironment. In tumor cells, LDs are sites of PGE2 (prostaglandin E2) synthesis, an important immune suppressive eicosanoid, and are associated with proliferation and activation of cancer stemness pathways. Adipocytes release cytokines and fatty acids to fuel metastasis and aggressiveness. In myeloids derived cells, LDs were associated with polarization of TAM (tumor-associated macrophage), a modulatory phenotype of MDSCs (myeloid-derived suppressor cells) and in dendritic cell, LDs enriched with oxidized triacylglycerol species were associated with antigen presentation dysfunction. ROS reactive oxygen species, MHCI major histocompatibility complex class I protein, COX-2 cyclooxygenase-2, cPLA2 cytosolic phospholipase A2, AA arachidonic acid, FA fatty acid, CD36 fatty acid translocase.
The tumor microenvironment (TME) is a term that refers to a heterogeneous cell population of cancer cells and host resident and recruited cells, secreted factors and extracellular matrix that constitute the tumor mass82. Furthermore, the TME plays a critical role in the prognosis of tumor development and therapeutic responses83. Recent studies indicate that LDs modulate the cross-talk between tumors and phenotypic modulation of immune cells, mainly myeloid cells, such as tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and dendritic cells (DCs)84,85,86,87,88,89 (Fig. 3). Indeed, as mentioned above, LDs are involved in increased eicosanoid synthesis in tumor cell LDs and inflammatory cells8,71,90, suggesting roles of LDs as an eicosanoid production site in modulating TME cells. In addition to eicosanoids synthesis, LDs are emerging as sites for regulation of different signaling pathways with potential functions in TME regulation.
TAMs are the major tumor-infiltrating leukocytes and promote malignant progression by stimulating angiogenesis and tissue remodeling and preventing immune destruction91. The role of LDs in these cells are still not fully established, since the literature demonstrates both protumorigenic and antitumorigenic functions. FA-binding protein E (E-FABP), a lipid-binding chaperone, is highly expressed in TAMs presenting anticancer activities84. In this scenario, IFN-β signaling is regulated through E-FABP-mediated LD biogenesis, recruiting natural killer cells to the tumor stroma84. In contrast, cancer cells stimulate TAM differentiation toward a protumorigenic phenotype by a caspase-1-dependent, nonconventional cleavage of PPARγ87. Cleaved PPARγ then translocates to mitochondria and reduces FA oxidation, increasing LDs and promoting protumor TAM differentiation87. Also, Wu et al.92 showed that unsaturated FA, oleate, polarizes bone marrow-derived myeloid cells into an immunosuppressive TAM. This phenotype is dependent on mTOR signaling, LD formation, and utilization by mitochondrial respiration. Moreover, LDs from TAMs were shown as an effective target to avoid TAM polarization and tumor growth92. Further work to elucidate the functions of the LDs in TAM differentiation and the origin of its lipids are necessary.
MDSCs are a diverse group of immune cells from the myeloid lineage with strong immunoregulatory properties93, which are shown to depend on both oxidative stress and lipid metabolism94,95,96. Tumor-infiltrating MDSCs increase FA uptake and oxidation, mitochondrial mass, and oxygen consumption rate. The inhibition of FA oxidation blocks the immunosuppressive functions of MDSCs, enhancing the efficacy of cancer immune therapy94. These findings encouraged other studies focused on the role of lipids in the induction of the regulatory phenotype of MDSCs86,88. Wu et al.88 demonstrated that unsaturated FAs, but not saturated FAs, are capable of inducing a modulatory phenotype in these cells paralleled by increased LD formation. The inhibition of LD formation by DGAT blockade abrogated the MDSC phenotype, while the inhibition of de novo FA synthesis had no effect, suggesting a critical role for exogenous FA and LD biogenesis. In addition, Al-Khami et al.86 reached similar conclusions when evaluating a tumor-bearing mouse model. They observed that the tumor-released cytokines G-CSF and GM-CSF triggered lipid influx and LD biogenesis, oxidative metabolism and T-cell suppression. They verified that exogenous lipoproteins and unsaturated FAs, but not saturated FAs, enhanced the generation of immunosuppressive MDSCs. These results showed that the LD biogenesis necessary to regulate phenotype MDSCs in cancer was triggered by exogenous lipids. Although the source of lipids in the TME was not evaluated, the specific induction by unsaturated FAs may provide clues about the mechanisms similar to that of the DC regulation described below.
DCs are central in the anticancer response due to cross-presentation of tumor-associated antigens via MHC-I complexes to CD8+ cytotoxic T cells93. Although the presence of DCs is associated with a better prognosis, studies in tumor-bearing mice showed impaired cross-presentation by DCs in the TME97,98,99,100. There are conflicting data on the role of LDs, which are associated with both promotion and inhibition of cross-presentation in tumor-infiltrating DCs85,101,102,103. These differences may be caused by LD quality, not quantity, and related to DC antigen presentation dysfunction103. Veglia et al.89 showed that LDs from tumor-infiltrating DCs are enriched with oxidized triacylglycerol species. In addition, oxidized LDs sequestrated HSP70, which directed pMHCI localization to lysosomes rather than to the plasma membrane89. Though the authors did not confirm the TME lipid source, it would reasonable to suggest the involvement of cancer lipogenesis. Thereafter, Jiang et al. confirmed that FASN overexpression of tumor cells was responsible for elevated levels of LDs and subsequent inhibition of DCs in an ovarian cancer mouse model104. FASN silencing in cancer cells decreases LDs in DCs, consequently increasing infiltrative T cells and delaying tumor growth, which suggests that tumor cell lipogenesis could be involved in anticancer immunity104.
In conclusion, these data demonstrate that LDs are associated with the immunometabolic modulation phenotype of myeloid cells, which largely culminate in cancer immune evasion. However, more research is necessary to understand the exact mechanisms of how LDs are involved in phenotype modulation89. In the DC studies, the combination of a lipid-enriched microenvironment and oxidative stress was necessary to trigger modulation. High levels of circulating lipids and oxidative stress are widely described in many tumors and are associated with a poor prognosis105,106,107. The identification of the lipid source used in LD biogenesis may also be an important aspect in the signaling in which these organelles are involved, since these lipids may come from both external sources, such as tumor cells and adipose tissue, and from intracellular sources, such as de novo synthesis or autophagy. Surprisingly, cell free LDs were described in a 3D bioengineered brain tumor glioblastoma tissue platform, where it was suggested may participate in drug response, however, the role and mechanism remain unclear108. In addition, it is necessary to determine how LDs are involved in the exclusion of T cells from the TME, since this may be an intriguing target in immune cancer therapy.
Lipid droplets in cell proliferation
Accumulating evidence have shown that an increase in LD numbers occurs in cells undergoing proliferation, which is a common feature in many neoplastic processes, suggesting LD may contribute to cell proliferation109. Although no definitive studies establish a causal link between the increase in LD numbers and cancer development, recent studies are starting to shed light in this process. Indeed, emerging data associates increased LD biosynthesis and cell cycle progression. It was recently described that cell cycle progression regulates the number and cellular localization of LDs in nontransformed cells, with an increase in LDs numbers and dispersed subcellular localization upon entering S phase110. Moreover, detailed analysis of the distribution of lipid droplets during mitosis showed their polarization before cell division110. In addition, it was observed that the yeast lipase Tgl4 (human ATGL analog) is a target for phosphorylation by the major cell cycle regulator Cdc28 (human CDK1 analog), which is necessary for Tgl4 activity and cell cycle progression111. In mammals, a lipid-mediated PTEN-dependent late G1 checkpoint was recently described112. In this work, lipid deprivation in culture media was able to induce G1 arrest in several cancer cells, except in clear-cell renal carcinoma cells, where it is suggested that an increase in LDs contributes to bypass this checkpoint112. Collectively, these results suggest that cell cycle progression and lipid homeostasis are coordinated by a shared mechanism acting at the G1/S transition, thus suggesting that lipid droplet maintenance, biogenesis, or consumption is involved in cell cycle progression through S phase.
Activation of specific signaling pathways in colon cancer cells is linked with LD formation and cell growth modulation8. FOXO3 plays a pivotal role in inhibiting colon cancer cell proliferation, mainly through upregulation of the cell cycle inhibitor p27kip1113. However, FOXO3 activity is dependent on LD density, and an increase in LD numbers induced the loss of FOXO3 and p27kip1 expression114. Increased LD density promoted proliferation of colon cancer cells in a FOXO3 loss-dependent manner114. In addition, the mouse model for FOXO3 deficiency resulted in a decrease in Sirtuin6, a negative regulator of lipid metabolism, revealing the existence of a regulatory network between LD biogenesis and FOXO3 activity114 Interestingly, in vitro cellular transformation using H-RasV12 oncoprotein was associated with LD accumulation8,110 and an increase in perilipin-2 protein levels110, although perilipin-2 overexpression alone was not enough to induce cell transformation in murine fibroblasts110.
Of note, a variety of signaling-associated proteins have been found within LDs, suggesting a key role for this organelle as a cytoplasmic hub favoring quick proliferative signaling. Proteins with well-established roles in oncogenic transformation, tumorigenesis, and metastasis, including PI3K, ERK1, ERK2, p38, PKC, and caveolin, were shown to localize to LDs in a variety of cell types69,115,116. Nevertheless, until now, no study has unraveled the actual role of lipid droplet-resident kinases in cell proliferation. Hence, LDs could be a potential downstream target against uncontrolled triggering of the membrane receptor signaling cascade. Despite all suggestive data linking LDs and cell cycle progression, further studies are necessary to characterize whether LDs play a direct role in cell proliferation.
Lipid droplets in apoptosis and cell death
It has been reported that apoptosis induction leads to an early onset and subsequent accumulation of LDs117,118,119. Indeed, LD formation during apoptosis may delay the accumulation of toxic FAs120,121. Apoptosis-induced activation of p53 and inhibition of mTOR122 and MYC123 in tumor cells leads to lipid accumulation due to inhibition of FA β-oxidation and redirection of FAs to de novo lipogenesis. In addition, the increase in LD content has been used as an in vivo marker of post treatment tumor cell death through 1H nuclear magnetic resonance spectroscopy, a noninvasive diagnostic technique to detect the earliest signs of cell death following cancer treatment119,124.
Increased LDs in cancer cells may play an indirect role in maintaining cell survival during cancer therapy. An increase in LD numbers was previously observed in drug-resistant cancer cells, and thus, these organelles were postulated to sequester hydrophobic therapeutic agents, reducing drug effectiveness125,126. The development of drug-resistant cells derived from myeloid leukemia also revealed a positive correlation between increased LD content and resistance to an aminopeptidase inhibitor, along with activation of the ERK/Akt/mTOR survival pathway127. These data prompt the idea of hampering LD biogenesis to improve cancer therapy efficiency. Interestingly, reduction of LD formation by inhibition of cPLA2α enhanced the effectiveness of the anticancer agent curcumin in glioblastoma cells128. Hence, impairing LD drug sequestration could be interesting in a variety of other multidrug resistance scenarios to improve cell death upon antineoplastic drug administration.
Increased neutral lipids may enable further survival of cancer cells through other mechanisms. A recent study described a protective role of LDs in colorectal cancer cells against chemotherapy-induced cell death. LD biogenesis mediated by lysophosphatidylcholine acyltransferase 2 (LPCAT2) in these cells impaired ER stress pathways, resulting in diminished calreticulin (CRT) membrane exposure and, consequently, a reduction in immunogenic cell death after treatment129. Cell-surface CRT exposure is a key feature in anticancer immune responses130, and interestingly, CRT appeared to be sequestrated in LDs in these chemoresistant colorectal cells129. Other studies also revealed altered lipid metabolism in other resistant cancer cell lines, such as breast125,131,132 and ovarian cancer133; thus, LD formation may be involved in drug resistance. An interesting example was recently described in hypoxic conditions, where increased expression of acylglycerol-3-phosphate acyltransferase 2 (AGPAT2) was directly involved in LD accumulation and cell survival upon etoposide treatment in different cancer cell lines134.
Lipid droplets in hypoxia and angiogenesis
Hypoxia is defined as a reduction of oxygen (O2) concentration, a condition encountered in a variety of pathological conditions. Hypoxia responses are transcriptionally regulated by the hypoxia-inducible factor (HIF) family, which includes heterodimeric transcription factors consisting of an oxygen-regulated α-subunit (HIF-1α or HIF-2α) and a constitutively expressed β-subunit (HIF-β/ARNT)135,136,137. Hanahan and Weinberg suggest that the hypoxia response system not only causes an induction of the “angiogenic switch” in cancer138, but is also one of the factors that acts in the reprogramming of cancer cell metabolism10,139. In fact, evidence points out important changes in lipid metabolism in these instances. Increased FA synthesis is thought to be stimulated in low O2 conditions; for example, FA synthase is upregulated during hypoxia through the HIF-1α/Akt/SREBP-1 signaling pathway140, whereas HIF-1α activation increases glycolysis and free FA uptake by upregulating PPARγ expression141. This supports the idea that hypoxia responses can significantly alter cell lipid metabolism in several pathologies, including cancer, and that LDs would accumulate in hypoxic cells. Indeed, an inverse correlation between the oxygen concentration and LD levels was first observed in endothelial cells derived from bovine aortic or pulmonary vascular beds, emerging as a specific rather than a standardized response for any variety of cellular stresses142. Accordingly, perilipin-2 expression was increased in cancer cell lines under hypoxic conditions, as well as in the liver of mice treated with CoCl2, an inducer of hypoxia-like responses143. LD staining was also found in highly hypoxic cells located at the periphery of necrotic areas of intracerebral glioma144. In this scenario, HIF transcription factors seem to portray specific roles in LD regulation. Interestingly, HIF-2α—but not HIF-1α—promotes PLIN2 gene overexpression in clear-cell renal cell carcinoma, and perilipin-2 accumulation in these cells was associated with increased cancer cell viability145. Likewise, expression of both HIF-1α and HIF-2α was found to be important for establishing an invasive and metastatic phenotype in triple-negative breast cancer cells146, but single inhibition of HIF-2α expression alone resulted in altered metabolism and reduced formation of LDs in these cells146. In addition, in a mouse glioblastoma xenograft model, hypoxia induced LD accumulation in an HIF-1α-dependent manner due to increased FA uptake but not de novo lipid synthesis147. FABP3, FABP7, and perilipin-2 were essential for the formation of LDs under hypoxic conditions in this work147, and disruption of FABP3, FABP7, or perilipin-2 expression in this model reduced ATP production and increased ROS levels, which were accompanied by a decrease in cell growth and survival both in vitro and in vivo147. This evidence highlights the importance of lipid modulation during hypoxia as an important mechanism that directly regulates cell survival and aggressiveness.
Lipid droplets in cancer aggressiveness, invasion, and metastasis
Epithelial–mesenchymal transition (EMT) is the first of several steps toward a carcinoma metastasis event. In this process, carcinoma cells lose epithelial traits, such as apical–basal polarity and epithelial cell junctions, and display mesenchymal cell morphology and increased cell migration potential148. It was recently shown that the lipid profile differs between epithelial and mesenchymal breast cancer cells, revealing that monounsaturated lipids and de novo FA synthesis are markedly characteristic of epithelial cells, whereas reduced lipogenesis, increased polyunsaturated FA levels, and the expression of genes involved in TAG synthesis and LD formation were mainly traits of mesenchymal breast cancer cells149. In parallel, loss of CGI-58/ABHD5 promotes invasion and proliferation in prostate cancer through an ATGL-independent mechanism and correlated with increased aerobic glycolysis and loss of E-cadherin expression and Snail accumulation, markers of the EMT process34. However, the importance of LD accumulation in cell invasion and metastasis is still debatable, particularly why and when lipid mobilization from LDs would be necessary to trigger a more aggressive phenotype in cancer cells. Wright and collaborators discuss the necessity of an LD increase in primary cancer cells prior to a metastasis event. In these observations, in vitro accumulation of prometastatic protein CDCP-1 (CUB-domain containing protein 1) decreased the lipid content in triple-negative breast cancer cells and was correlated with augmented invasion in 3D culture150. Interestingly, primary tumors in vivo displayed increased LDs, along with decreased CDCP-1 activity, when compared with their corresponding metastatic nodules150.
Some studies also indicate that fat mobilization between stromal and cancer cells is required for metastasis and cancer aggressiveness. Metastasis of ovarian cancer to the omentum was shown to be mediated by local adipocytes that released cytokines and provided FAs to cancer cells, which displayed increased LD formation and β-oxidation, a mechanism dependent on FABP4125. Adipocyte-induced FA translocator CD36 expression confers a more aggressive phenotype in ovarian cancer cells. In addition, CD36 inhibition was sufficient to reduce LD accumulation in cocultured cancer cells and limit tumor growth and invasion both in vitro and in vivo151. Indeed, CD36+ cells were previously highlighted as initiators of metastasis in mouse oral squamous cell carcinomas, and CD36 expression correlated drastically with poor prognosis in lung squamous cell cancer, bladder cancer, or luminal A breast cancer152. Moreover, ATGL accumulation and increased activity were observed predominately in aggressive breast cancer cell lines, and its expression increased in cells with direct contact with adipocytes in primary human breast cancer samples153. These findings add a new layer of complexity to the implications of the TME in cancer aggressiveness.
Lipid droplets and cancer stem cells
Cancer stem cells (CSCs) comprise a small subpopulation of malignant cells that can propagate clones indefinitely as a self-renewal feature and also maintain the tumor by generating heterogeneous cancer cells that compose the bulk of the tumor mass154,155. Consequently, a single CSC holds an inherent potential for cancer initiation155,156,157 and is directly involved in cancer therapy resistance158 and efficiency155. The identification and isolation of CSCs from different cancer types are still matters of discussion155,159,160,161, and currently, the idea that stemness is a flexible, reversible trait of some cancer cells is being upheld160.
Current data show that CSCs have higher LD contents than differentiated tumor cells162,163,164. In a colorectal cancer model, cells with a high LD content showed CSC tumorigenic features in vitro and in vivo. In addition, there was a positive correlation between a high LD content, CD133 expression, and Wnt/β-catenin upregulation162. In the ovarian cancer cell population, the activity of stearoyl-CoA desaturase 1 (SCD1), an enzyme involved in monounsaturated FA synthesis, was strongly associated with LD levels and cancer stemness163. The ovarian CSC population had higher levels of LDs and unsaturated FAs. SCD1 blockade decreased LDs and impaired cancer stemness by inactivating the NF-кB pathway163. In addition, some reported that CSCs accumulate LDs to use as lipid reserves for energy supply164,165. Singh et al.165 showed that blocking lipolysis by targeting vesicle-mediated COPI complex, which transports lipases to the LD surface, starves CSCs to death. In addition, a CSC glioblastoma population showed a strong dependence on oxidative metabolism, FA uptake and high LD content, which is preferentially metabolized under glucose-deprived conditions164,166,167. In summary, the role of LDs in CSCs was associated with both energy demands and activation of cancer stemness pathways, such as Wnt/β-catenin and NF-κB signaling162,163,164,165. These data demonstrate that LDs are important for CSC maintenance, but further studies are needed to clarify their role and possible application as CSC-targeted therapy.
Lipid droplets as a cancer biomarker
From the first observations in the late 1960s6,7, the indication of the increased LD numbers in cancer has raised the possibility of using the detection of LDs as biomarkers for diagnosis and prognosis (Table 1). Most studies correlate PAT proteins expression, mostly perilipin-2, with clinical–pathological features, since RNA from tumor samples or paraffin-embedded tissues is a readily available resource in biomarker studies. Overexpression of PAT proteins has been correlated with the differentiation between malignant and benign tissues168,169,170,171,172, clinical staging171,172, invasion171,172,173,174, and survival in several tumors172,174,175,176.
Recent advances in lipidomic detection, particularly in hyperspectral-stimulating Raman scattering microscopy, allowed the evaluation of the individual LD composition in a single cell177. This raised the possibility that LD lipid composition could have prognostic value in cancer. Indeed, an aberrant accumulation of CEs in LDs was demonstrated in high-grade and metastatic prostate cancers but not in benign lesions or normal tissues17,21,50. Previous data that evaluated the total lipid composition of tumors also showed that CE accumulation was able to differentiate normal from tumor tissue in prostate cancer, leukemia cells, and clear renal cell carcinoma48,49,178.
The potential of LDs as a biomarker also came directly from clinical practice in endoscopy for cancer diagnosis. Some works have evaluated whether microvascular patterns on magnifying endoscopy could be used to diagnose benign and malignant lesions, since tumor vessels are structurally and functionally abnormal179,180. The presence of a white opaque substance (WOS) that prevented the identification of the microvasculature pattern has been reported181 to discriminate benign and malignant lesions. Later, it was shown that the WOS was strongly associated with the presence of LDs, as evidenced by perilipin-2 immunohistochemical and/or Oil red O staining, in gastric, esophageal, and colorectal tumors182,183,184,185,186,187,188,189.
Concluding remarks
Although in the past, the presence of LDs was solely implicated in storage and lipid trafficking, it is currently recognized that these organelles may partake in several cellular functions through a variety of mechanisms. Still, to a great extent, these mechanisms have not yet been fully elucidated. Throughout this review, we discussed features that involve LDs in cancer establishment, pointing out recent evidence that associates these organelles with some of the currently accepted hallmarks of cancer. This draws attention to a potential role of LDs during cancer development, wherein the intricate regulation of LDs could be targeted for drug development or their increased biogenesis inspected as a potential biomarker for diseases. Nevertheless, there are still open questions of fundamental importance to determine a causal relationship between these organelles and tumorigenesis. First, how important is LD biogenesis or lipid metabolism during the distinct phases of tumor development? Second, why is LD accumulation or PAT protein expression in cancer an event that seems to be cell and tissue specific? As discussed above, LDs may have a role during either the initial tumor promotion, by converging mitogenic signaling pathways, partaking in cancer cell metabolism, and providing proinflammatory signaling molecules for TME establishment, or during more advanced cancerous stages, protecting cancer cells from hypoxia and or boosting cells for an aggressive, metastatic phenotype. These points are currently under intense investigation, and therefore, LDs might be suitable candidates for future anticancer therapies.
References
Walther, T. C. & Farese, R. V. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. https://doi.org/10.1146/annurev-biochem-061009-102430 (2012).
Tauchi-Sato, K., Ozeki, S., Houjou, T., Taguchi, R. & Fujimoto, T. The surface of lipid droplets is a phospholipid monolayer with a unique fatty acid composition. J. Biol. Chem. 277, 44507–44512 (2002).
Bozza, P. T., Magalhaes, K. G. & Weller, P. F. Leukocyte lipid bodies—biogenesis and functions in inflammation. Biochim. Biophys. Acta 1791, 540–551 (2009).
Murphy, D. J. The dynamic roles of intracellular lipid droplets: from archaea to mammals. Protoplasma 249, 541–585 (2012).
Jackson, C. L. Lipid droplet biogenesis. Curr. Opin. Cell Biol. https://doi.org/10.1016/j.ceb.2019.03.018 (2019).
Aboumrad, M. H., Horn, R. C. & Fine, G. Lipid-secreting mammary carcinoma. Report of a case associated with paget’s disease of the nipple. Cancer 16, 521–525 (1963).
Wright, D. H. Lipid content of malignant lymphomas. J. Clin. Pathol. https://doi.org/10.1136/jcp.21.5.643 (1968).
Accioly, M. T. et al. Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res 68, 1732–1740 (2008).
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell. https://doi.org/10.1016/S0092-8674(00)81683-9 (2000).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Röhrig, F. & Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer https://doi.org/10.1038/nrc.2016.89 (2016).
Huang, W.-C., Li, X., Liu, J., Lin, J. & Chung, L. W. K. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Mol. Cancer Res. https://doi.org/10.1158/1541-7786.mcr-11-0206 (2012).
Yu, M. et al. Expression of CIDE proteins in clear cell renal cell carcinoma and their prognostic significance. Mol. Cell Biochem. 378, 145–151 (2013).
Audet-Walsh, É. et al. SREBF1 activity is regulated by an AR/mTOR nuclear axis in prostate cancer. Mol. Cancer Res. https://doi.org/10.1158/1541-7786.mcr-17-0410 (2018).
Chakraborty, P. K. et al. Role of cystathionine beta synthase in lipid metabolism in ovarian cancer. Oncotarget https://doi.org/10.18632/oncotarget.5424 (2015).
Morrison, S. F., Nakamura, K. & Madden, C. J. Central control of thermogenesis in mammals. Exp. Physiol. 93, 773–797 (2008).
Li, J. et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene https://doi.org/10.1038/onc.2016.168 (2016).
Valli, A. et al. Hypoxia induces a lipogenic cancer cell phenotype via HIF1α-dependent and -independent pathways. Oncotarget 6, 1920–1941 (2015).
Gang, X. et al. P300 acetyltransferase regulates fatty acid synthase expression, lipid metabolism and prostate cancer growth. Oncotarget https://doi.org/10.18632/oncotarget.7715 (2016).
Guri, Y. et al. mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell 32, 807–823.e12 (2017).
Yue, S. et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 19, 393–406 (2014).
O’Malley, J. et al. Lipid quantification by Raman microspectroscopy as a potential biomarker in prostate cancer. Cancer Lett. https://doi.org/10.1016/j.canlet.2017.03.025 (2017).
Shimano, H. & Sato, R. SREBP-regulated lipid metabolism: convergent physiology-divergent pathophysiology. Nat. Rev. Endocrinol. https://doi.org/10.1038/nrendo.2017.91 (2017).
Kim, J. & Guan, K. L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. https://doi.org/10.1038/s41556-018-0205-1 (2019).
Li, J. et al. CD147 reprograms fatty acid metabolism in hepatocellular carcinoma cells through Akt/mTOR/SREBP1c and P38/PPARα pathways. J. Hepatol. https://doi.org/10.1016/j.jhep.2015.07.039 (2015).
Lee, G. et al. Post-transcriptional Regulation of De Novo Lipogenesis by mTORC1-S6K1-SRPK2 Signaling. Cell https://doi.org/10.1016/j.cell.2017.10.037 (2017).
Fazolini, N. P. et al. Leptin activation of mTOR pathway in intestinal epithelial cell triggers lipid droplet formation, cytokine production and increased cell proliferation. Cell Cycle 14, 2667–2676 (2015).
Ackerman, D. et al. Triglycerides promote lipid homeostasis during hypoxic stress by balancing fatty acid saturation. Cell Rep. https://doi.org/10.1016/j.celrep.2018.08.015 (2018).
Nardi, F. et al. DGAT1 inhibitor suppresses prostate tumor growth and migration by regulating intracellular lipids and non-centrosomal MTOC protein GM130. Sci. Rep. https://doi.org/10.1038/s41598-019-39537-z (2019).
Lu, X., Yang, X. & Liu, J. Differential control of ATGL-mediated lipid droplet degradation by CGI-58 and G0S2. Cell Cycle https://doi.org/10.4161/cc.9.14.12181 (2010).
Al-Zoughbi, W. et al. Loss of adipose triglyceride lipase is associated with human cancer and induces mouse pulmonary neoplasia. Oncotarget 7, 33832–33840 (2016).
Gindlhuber, J. et al. Deletion of adipose triglyceride lipase links triacylglycerol accumulation to a more-aggressive phenotype in A549 lung carcinoma cells. J. Proteome Res. 17, 1415–1425 (2018).
Ou, J. et al. Loss of Abhd5 promotes colorectal tumor development and progression by inducing aerobic glycolysis and epithelial-mesenchymal transition. Cell Rep. https://doi.org/10.1016/j.celrep.2014.11.016 (2014).
Chen, G. et al. Loss of ABHD5 promotes the aggressiveness of prostate cancer cells. Sci. Rep. 7, 1–10 (2017).
Zagani, R., El-Assaad, W., Gamache, I. & Teodoro, J. G. Inhibition of adipose triglyceride lipase (ATGL) by the putative tumor suppressor G0S2 or a small molecule inhibitor attenuates the growth of cancer cells. Oncotarget https://doi.org/10.18632/oncotarget.5061 (2015).
Nomura, D. K. et al. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 140, 49–61 (2010).
Hu, W. R. et al. Monoacylglycerol lipase promotes metastases in nasopharyngeal carcinoma. Int. J. Clin. Exp. Pathol. 7, 3704–3713 (2014).
Pagano, E. et al. Pharmacological inhibition of MAGL attenuates experimental colon carcinogenesis. Pharmacol. Res. https://doi.org/10.1016/j.phrs.2017.02.002 (2017).
Zhang, J. et al. Monoacylglycerol lipase: a novel potential therapeutic target and prognostic indicator for hepatocellular carcinoma. Sci. Rep. https://doi.org/10.1038/srep35784 (2016).
Ye, L. et al. Monoacylglycerol lipase (MAGL) knockdown inhibits tumor cells growth in colorectal cancer. Cancer Lett. https://doi.org/10.1016/j.canlet.2011.03.007 (2011).
Nomura, D. K. et al. Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chem. Biol. https://doi.org/10.1016/j.chembiol.2011.05.009 (2011).
Tisdale, M. J. Mechanisms of cancer cachexia. Physiol. Rev. 89, 381–410 (2009).
Das, S. K. et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science 333, 233–238 (2011).
Chang, T.-Y., Li, B.-L., Chang, C. C. Y. & Urano, Y. Acyl-coenzyme A: cholesterol acyltransferases. Am. J. Physiol. Metab. https://doi.org/10.1152/ajpendo.90926.2008 (2009).
Chang, T.-Y., Chang, C. C. Y., Ohgami, N. & Yamauchi, Y. Cholesterol Sensing, Trafficking, and Esterification. Annu. Rev. Cell Dev. Biol. https://doi.org/10.1146/annurev.cellbio.22.010305.104656 (2006).
Geng, F. et al. Inhibition of SOAT1 suppresses glioblastoma growth via blocking SREBP-1-mediated lipogenesis. Clin. Cancer Res. 22, 5337–5348 (2016).
de Gonzalo-Calvo, D. et al. Intratumor cholesteryl ester accumulation is associated with human breast cancer proliferation and aggressive potential: a molecular and clinicopathological study. BMC Cancer 15, 460 (2015).
Li, J. et al. Integration of lipidomics and transcriptomics unravels aberrant lipid metabolism and defines cholesteryl oleate as potential biomarker of prostate cancer. Sci. Rep. https://doi.org/10.1038/srep20984 (2016).
Saito, K. et al. Lipidomic signatures and associated transcriptomic profiles of clear cell renal cell carcinoma. Sci. Rep. https://doi.org/10.1038/srep28932 (2016).
Lee, H. J. et al. Cholesterol esterification inhibition suppresses prostate cancer metastasis by impairing the Wnt/β-catenin pathway. Mol. Cancer Res. https://doi.org/10.1158/1541-7786.mcr-17-0665 (2018).
Wang, C. W. Lipid droplets, lipophagy, and beyond. Biochim. Biophys. Acta 1861, 793–805 (2016).
Petan, T., Jarc, E. & Jusović, M. Lipid droplets in cancer: guardians of fat in a stressful world. Molecules 23, 11–15 (2018).
Singh, R. et al. Autophagy regulates lipid metabolism. Nature 458, 1131–1135 (2009).
Mayatepek, E., Seedorf, U., Wiebusch, H., Lenhartz, H. & Assmann, G. Fatal genetic defect causing Wolman disease. J. Inherit. Metab. Dis. 22, 93–94 (1999).
Du, H. et al. Lysosomal acid lipase-deficient mice: depletion of white and brown fat, severe hepatosplenomegaly, and shortened life span. J. Lipid Res. 42, 489–500 (2001).
Chiang, H. L., Terlecky, S. R., Plant, C. P. & Dice, J. F. A role for a 70-kilodaton heat shock protein in lysosomal degradation of intracellular proteins. Science 246, 382–385 (1989).
Cuervo, A. M. & Dice, J. F. A receptor for the selective uptake and degradation of proteins by lysosomes. Science 273, 501–503 (1996).
Kaushik, S. & Cuervo, A. M. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol. 17, 759–770 (2015).
Kaushik, S. & Cuervo, A. M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 19, 365–381 (2018).
Kounakis, K., Chaniotakis, M., Markaki, M. & Tavernarakis, N. Emerging roles of lipophagy in health and disease. Front. Cell Dev. Biol. 7, 1–8 (2019).
Lu, G. D. et al. CCAAT/enhancer binding protein α predicts poorer prognosis and prevents energy starvation-induced cell death in hepatocellular carcinoma. Hepatology 61, 965–978 (2015).
Kaini, R. R., Sillerud, L. O., Zhaorigetu, S. & Hu, C. A. A. Autophagy regulates lipolysis and cell survival through lipid droplet degradation in androgen-sensitive prostate cancer cells. Prostate 72, 1412–1422 (2012).
Assumpção, J. A. F., Magalhães, K. G. & Corrêa, J. R. The role of pparγ and autophagy in ros production, lipid droplets biogenesis and its involvement with colorectal cancer cells modulation. Cancer Cell Int. 17, 1–12 (2017).
Mukhopadhyay, S. et al. ATG14 facilitated lipophagy in cancer cells induce ER stress mediated mitoptosis through a ROS dependent pathway. Free Radic. Biol. Med. 104, 199–213 (2017).
Bozza, P. T., Bakker-Abreu, I., Navarro-Xavier, R. A. & Bandeira-Melo, C. Lipid body function in eicosanoid synthesis: an update. Prostaglandins Leukot. Ess. Fat. Acids 85, 205–213 (2011).
Wang, D. & Dubois, R. N. Eicosanoids and cancer. Nat. Rev. Cancer https://doi.org/10.1038/nrc2809 (2010).
Silva, A. R. et al. Lipid bodies in oxidized LDL-induced foam cells are leukotriene-synthesizing organelles: a MCP-1/CCL2 regulated phenomenon. Biochim. Biophys. Acta https://doi.org/10.1016/j.bbalip.2009.06.004 (2009).
Bozza, P. T. et al. Eosinophil lipid bodies: specific, inducible intracellular sites for enhanced eicosanoid formation. J. Exp. Med. 186, 909–920 (1997).
Yu, W. et al. Co-compartmentalization of MAP kinases and cytosolic phospholipase A2 at cytoplasmic arachidonate-rich lipid bodies. Am. J. Pathol. 152, 759–769 (1998).
Dvorak, A. M. et al. Ultrastructural immunogold localization of prostaglandin endoperoxide synthase (cyclooxygenase) to non-membrane-bound cytoplasmic lipid bodies in human lung mast cells, alveolar macrophages, type II pneumocytes, and neutrophils. J. Histochem. Cytochem. 40, 759–769 (1992).
Dvorak, A. M., Weller, P. F., Harvey, V. S., Morgan, E. S. & Dvorak, H. F. Ultrastructural localization of prostaglandin endoperoxide synthase (Cyclooxygenase) to isolated, purified fractions of guinea pig peritoneal macrophage and line 10 hepatocarcinoma cell lipid bodies. Int. Arch. Allergy Immunol. 101, 136–142, https://doi.org/10.1159/000236511 (1993).
Bandeira-Melo, C., Paiva, L. A., Amorim, N. R. T., Weller, P. F. & Bozza, P. T. Eicosacell: an imaging-based assay to identify spatiotemporal eicosanoid synthesis. Methods Mol. Biol. https://doi.org/10.1007/978-1-4939-6759-9_6 (2017).
Penrose, H. et al. Epidermal growth factor receptor mediated proliferation depends on increased lipid droplet density regulated via a negative regulatory loop with FOXO3/Sirtuin6. Biochem Biophys. Res. Commun. 469, 370–376 (2016).
D’Avila, H. et al. Mycobacterium bovis bacillus Calmette-Guerin induces TLR2-mediated formation of lipid bodies: intracellular domains for eicosanoid synthesis in vivo. J. Immunol. 176, 3087–3097 (2006).
Plotkowski, M. C. et al. Lipid body mobilization in the ExoU-induced release of inflammatory mediators by airway epithelial cells. Micro. Pathog. 45, 30–37 (2008).
Pacheco, P. et al. Monocyte chemoattractant protein-1/CC chemokine ligand 2 controls microtubule-driven biogenesis and leukotriene B4-synthesizing function of macrophage lipid bodies elicited by innate immune response. J. Immunol. 179, 8500–8508 (2007).
Kim, S. H. et al. The COX2 effector microsomal PGE2 synthase 1 is a regulator of immunosuppression in cutaneous melanoma. Clin. Cancer Res. https://doi.org/10.1158/1078-0432.CCR-18-1163 (2019).
Sanin, D. E. et al. Mitochondrial membrane potential regulates nuclear gene expression in macrophages exposed to prostaglandin E2. Immunity https://doi.org/10.1016/j.immuni.2018.10.011 (2018).
Veglia, F. et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature https://doi.org/10.1038/s41586-019-1118-2 (2019).
Gorchs, L. et al. Human pancreatic carcinoma-associated fibroblasts promote expression of co-inhibitory markers on CD4+ and CD8+ T-cells. Front. Immunol. https://doi.org/10.3389/fimmu.2019.00847 (2019).
Böttcher, J. P. et al. NK Cells Stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell https://doi.org/10.1016/j.cell.2018.01.004 (2018).
Binnewies, M. et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. https://doi.org/10.1038/s41591-018-0014-x (2018).
Hirata, E. & Sahai, E. Tumor microenvironment and differential responses to therapy. Cold Spring Harb. Perspect. Med. https://doi.org/10.1101/cshperspect.a026781 (2017).
Zhang, Y. et al. Fatty acid-binding protein E-FABP restricts tumor growth by promoting IFN-responses in tumor-associated macrophages. Cancer Res. 74, 2986–2998 (2014).
den Brok, M. H. et al. Saponin-based adjuvants induce cross-presentation in dendritic cells by intracellular lipid body formation. Nat. Commun. 7, 13324 (2016).
Al-Khami, A. A. et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology 6, e1344804 (2017).
Niu, Z. et al. Caspase-1 cleaves PPARγ for potentiating the pro-tumor action of TAMs. Nat. Commun. 8, 766 (2017).
Wu, H. et al. Oleate but not stearate induces the regulatory phenotype of myeloid suppressor cells. Sci. Rep. 7, 7498 (2017).
Veglia, F. et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat. Commun. 8, 2122 (2017).
Goswami, S. & Sharma-Walia, N. Crosstalk between osteoprotegerin (OPG), fatty acid synthase (FASN) and, cycloxygenase-2 (COX-2) in breast cancer: implications in carcinogenesis. Oncotarget https://doi.org/10.18632/oncotarget.9835 (2016).
Mantovani, A., Marchesi, F., Malesci, A., Laghi, L. & Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 14, 399–416 (2017).
Wu, H. et al. Lipid droplet‐dependent fatty acid metabolism controls the immune suppressive phenotype of tumor‐associated macrophages. EMBO Mol. Med. https://doi.org/10.15252/emmm.201910698 (2019).
Veglia, F., Perego, M. & Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 19, 108–119 (2018).
Hossain, F. et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol. Res. 3, 1236–1247 (2015).
Condamine, T. et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci. Immunol. 1, aaf8943–aaf8943 (2016).
Ohl, K. & Tenbrock, K. Reactive oxygen species as regulators of MDSC-mediated immune suppression. Front. Immunol. 9, 2499 (2018).
Goc, J. et al. Dendritic cells in tumor-associated tertiary lymphoid structures signal a Th1 cytotoxic immune contexture and license the positive prognostic value of infiltrating CD8+ T cells. Cancer Res. 74, 705–715 (2014).
Shi, Y. et al. Suppression of vascular endothelial growth factor abrogates the immunosuppressive capability of murine gastric cancer cells and elicits antitumor immunity. FEBS J. 281, 3882–3893 (2014).
McDonnell, A. M. et al. Tumor-infiltrating dendritic cells exhibit defective cross-presentation of tumor antigens, but is reversed by chemotherapy. Eur. J. Immunol. 45, 49–59 (2015).
Truxova, I. et al. Mature dendritic cells correlate with favorable immune infiltrate and improved prognosis in ovarian carcinoma patients. J. Immunother. Cancer 6, 139 (2018).
Bougnères, L. et al. A role for lipid bodies in the cross-presentation of phagocytosed antigens by MHC class I in dendritic cells. Immunity https://doi.org/10.1016/j.immuni.2009.06.022 (2009).
Herber, D. L. et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 16, 880–886 (2010).
Ramakrishnan, R. et al. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J. Immunol. 192, 2920–2931 (2014).
Jiang, L., Fang, X., Wang, H., Li, D. & Wang, X. Ovarian cancer-intrinsic fatty acid synthase prevents anti-tumor immunity by disrupting tumor-infiltrating dendritic cells. Front. Immunol. 9, 2927 (2018).
Fiaschi, T. & Chiarugi, P. Oxidative stress, tumor microenvironment, and metabolic reprogramming: a diabolic liaison. Int. J. Cell Biol. 2012, 1–8 (2012).
Chen, P. et al. Preoperative serum lipids as prognostic predictors in esophageal squamous cell carcinoma patients with esophagectomy. Oncotarget 8, 41605–41619 (2017).
Zhang, Y. et al. Association of serum lipids and severity of epithelial ovarian cancer: an observational cohort study of 349 Chinese patients. J. Biomed. Res. 32, 336–342 (2018).
Sood, D. et al. 3D extracellular matrix microenvironment in bioengineered tissue models of primary pediatric and adult brain tumors. Nat. Commun. https://doi.org/10.1038/s41467-019-12420-1 (2019).
Bozza, P. T. & Viola, J. P. Lipid droplets in inflammation and cancer. Prostaglandins Leukot. Essent. Fat. Acids 82, 243–250 (2010).
Cruz, A. L. S. et al. Cell cycle progression regulates biogenesis and cellular localization of lipid droplets. Mol. Cell. Biol. 1–20 https://doi.org/10.1128/MCB.00374-18 (2019).
Kurat, C. F. et al. Cdk1/Cdc28-dependent activation of the major triacylglycerol lipase Tgl4 in yeast links lipolysis to cell-cycle progression. Mol. Cell https://doi.org/10.1016/j.molcel.2008.12.019 (2009).
Patel, D. et al. A late G1 lipid checkpoint that is dysregulated in clear cell renal carcinoma cells. J. Biol. Chem. 292, 936–944 (2017).
Qi, W. et al. Tumor suppressor FOXO3 mediates signals from the EGF receptor to regulate proliferation of colonic cells. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G264–G272 (2010).
Qi, W. et al. FOXO3 growth inhibition of colonic cells is dependent on intraepithelial lipid droplet density. J. Biol. Chem. 288, 16274–16281 (2013).
Yu, W., Cassara, J. & Weller, P. F. Phosphatidylinositide 3-kinase localizes to cytoplasmic lipid bodies in human polymorphonuclear leukocytes and other myeloid-derived cells. Blood 95, 1078–1085 (2000).
Fujimoto, T., Kogo, H., Ishiguro, K., Tauchi, K. & Nomura, R. Caveolin-2 is targeted to lipid droplets, a new ‘membrane domain’ in the cell. J. Cell Biol. 152, 1079–1085 (2001).
Callies, R., Sri-Pathmanathan, R. M., Ferguson, D. Y. & Brindle, K. M. The appearance of neutral lipid signals in the 1H NMR spectra of a myeloma cell line correlates with the induced formation of cytoplasmic lipid droplets. Magn. Reson. Med. 29, 546–550 (1993).
Hakumaki, J. M. & Kauppinen, R. A. 1H NMR visible lipids in the life and death of cells. Trends Biochem. Sci. 25, 357–362 (2000).
Blankenberg, F. G. In vivo detection of apoptosis. J. Nucl. Med. 49(Suppl 2), 81S–95S (2008).
Henique, C. et al. Increased mitochondrial fatty acid oxidation is sufficient to protect skeletal muscle cells from palmitate-induced apoptosis. J. Biol. Chem. 285, 36818–36827 (2010).
Choi, S. E. et al. Stimulation of lipogenesis as well as fatty acid oxidation protects against palmitate-induced INS-1 beta-cell death. Endocrinology 152, 816–827 (2011).
Boren, J. & Brindle, K. M. Apoptosis-induced mitochondrial dysfunction causes cytoplasmic lipid droplet formation. Cell Death Differ. 19, 1561–1570 (2012).
Zirath, H. et al. MYC inhibition induces metabolic changes leading to accumulation of lipid droplets in tumor cells. Proc. Natl Acad. Sci. USA 110, 10258–10263 (2013).
Hakumaki, J. M., Poptani, H., Sandmair, A. M., Yla-Herttuala, S. & Kauppinen, R. A. 1H MRS detects polyunsaturated fatty acid accumulation during gene therapy of glioma: implications for the in vivo detection of apoptosis. Nat. Med. 5, 1323–1327 (1999).
Schlaepfer, I. R. et al. Progestin modulates the lipid profile and sensitivity of breast cancer cells to docetaxel. Mol. Cell Endocrinol. 363, 111–121 (2012).
Rak, S. et al. FTIR spectroscopy reveals lipid droplets in drug resistant laryngeal carcinoma cells through detection of increased ester vibrational bands intensity. Analyst 139, 3407–3415 (2014).
Verbrugge, S. E. et al. Multifactorial resistance to aminopeptidase inhibitor prodrug CHR2863 in myeloid leukemia cells: down-regulation of carboxylesterase 1, drug sequestration in lipid droplets and pro-survival activation ERK/Akt/mTOR. Oncotarget 7, 5240–5257 (2016).
Zhang, I. et al. Pharmacological inhibition of lipid droplet formation enhances the effectiveness of curcumin in glioblastoma. Eur. J. Pharm. Biopharm. 100, 66–76 (2016).
Cotte, A. K. et al. Lysophosphatidylcholine acyltransferase 2-mediated lipid droplet production supports colorectal cancer chemoresistance. Nat. Commun. 9, 322 (2018).
Obeid, M. et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 13, 54–61 (2007).
Hultsch, S. et al. Association of tamoxifen resistance and lipid reprogramming in breast cancer. BMC Cancer 18, 850 (2018).
Lettiero, B., Inasu, M., Kimbung, S. & Borgquist, S. Insensitivity to atorvastatin is associated with increased accumulation of intracellular lipid droplets and fatty acid metabolism in breast cancer cells. Sci. Rep. 8, 1–10 (2018).
Montopoli, M. et al. “Metabolic Reprogramming” in ovarian cancer cells resistant to cisplatin. Curr. Cancer Drug Targets 11, 226–235 (2011).
Triantafyllou, E.-A., Georgatsou, E., Mylonis, I., Simos, G. & Paraskeva, E. Expression of AGPAT2, an enzyme involved in the glycerophospholipid/triacylglycerol biosynthesis pathway, is directly regulated by HIF-1 and promotes survival and etoposide resistance of cancer cells under hypoxia. Biochim. Biophys. Acta 1863, 1142–1152 (2018).
Wenger, R. H. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. Faseb J. 16, 1151–1162 (2002).
Kaelin, W. G. Jr. & Ratcliffe, P. J. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell 30, 393–402 (2008).
Bertout, J. A., Patel, S. A. & Simon, M. C. The impact of O2 availability on human cancer. Nat. Rev. Cancer 8, 967–975 (2008).
Hanahan, D. & Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86, 353–364 (1996).
Huang, D., Li, C. & Zhang, H. Hypoxia and cancer cell metabolism. Acta Biochim. Biophys. Sin. 46, 214–219 (2014).
Furuta, E. et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 68, 1003–1011 (2008).
Krishnan, J. et al. Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 9, 512–524 (2009).
Scarfo, L. M., Weller, P. F. & Farber, H. W. Induction of endothelial cell cytoplasmic lipid bodies during hypoxia. Am. J. Physiol. Heart Circ. Physiol. 280, H294–H301 (2001).
Saarikoski, S. T., Rivera, S. P. & Hankinson, O. Mitogen-inducible gene 6 (MIG-6), adipophilin and tuftelin are inducible by hypoxia. FEBS Lett. 530, 186–190 (2002).
Zoula, S. et al. Pimonidazole binding in C6 rat brain glioma: relation with lipid droplet detection. Br. J. Cancer 88, 1439–1444 (2003).
Qiu, B. et al. HIF2alpha-dependent lipid storage promotes endoplasmic reticulum homeostasis in clear-cell renal cell carcinoma. Cancer Discov. 5, 652–667 (2015).
Shah, T. et al. HIF isoforms have divergent effects on invasion, metastasis, metabolism and formation of lipid droplets. Oncotarget 6, 28104–28119 (2015).
Bensaad, K. et al. Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 9, 349–365 (2014).
Yilmaz, M. & Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 28, 15–33 (2009).
Giudetti, A. M. et al. A specific lipid metabolic profile is associated with the epithelial mesenchymal transition program. Biochim. Biophys. Acta 1864, 344–357 (2019).
Wright, H. J. et al. CDCP1 drives triple-negative breast cancer metastasis through reduction of lipid-droplet abundance and stimulation of fatty acid oxidation. Proc. Natl Acad. Sci. 114, E6556–E6565 (2017).
Ladanyi, A. et al. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 37, 2285–2301 (2018).
Pascual, G. et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 541, 41–45 (2017).
Wang, Y. Y. et al. Mammary adipocytes stimulate breast cancer invasion through metabolic remodeling of tumor cells. JCI Insight 2, e87489 (2017).
Clarke, M. F. et al. Cancer stem cells—perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res. 66, 9339–9344 (2006).
Valent, P. et al. Cancer stem cell definitions and terminology: the devil is in the details. Nat. Rev. Cancer 12, 767–775 (2012).
Lapidot, T. et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367, 645–648 (1994).
Collins, A. T., Berry, P. A., Hyde, C., Stower, M. J. & Maitland, N. J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 65, 10946–10951 (2005).
Carnero, A. et al. The cancer stem-cell signaling network and resistance to therapy. Cancer Treat. Rev. 49, 25–36 (2016).
Hill, R. P. Identifying cancer stem cells in solid tumors: case not proven. Cancer Res. 66, 1891–1896 (2006).
Medema, J. P. Cancer stem cells: the challenges ahead. Nat. Cell Biol. 15, 338–344 (2013).
Abbaszadegan, M. R. et al. Isolation, identification, and characterization of cancer stem cells: a review. J. Cell. Physiol. 232, 2008–2018 (2017).
Tirinato, L. et al. Lipid droplets: a new player in colorectal cancer stem cells unveiled by spectroscopic imaging. Stem Cells https://doi.org/10.1002/stem.1837 (2015).
Li, J. et al. Lipid desaturation is a metabolic marker and therapeutic target of ovarian cancer stem cells article lipid desaturation is a metabolic marker and therapeutic target of ovarian cancer stem cells. Stem Cell 20, 1–12 (2017).
Hoang‐Minh, L. B. et al. Infiltrative and drug‐resistant slow‐cycling cells support metabolic heterogeneity in glioblastoma. EMBO J. https://doi.org/10.15252/embj.201798772 (2018).
Singh, S. R. et al. The lipolysis pathway sustains normal and transformed stem cells in adult Drosophila. Nature https://doi.org/10.1038/nature19788 (2016).
Deleyrolle, L. P. et al. Evidence for label-retaining tumour-initiating cells in human glioblastoma. Brain https://doi.org/10.1093/brain/awr081 (2011).
Deleyrolle, L. P., Rohaus, M. R., Fortin, J. M., Reynolds, B. A. & Azari, H. Identification and isolation of slow-dividing cells in human glioblastoma using carboxy fluorescein succinimidyl ester (CFSE). J. Vis. Exp. https://doi.org/10.3791/3918 (2012).
Zhang, X. D. et al. Identification of adipophilin as a potential diagnostic tumor marker for lung adenocarcinoma. Int J. Clin. Exp. Med. 7, 1190–1196 (2014).
Gushima, R. et al. Expression of adipophilin in gastric epithelial neoplasia is associated with intestinal differentiation and discriminates between adenoma and adenocarcinoma. Virchows Arch. https://doi.org/10.1007/s00428-015-1870-0 (2016).
Matsubara, J. et al. Identification of adipophilin as a potential plasma biomarker for colorectal cancer using label-free quantitative mass spectrometry and protein microarray. Cancer Epidemiol. Biomarkers Prev. https://doi.org/10.1158/1055-9965.EPI-11-0400 (2011).
Fujimoto, M. et al. Adipophilin expression in cutaneous malignant melanoma. J. Cutan. Pathol. https://doi.org/10.1111/cup.12868 (2017).
Wang, K. et al. PLIN3 is up-regulated and correlates with poor prognosis in clear cell renal cell carcinoma. Urol. Oncol. Semin. Orig. Investig. https://doi.org/10.1016/j.urolonc.2018.04.006 (2018).
Szigeti, A. et al. Preliminary study of TIP47 as a possible new biomarker of cervical dysplasia and invasive carcinoma. Anticancer Res. 29, 717–724 (2009).
Fujimoto, M. et al. Adipophilin expression in lung adenocarcinoma is associated with apocrine-like features and poor clinical prognosis: an immunohistochemical study of 328 cases. Histopathology 70, 232–241 (2017).
Yao, M. et al. Gene expression analysis of renal carcinoma: adipose differentiation-related protein as a potential diagnostic and prognostic biomarker for clear-cell renal carcinoma. J. Pathol. 205, 377–387 (2005).
Yao, M. et al. Expression of adipose differentiation-related protein: a predictor of cancer-specific survival in clear cell renal carcinoma. Clin. Cancer Res 13, 152–160 (2007).
Zhang, C., Li, J., Lan, L. & Cheng, J. X. Quantification of lipid metabolism in living cells through the dynamics of lipid droplets measured by stimulated raman scattering imaging. Anal. Chem. https://doi.org/10.1021/acs.analchem.6b04699 (2017).
Mulas, M. F. et al. Cholesterol esters as growth regulators of lymphocytic leukaemia cells. Cell Prolif. https://doi.org/10.1111/j.1365-2184.2011.00758.x (2011).
Gono, K. Narrow band imaging: technology basis and research and development history. Clin. Endosc. https://doi.org/10.5946/ce.2015.48.6.476 (2015).
East, J. E. et al. Advanced endoscopic imaging: European Society of Gastrointestinal Endoscopy (ESGE) Technology Review. Endoscopy https://doi.org/10.1055/s-0042-118087 (2016).
Yao, K. et al. White opaque substance within superficial elevated gastric neoplasia as visualized by magnification endoscopy with narrow-band imaging: a new optical sign for differentiating between adenoma and carcinoma. Gastrointest. Endosc. https://doi.org/10.1016/j.gie.2008.04.011 (2008).
Yao, K. et al. Nature of white opaque substance in gastric epithelial neoplasia as visualized by magnifying endoscopy with narrow-band imaging. Dig. Endosc. https://doi.org/10.1111/j.1443-1661.2012.01314.x (2012).
Ueyama, H. et al. A white opaque substance-positive gastric hyperplastic polyp with dysplasia. World J. Gastroenterol. https://doi.org/10.3748/wjg.v19.i26.4262 (2013).
Ueo, T. et al. Histologic differentiation and mucin phenotype in white opaque substance-positive gastric neoplasias. Endosc. Int. Open https://doi.org/10.1055/s-0034-1393177 (2015).
Yoshii, S. et al. Esophageal adenocarcinoma with white opaque substance observed by magnifying endoscopy with narrow band imaging. Dig. Endosc. 27, 392–396 (2015).
Hisabe, T. et al. Novel endoscopic findings as visualized by magnifying endoscopy with narrow-band imaging: white opaque substance is present in colorectal hyperplastic polyps. Digestion https://doi.org/10.1159/000441841 (2016).
Imamura, K. et al. The nature of the white opaque substance within colorectal neoplastic epithelium as visualized by magnifying endoscopy with narrow-band imaging. Endosc. Int. Open https://doi.org/10.1055/s-0042-116487 (2016).
Kawasaki, K. et al. Association between white opaque substance under magnifying colonoscopy and lipid droplets in colorectal epithelial neoplasms. World J. Gastroenterol. https://doi.org/10.3748/wjg.v23.i47.8367 (2017).
Ueo, T. et al. White opaque substance represents an intracytoplasmic accumulation of lipid droplets: Immunohistochemical and immunoelectron microscopic investigation of 26 cases. Dig. Endosc. https://doi.org/10.1111/j.1443-1661.2012.01364.x (2013).
Opstad, K. S., Bell, B. A., Griffiths, J. R. & Howe, F. A. An investigation of human brain tumour lipids by high-resolution magic angle spinning 1H MRS and histological analysis. NMR Biomed. 21, 677–685 (2008).
Lu, F. K. et al. Label-free neurosurgical pathology with stimulated Raman imaging. Cancer Res. 76, 3451–3462 (2016).
Straub, B. K. et al. Lipid droplet-associated PAT-proteins show frequent and differential expression in neoplastic steatogenesis. Mod. Pathol. 23, 480–492 (2010).
Moritani, S. et al. Intracytoplasmic lipid accumulation in apocrine carcinoma of the breast evaluated with adipophilin immunoreactivity: a possible link between apocrine carcinoma and lipid-rich carcinoma. Am. J. Surg. Pathol. 35, 861–867 (2011).
Piris, A. et al. Cutaneous and mammary apocrine carcinomas have different immunoprofiles. Hum. Pathol. 45, 320–326 (2014).
Guan, B. et al. Lipid-rich carcinoma of the breast clinicopathologic analysis of 17 cases. Ann. Diagn. Pathol. 15, 225–232 (2011).
Mellick, A. S., Day, C. J., Weinstein, S. R., Griffiths, L. R. & Morrison, N. A. Differential gene expression in breast cancer cell lines and stroma-tumor differences in microdissected breast cancer biopsies revealed by display array analysis. Int J. Cancer 100, 172–180 (2002).
Than, N. G. et al. Lipid droplet and milk lipid globule membrane associated placental protein 17b (PP17b) is involved in apoptotic and differentiation processes of human epithelial cervical carcinoma cells. Eur. J. Biochem. 270, 1176–1188 (2003).
Hisabe, T. et al. Novel endoscopic findings as visualized by magnifying endoscopy with narrow-band imaging: white opaque substance is present in colorectal hyperplastic polyps. Digestion 93, 127–131 (2015).
Mariano, F. V. et al. Mammary analogue secretory carcinoma of salivary glands is a lipid-rich tumour, and adipophilin can be valuable in its identification. Histopathology 63, 558–567 (2013).
Oshiro, H. et al. Primary sebaceous carcinoma of the tongue. Med. Mol. Morphol. 43, 246–252 (2010).
Rae, F. K., Stephenson, S. A., Nicol, D. L. & Clements, J. A. Novel association of a diverse range of genes with renal cell carcinoma as identified by differential display. Int J. Cancer 88, 726–732 (2000).
Mentrikoski, M. J., Wendroth, S. M. & Wick, M. R. Immunohistochemical distinction of renal cell carcinoma from other carcinomas with clear-cell histomorphology: utility of CD10 and CA-125 in addition to PAX-2, PAX-8, RCCma, and adipophilin. Appl Immunohistochem. Mol. Morphol. 22, 635–641 (2014).
Cao, Q. et al. Overexpression of PLIN2 is a prognostic marker and attenuates tumor progression in clear cell renal cell carcinoma. Int. J. Oncol. https://doi.org/10.3892/ijo.2018.4384 (2018).
Kurokawa, Y. et al. PCR-array gene expression profiling of hepatocellular carcinoma. J. Exp. Clin. Cancer Res 23, 135–141 (2004).
Kanno, T. et al. Implications of hyperechoic lesions in small hepatocellular carcinoma. Gastroenterol. Jpn 24, 528–534 (1989).
Yang, S. H., Watanabe, J., Nakashima, O. & Kojiro, M. Clinicopathologic study on clear cell hepatocellular carcinoma. Pathol. Int. 46, 503–509 (1996).
Zen, Y. et al. Hepatocellular carcinoma arising in non-alcoholic steatohepatitis. Pathol. Int. 51, 127–131 (2001).
Arai, K. et al. Adrenal rest tumor of the liver: a case report with immunohistochemical investigation of steroidogenesis. Pathol. Int. 50, 244–248 (2000).
Ambrosio, M. R. et al. The alteration of lipid metabolism in Burkitt lymphoma identifies a novel marker: adipophilin. PLoS One 7, e44315 (2012).
Nieman, K. M. et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 17, 1498–1503 (2011).
Ohkawa, K., Amasaki, H., Terashima, Y., Aizawa, S. & Ishikawa, E. Clear cell carcinoma of the ovary: light and electron microscopic studies. Cancer 40, 3019–3029 (1977).
Abbey-Toby, A. et al. A clear cell malignant gastrinoma of the pancreas with cytoplasmic accumulation of lipid droplets. Virchows Arch. 448, 105–106 (2006).
Boussahmain, C., Mochel, M. C. & Hoang, M. P. Perilipin and adipophilin expression in sebaceous carcinoma and mimics. Hum. Pathol. 44, 1811–1816 (2013).
Ostler, D. A. et al. Adipophilin expression in sebaceous tumors and other cutaneous lesions with clear cell histology: an immunohistochemical study of 117 cases. Mod. Pathol. 23, 567–573 (2010).
Milman, T., Schear, M. J. & Eagle, R. C. Jr Diagnostic utility of adipophilin immunostain in periocular carcinomas. Ophthalmology 121, 964–971 (2014).
Muthusamy, K., Halbert, G. & Roberts, F. Immunohistochemical staining for adipophilin, perilipin and TIP47. J. Clin. Pathol. 59, 1166–1170 (2006).
Tjarks, B. J. et al. Evaluation and comparison of staining patterns of factor XIIIa (AC-1A1), adipophilin and GATA3 in sebaceous neoplasia. J. Cutan. Pathol. 45, 1–7 (2017).
Takada, N. et al. Cytoplasmic lipid accumulation characteristic of the cribriform variant of papillary thyroid carcinoma. Pathobiology 84, 251–257 (2017).
Acknowledgements
We would like to recognize present and past members of the Laboratory of Immunopharmacology for their valuable contributions. We apologize to investigators whose relevant work has not been cited because of space constraints. The authors are indebt with Filipe Pereira Dutra for figures art work. The work of the authors is supported by Fundação de Amparo à Pesquisa do Rio de Janeiro (FAPERJ, Brasil); Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brasil); Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Capes).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by G. Melino
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cruz, A.L.S., Barreto, E.d.A., Fazolini, N.P.B. et al. Lipid droplets: platforms with multiple functions in cancer hallmarks. Cell Death Dis 11, 105 (2020). https://doi.org/10.1038/s41419-020-2297-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-020-2297-3
This article is cited by
-
Cancer radioresistance is characterized by a differential lipid droplet content along the cell cycle
Cell Division (2024)
-
Extracellular lipidosomes containing lipid droplets and mitochondria are released during melanoma cell division
Cell Communication and Signaling (2024)
-
Potential role of lipophagy impairment for anticancer effects of glycolysis-suppressed pancreatic ductal adenocarcinoma cells
Cell Death Discovery (2024)
-
PIM1 drives lipid droplet accumulation to promote proliferation and survival in prostate cancer
Oncogene (2024)
-
Are lipid droplets the picnic basket of brain tumours?
Cell Death Discovery (2024)
