
Abstract
In eukaryotic cells, macromolecular homeostasis requires selective degradation of damaged units by the ubiquitin-proteasome system (UPS) and autophagy. Thus, dysfunctional degradation systems contribute to multiple pathological processes. Ferroptosis is a type of iron-dependent oxidative cell death driven by lipid peroxidation. Various antioxidant systems, especially the system xc−-glutathione-GPX4 axis, play a significant role in preventing lipid peroxidation-mediated ferroptosis. The endosomal sorting complex required for transport-III (ESCRT-III)–dependent membrane fission machinery counteracts ferroptosis by repairing membrane damage. Moreover, cellular degradation systems play a dual role in regulating the ferroptotic response, depending on the cargo they degrade. The key ferroptosis repressors, such as SLC7A11 and GPX4, are degraded by the UPS. In contrast, the overactivation of selective autophagy, including ferritinophagy, lipophagy, clockophagy and chaperone-mediated autophagy, promotes ferroptotic death by degrading ferritin, lipid droplets, circadian proteins, and GPX4, respectively. Autophagy modulators (e.g., BECN1, STING1/TMEM173, CTSB, HMGB1, PEBP1, MTOR, AMPK, and DUSP1) also determine the ferroptotic response in a context-dependent manner. In this review, we provide an updated overview of the signals and mechanisms of the degradation system regulating ferroptosis, opening new horizons for disease treatment strategies.
Similar content being viewed by others

Facts
-
Ferroptosis is a type of regulated cell death driven by iron accumulation and lipid peroxidation.
-
Various oxidative and antioxidant systems converge on cellular degradation machinery to shape the ferroptotic response.
-
The interaction between the ubiquitin-proteasome system and autophagy is complex and can either cause or abolish ferroptosis, depending on the cargo that is degraded.
-
Several types of selective autophagy drive the development of ferroptosis by inducing oxidative damage.
Open questions
-
How can the ubiquitin-proteasome system and autophagy machinery fine-tune the intrinsic or extrinsic ferroptotic pathway to regulate cell survival or cell death?
-
What are the key substrates for E3 ubiquitin ligases or autophagy receptors that regulate ferroptosis?
-
How can we optimally target the degradation pathways for ferroptosis modulation and apply this knowledge to translational medicine?
Introduction
Cell death can be divided into accidental cell death and regulated cell death (RCD) [1]. RCD is further classified in several subtypes that can be distinguished according to their signaling and degradation pathways, as well as the biochemical end morphological characteristics of dead cells [2]. Ferroptosis is a type of non-apoptotic RCD, which is characterized by lipid peroxidation and plasma membrane damage [3, 4]. With the discovery of various regulators and pathways, basic research in ferroptosis is rapidly expanding, thus providing new opportunities to treat various pathological conditions and diseases [5, 6].
In mammalian cells, protein degradation is mainly mediated by two proteolytic systems: the ubiquitin-proteasome system (UPS) and the autophagy-lysosome system. In the UPS, proteins (especially short-lived proteins) are selectively labeled with ubiquitin tags for destruction by the 26S proteasome, which represents the major extra-lysosomal system for protein degradation [7]. In contrast, autophagy not only degrades proteins (especially long-lived proteins), but also destroys other macromolecules (lipids, carbohydrates, RNA, and DNA) [8]. There are three types of autophagy, namely macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA), which ultimately degrade cellular components through lysosomes containing digestive enzymes [9]. Macroautophagy (hereafter referred to as autophagy) is the best studied form of autophagy and plays a crucial role in sustaining cellular homeostasis [10]. Under environmental pressure, the upregulation of autophagy usually promotes survival [11]. However, excessive autophagy may lead to cell death, which then is referred to as autophagy-dependent cell death [12].
In this review, we first introduce the basic process of ferroptosis and then focus on the integrated role of the UPS and autophagy in shaping the ferroptotic response in various pathological processes.
Core mechanisms of ferroptosis
The mechanisms of ferroptosis vary as a function of the stressors and drugs causing the process [5, 13]. Ferroptosis may be considered as a cell death modality driven by the imbalance between oxidative stress and the antioxidant system [14]. Ferroptosis can occur through two major pathways, the extrinsic or transporter-dependent pathway (e.g., via inhibition of system xc−), and the intrinsic or enzyme-regulated pathway (e.g., via direct inhibition of glutathione peroxidase 4 [GPX4], induction of lipid peroxidation, or induction of mitochondrial dysfunction) [3]. The extrinsic and intrinsic pathways are closely linked through various metabolic pathways and subcellular organelles (e.g., mitochondria [15], endoplasmic reticulum [16], Golgi apparatus [17], nucleus [18], lysosomes [19, 20], and peroxisomes [21, 22]), which have been well-described in previous reviews or studies. As it is beyond the scope of this review to cover all mechanisms of ferroptosis, we only summarize the two core redox mechanisms that regulate ferroptosis.
Blocking the SLC7A11-GSH-GPX4 antioxidant axis
System xc− is an amino acid transporter on the cell membrane that imports cystine and exports glutamate, leading to glutathione (GSH) synthesis. As an antioxidant tripeptide, GSH is used as an obligatory cofactor of GPX4, an enzyme that reduces lipid hydroperoxide to a nontoxic lipid alcohol. As a result, pharmacologically inhibiting solute carrier family 7 member 11 (SLC7A11, a key subunit of system xc−) by drugs (e.g., erastin [4], sorafenib [16, 23], or sulfasalazine [16]) leads to GSH depletion and the subsequent inactivation of GPX4, thereby causing lipid peroxidation-mediated ferroptotic cell death. Alternatively, pharmacologically blocking GPX4 (e.g., with RSL3 [24] or ML210 [25]) or inducing GPX4 degradation (e.g., with FIN56 [26] or PdPT [27]) also triggers ferroptosis. The genetic depletion of Slc7a11 or Gpx4 in mouse embryonic fibroblasts leads to ferroptotic cell death [28, 29]. In vivo, the deletion of Slc7a11 or conditional loss of Gpx4 in mice also causes ferroptotic-like damage [30], although other types of RCD (e.g., pyroptosis and necroptosis) may be involved in tissue damage as well [31, 32]. These findings establish the cardinal importance of the SLC7A11-GSH-GPX4 antioxidant axis in ferroptosis.
GPX4-independent pathways, such as the apoptosis-inducing factor mitochondria-associated 2-CoQ system [33, 34], the GTP cyclohydrolase 1-tetrahydrobiopterin system [35, 36] and the endosomal sorting complex required for transport-III (ESCRT-III) membrane repair system [37, 38], also limit ferroptotic damage in some cases. Thus, GPX4-dependent and -independent regulatory pathways may complement each other in an integrated antioxidant system (rather than in a single pathway) that suppress ferroptotic cell death.
Activating iron-dependent lipid peroxidation
The lipid peroxidation of polyunsaturated fatty acids (PUFAs) seems to be the main event leading to ferroptosis [39], although other types of lipids (e.g., monounsaturated fatty acids [MUFAs]) may have a synergistic or antagonistic effect on ferroptosis induced by RSL3 and erastin [40, 41]. The production of PUFA-related metabolites (arachidonic acid-CoA and adrenic acid-CoA) for ferroptosis requires the activation of acyl-CoA synthase long-chain family member 4 (ACSL4) [42,43,44], although ACSL4-independent pathways exist in some cases [45]. These PUFA-related metabolites undergo further oxidation for ferroptosis through either enzymatic (e.g., arachidonate lipoxygenase [ALOX] or cytochrome P450 oxidoreductase [POR])-dependent or non-enzymatic (e.g., Fenton reaction) mechanisms [13].
ALOXs are iron-containing lipid dioxygenases that catalyze the insertion of oxygen into PUFAs, such as arachidonic acid and linoleic acid. Among the six member of the ALOX family in humans, ALOX5 [39, 46], ALOX12 [45], ALOX15 [47], ALOX15B [39], and ALOXE3 [39] promote ferroptosis in a cell type-dependent manner. Therefore, the basal expression of ALOX family members may inform about the sensitivity of cells to ferroptosis. In certain ALOX-deficient or low-expressing cells, the activation of POR is necessary for ALOX-independent ferroptosis made possible by POR donating electrons to cytochrome P450 (CYP) or other redox partners [48].
Excess reactive oxygen species (ROS) from different sources can cause oxidative damage to various structural components of cells, including lipids and DNA. Either mitochondria or superoxide-producing enzymes from the NADPH oxidase (NOX) family generate ROS for the induction of ferroptosis in a context-dependent manner. For example, NOX1 [4, 49], cytochrome B-245 beta chain (CYBB/NOX2) [50], and NOX4 [51] mediate ROS production responsible for lipid peroxidation during the erastin-induced ferroptotic demise of human cancer cells. The function of NOX in ferroptosis is further enhanced by its binding proteins, such as dipeptidyl peptidase 4 (a membrane-bound protein with catalytic activity) [49].
Redox-active iron is involved in the initiation and amplification of free radical-mediated lipid peroxidation reactions in ferroptosis [52]. In addition to iron being a generator of ROS through the Fenton reaction, the importance of iron in ferroptosis is supported by the fact that several heme and nonheme iron-containing enzymes (e.g., ALOX, CYP, and NOX) promote lipid peroxidation during ferroptosis [52]. While iron-containing enzymes are a large family with either anti-death or death-promoting properties, it remains to be clarified how iron selectively activates these enzymes to promote lipid peroxidation during ferroptosis.
UPS in ferroptosis
Ubiquitination, the first step of UPS, consists in a reversible covalent modification introducing an isopeptide bond between the carboxy terminus of ubiquitin and lysine residues on the target protein. The process of ubiquitination is a cascade mechanism mediated by three different classes of enzymes, namely E1 (a ubiquitin-activating enzyme), E2 (a ubiquitin-conjugating enzyme), and E3 (a ubiquitin-protein ligase) (Fig. 1) [53]. Repeating the ubiquitination process leads to the formulation of polyubiquitin chains. All seven lysine residues of ubiquitin (K6, K11, K27, K29, K33, K48, and K63) can be used to extend chains from different types of linkage as well as the linear ubiquitin chain linkage, giving rise to functionally distinct ubiquitination tags. Finally, the ubiquitin-conjugated proteins are degraded in the proteasome, which is a large multi-subunit complex that functions in an ATP-dependent manner. Deubiquitinase (DUB) can remove ubiquitin from the target protein, a process called deubiquitination. The crosstalk between ubiquitination and deubiquitination sets the threshold of cellular protein degradation for the proteasome, and regulates various ferroptosis-related cellular processes (Fig. 2).

Ubiquitination is a highly regulated process catalyzed by three types of enzymes, namely E1 (ubiquitin-activating enzyme), E2 (ubiquitin-conjugating enzyme), and E3 (ubiquitin-protein ligase). Ubiquitinated proteins are degraded in the proteasome. Deubiquitinase (DUB) can remove the ubiquitin (Ub) from substrates, thus preventing protein degradation and recycling of ubiquitin.

Autophagy is mainly regulated by ATG proteins, which can form different protein complexes. The initial induction of autophagy requires the activity of the ULK1/ATG1 complex and subsequent class III PtdIns3K complex. The formation of an autophagosome requires two ubiquitin-like conjugation systems, namely the MAP1LC3/ATG8 and ATG12 systems. At the final step of this process, the autophagosome fuses with a lysosome, which contains the hydrolytic enzymes for the degradation of the engulfed material. ATG autophagy-related, BECN1 beclin 1, MAP1LC3 microtubule-associated protein 1 light chain 3, PE phosphatidylethanolamine, PIK3C3 phosphatidylinositol 3-kinase catalytic subunit type 3, PIK3R4 phosphoinositide 3-kinase regulatory subunit 4, PtdIns3K phosphatidylinositol 3-kinase, RB1CC1/FIP200 RB1 inducible coiled-coil 1, ULK1 Unc-51–like autophagy activating kinase 1.
Regulation of antioxidant defense
OTUB1-SLC7A11
OTU deubiquitinase, ubiquitin aldehyde binding 1 (OTUB1) improves SLC7A11 protein stability by removing ubiquitin moieties that otherwise would target SLC7A11 for destruction [54]. Therefore, OTUB1 inhibition leads to proteasome-dependent degradation of SLC7A11, enhancing the sensitivity of cells to ferroptosis induced by erastin in vitro [54]. OTUB1-mediated SLC7A11 protein stability is further enhanced by CD44 cellular adhesion molecules [54]. The identification of the key E3 ligases responsible for the degradation of SLC7A11 in human cancers may yield additional information on this regulatory system.
DUB-GPX4
Palladium pyrithione is a broad-spectrum DUB inhibitor that induces ferroptosis by promoting GPX4 protein degradation in non-small cell lung cancer cells [27]. PdPT is able to inhibit multiple DUBs, including ubiquitin-specific peptidase (USP) family members (USP14, USP15, USP10, USP7, and USP25), as well as ubiquitin C-terminal hydrolase (UCH) member UCHL5, but fails to inhibit OTU members (OTUB1 and OTUD1) [27]. The proteasome inhibitor bortezomib reverses PdPT-induced GPX4 degradation, indicating that the proteasome is responsible for PdPT-induced GPX4 degradation [27]. Because autophagy also involves the degradation of GPX4 (discussed below), cells may use molecularly distinct approaches to achieve the same outcome on GPX4 degradation during ferroptosis.
KEAP1-NFE2L2
The nuclear factor, erythroid 2-like 2 (NFE2L2/NRF2) system is a transcriptional defense mechanism in ferroptosis [55]. Under quiescent conditions, the transcription factor NFE2L2 is constitutively degraded through the UPS, due to its binding with Kelch-like ECH-associated protein 1 (KEAP1). In response to oxidative stress and electrophilic stress, KEAP1 is inactivated, leading to the stabilization of NFE2L2 and subsequent transcriptional activation of the expression of many cytoprotective genes. Sequestosome 1 (SQSTM1/p62, an autophagy receptor) can interact with the NFE2L2-binding site on KEAP1, and then stabilize NFE2L2 [56]. Upon exposure to ferroptosis activators (erastin and sorafenib), the interaction between SQSTM1 and KEAP1 is increased [57], resulting in the accumulation of NFE2L2 and the enhanced nuclear transcription of NFE2L2 target genes, such as SLC7A11, NAD(P)H quinone dehydrogenase 1 (NQO1), heme oxygenase 1 (HMOX1), and ferritin heavy chain 1 (FTH1) [58]. Consistently, blocking NFE2L2 enhances the anticancer activity of ferroptosis inducers, including sorafenib (a first-line drug for patient with liver cancer) in vitro and in vivo [57, 59]. In contrast, the activation of NFE2L2 may prevent ferroptotic tissue damage. NFE2L2 also represses genes involved in GSH release (e.g., ATP-binding cassette subfamily C member 1 [ABCC1/MRP1]) to promote ferroptosis induced by RSL3, erastin, and ML162 in vitro [60]. All these findings indicate that NFE2L2 has a transcription-dependent function in ferroptosis, although little is known about its transcription-independent function in ferroptosis.
Regulation of oxidative damage by NEDD4-VDAC
The voltage-dependent anion channels (VDAC) are a family of multifunction channel proteins that mediate the transport of metabolites and ions in eukaryotic cells across the outer mitochondrial membrane. In addition to inhibiting the aforementioned plasma membrane transporter SLC7A11, erastin binds directly to VDAC2 for the induction of ferroptosis [61]. As a negative feedback mechanism, erastin induces UPS-dependent degradation of VDAC2 and VDAC3 in melanoma cells [62] and this degradation process is mediated by NEDD4 E3 ubiquitin protein ligase (NEDD4) [62]. Further research is needed to determine whether a specific mitochondrial E3 ligase is involved in the degradation of VDAC in ferroptosis.
Regulation of iron metabolism by NEDD4L-LTF
Lactotransferrin (LTF) is a member of the transferrin family of glycoproteins that specifically binds and transports iron. Similar to transferrin (TF), LTF promotes ferroptosis in pancreatic and ovarian cancer cells through increased iron uptake [63]. In contrast, NEDD4-like E3 ubiquitin protein ligase (NEDD4L)-mediated protein degradation of LTF diminishes iron accumulation and oxidative damage during ferroptotic cancer cell death induced by RSL3 and erastin in vitro [63]. The receptor for LTF-induced ferroptosis is still elusive but may be immune-related (e.g., toll-like receptors and the insulin receptor). This enigma needs to be elucidated in future studies.
Regulation of lipid metabolism by VHL-HIF
Hypoxia-inducible factors (HIFs) are a family of transcription factors activated by hypoxia, including three α subunits (hypoxia-inducible factor 1 subunit alpha [HIF1A/HIF1α], endothelial PAS domain protein 1 (EPAS1/HIF2α) and hypoxia-inducible factor 1 subunit alpha [HIF3A/HIF3α]) and one β subunit (aryl hydrocarbon receptor nuclear translocator [ARNT/HIF1β]). When oxygen is present, HIF1A is hydroxylated by Egl-9 family hypoxia-inducible factor (EGLN) prolyl hydroxylases, which target HIFs for ubiquitination by an E3 ubiquitin ligase complex that contains the von Hippel–Lindau tumor suppressor (VHL) protein [64]. Under hypoxic conditions, the absence of hydroxylation of HIF1A increases the stabilization and subsequent nuclear translocation and function of HIF1A. HIFs may play a dual role in ferroptosis, depending on the tumor type. HIF1A functions as a negative regulator of ferroptosis induced by RSL3 and FIN56 by transcriptional induction of the expression of fatty acid binding protein 3 and fatty acid binding protein 7, thereby promoting fatty acid uptake and storage in human HT1080 cells [65]. However, EPAS1 promotes RSL3-, ML162-, or ML210-induced ferroptosis by selectively enriching polyunsaturated lipids through transactivating the gene encoding hypoxia-inducible lipid droplet-associated in kidney cancer cells in vitro [66, 67]. Moreover, human lung adenocarcinomas confer resistance to hyperoxic tension through oxygen-dependent NFS1 regulation, thereby inhibiting ferroptotic cell death in vitro and in vivo [68]. It is certainly possible that targeting other signals related to hypoxia may be beneficial for ferroptosis-associated therapy.
Autophagy in ferroptosis
Autophagy relies on the formation of a series of membrane structures, such as phagophores, autophagosomes, and autolysosomes, which are regulated by a family of autophagy-related (ATG) proteins (Fig. 3). More than 40 ATG genes have been identified in yeast, and nearly half of them have othologues in mammalian cells [8]. ATG proteins can form several protein complexes (e.g., the Unc-51–like autophagy activating kinase 1 [ULK1] complex and the class III phosphatidylinositol 3-kinase [PtdIns3K] complex) to control the induction and nucleation of phagophores. Two ubiquitin-like (Ubl) conjugation systems, the ATG12-ATG5 conjugation system and the microtubule-associated protein 1 light chain 3 (MAP1LC3/LC3) conjugation systems, are responsible for the elongation and maturation of autophagosomes. Several lysosomal proteins (e.g., lysosomal-associated membrane proteins [LAMPs]) and membrane attachment proteins (e.g., soluble N-ethylmaleimide–sensitive factor attachment protein receptor) contribute to the formation of autolysosomes. Finally, autophagic cargos are degraded by lysosomal hydrolases in the autolysosomes, and the resulting cleavage products can be reused for protein synthesis or energy production. This dynamic ATG protein-mediated autophagy process is fine-tuned through posttranslational modification (especially acetylation and phosphorylation) of proteins [69] as well as lipid metabolism [70,71,72], which is important for multiple cellular processes. Autophagy is further divided into selective autophagy and nonselective autophagy, which plays a dual role in cell survival and cell death as a potential therapeutic modulator in diseases [73]. We discuss the complex role of selective autophagy and several autophagy regulators in the modulation of ferroptosis below using selected examples (Fig. 4).

Lipid peroxidation is the core mechanism of ferroptosis, which is caused mainly by inhibiting the SLC7A11-GSH-GPX4 antioxidant pathway. The ubiquitin-proteasome system is involved in the degradation of SLC7A11, GPX4, LTF, VDAC2/VDAC3, NFE2L2, or HIFs during ferroptosis. DUBs deubiquitinases, EGLNs Egl-9 family hypoxia-inducible factors, GPX4 glutathione peroxidase 4, GSH glutathione, HIFs hypoxia-inducible factors, KEAP1 Kelch-like ECH-associated protein 1, LTF lactotransferrin, NEDD4 NEDD4 E3 ubiquitin protein ligase, NEDD4L NEDD4-like E3 ubiquitin protein ligase, NFE2L2 nuclear factor, erythroid 2-like 2, OTUB1 OTU deubiquitinase, ubiquitin aldehyde binding 1, SLC3A2 solute carrier family 3 member 2, SLC7A11 solute carrier family 7 member 11, Ub ubiquitin, VDAC2 voltage-dependent anion channel 2, VDAC3 voltage-dependent anion channel 3, VHL von Hippel–Lindau tumor suppressor.

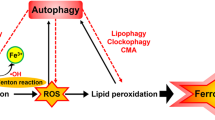
Selective autophagy (e.g., NCOA4-mediated ferritinophagy, RAB7A-related lipophagy, SQSTM1-dependent clockophagy, and HSP90-associated CMA) promotes ferroptotic cell death by inducing iron accumulation or lipid peroxidation. Several autophagy regulators, such as BECN1, MAP1LC3, MTOR, CTSB, STING1, HMGB1, and DUSP1 also regulate ferroptosis. ALOX15 arachidonate 15-lipoxygenase, BECN1 Beclin 1, CMA chaperone-mediated autophagy, CTSB cathepsin B, DUSP1 dual specificity phosphatase 1, FA fatty acid, FTH1 ferritin heavy chain 1, FTL ferritin light chain, GSH glutathione, HMGB1 high mobility group box 1, HSP90 heat shock protein 90, LAMP2A lysosome-associated membrane protein type 2A, LDs lipid droplets, MAP1LC3II microtubule-associated protein 1 light chain 3 II, MTOR mechanistic target of rapamycin kinase, NCOA4 nuclear receptor coactivator 4, PEBP1 phosphatidylethanolamine binding protein 1, PL phospholipid, PLOOH phospholipid hydroperoxides, RAB7A Rab7A, member RAS oncogene family, SLC3A2 solute carrier family 3 member 2, SLC7A11 solute carrier family 7 member 11, SQSTM1/P62 sequestosome 1, STAT3 signal transducer and activator of transcription 3, STING1 stimulator of interferon response cGAMP interactor 1.
Selective autophagy in ferroptosis
Ferritinophagy
The term ferritinophagy is used to describe the removal of the major iron storage protein ferritin by the autophagy machinery [74]. Ferritin, the major iron storage protein in most eukaryotic cell, is a complex consisting of FTH1 and ferritin light chain (FTL). Nuclear receptor coactivator 4 (NCOA4) is a cytosolic autophagy receptor used to bind ferritin for subsequent degradation by ferritinophagy [74, 75]. The knockdown of NCOA4 has an inhibitory effect on erastin-induced ferroptosis, while the overexpression of NCOA4 promotes ferroptosis by promoting ferrite degradation [76]. In parallel, the genetic suppression of ATG3, ATG5, ATG7, ATG13, or MAP1LC3 renders cancer cells or fibroblasts resistant to ferroptosis upon erastin treatment or cysteine depletion in vitro [76,77,78]. In conclusion, ferritinophagy leads to abnormal iron accumulation and eventually triggers ferroptotic death.
Ferritinophagy-mediated ferroptosis is further regulated by ELAV-like RNA-binding protein 1 (ELAVL1/HuR), which is required for sorafenib-induced liver fibrosis by inducing ferroptosis in human hepatic stellate cells [79]. Insufficient ferritinophagy limits ferroptosis induced by RSL3 and erastin in mouse senescent cells in vitro [80], but increased ferritinophagy facilitates ferroptosis-related inflammation [81]. The induction of ferritinophagy-dependent ferroptosis by dihydroartemisinin (a drug used to treat malaria) or JQ1 (a bromodomain containing 4 inhibitor) is also a potential anticancer approach [82, 83]. These findings support the pathological role of ferritinophagy-dependent ferroptosis in human diseases and therapy.
Lipophagy
Lipophagy is a type of selective autophagy involving the breakdown of lipid droplets (LDs) by autolysosomes [84]. The hydrophobic core of neutral lipids, including triacylglycerol (TG) and cholesteryl ester (CE) makes LDs the hub of lipid metabolism and energy homeostasis. Rab7a member RAS oncogene family (RAB7A), which belongs to the Ras superfamily of small GTPases, is not only a mediator of endosomal trafficking events, but also a central regulator of hepatic lipophagy [85]. LDs protect cells from the oxidation of PUFAs by transferring free fatty acids to their core, thereby providing a defense mechanism under oxidative stress [86]. This theoretical framework may explain why LDs limit ferroptosis. The protein levels of perilipin 2, an LD-specific marker, are increased in RSL3-treated human liver cancer cell lines in vitro at an early stage [87], suggesting that the accumulation of LDs may act as a negative feedback loop to limit lipid peroxidation. The upregulation of lipid storage mediated by tumor protein D52 also suppresses RSL3-induced ferroptosis in vitro [87]. At the late stage, increased lipophagy provides PUFAs for lipid peroxidation and subsequent ferroptosis, whereas the knockdown of RAB7A inhibits lipophagy-mediated ferroptosis in vitro [87]. These findings suggest a dynamic process involving lipid storage, degradation, release, and reuse in ferroptosis, although the specific autophagy receptor for lipophagy is still elusive.
Clockophagy
The selective degradation of the core circadian clock protein aryl hydrocarbon receptor nuclear translocator-like (ARNTL/BMAL1) by autophagy, namely clockophagy, promotes ferroptosis [65]. The ubiquitination-mediated protein degradation of ARNTL has been largely explored [88,89,90], but it was unclear whether it could be degraded by the autophagy system until clockophagy was identified [65]. The protein stability of ARNTL is decreased during ferroptosis caused by GPX4 inhibitor (e.g., RSL3 and FIN56), but not by SLC7A11 inhibitor (e.g., erastin, sulfasalazine, and sorafenib) in vitro [65]. SQSTM1 functions as an autophagy receptor for clockophagy to mediate ARNTL degradation, which is required for ferroptosis [65]. Mechanistically, ARNTL antagonizes ferroptosis by transcriptionally repressing the expression of Egl-9 family hypoxia- inducible factor 2 and sequentially activating the pro-survival HIF1A in vitro and in vivo [65]. These findings illustrate how the crosstalk between UPS and the autophagy pathway can promote ferroptosis.
The possible role of clockophagy in ferroptosis in vivo has also been explored using a pancreatitis animal model. Mice with a specific depletion of Arntl in the pancreas develop more severe acute pancreatitis triggered by l-arginine, and this pancreatic damage is inhibited by the administration of the ferroptosis inhibitor liproxstatin-1 [91], indicating that ferroptosis mediates the development of acute pancreatitis. These findings highlight that circadian rhythm disorders may be a risk factor for ferroptosis. The rhythmic expression of ferroptosis modulators needs to be further confirmed in animal studies.
Mitophagy
Mitophagy is the selective degradation of mitochondria through autophagy, which plays a role in maintaining the quantity and quality of mitochondria. Although the mechanism of this organelle-specific autophagy is complex, PTEN-induced kinase 1 (PINK1) and Parkin RBR E3 ubiquitin protein ligase (PRKN/PARK2) are major regulators of mitophagy [92]. The PINK1-PRKN pathway-mediated degradation of solute carrier family 25 member 37 (SLC25A37) and solute carrier family 25 member 37 (SLC25A28) induces accumulation of mitochondrial iron [93]. The effects of mitophagy in ferroptosis remain uncertain. On one hand, mitophagy mediates ferroptosis in human cancer cells that is induced by BAY87-2243 (a mitochondrial complex I inhibitor) [94] or by the expression of HMOX1 [95] in vitro. On the other hand, increased mitophagy may protect ferroptosis. For example, uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP)-induced mitophagy inhibits cysteine deprivation- or erastin-induced ferroptosis in PRKN-expressing human HT1080 cells in vitro [15]. However, other studies have shown that CCCP has no effect on ferroptosis caused by SLC7A11 inhibitor or GPX4 inhibitor in vitro [96]. More detailed insights are needed to understand potential off-target effects of CCCP and whether the mechanism of impaired mitochondrial recruitment in autophagosomes during ferroptosis requires a response to iron or lipid signals or the PINK1-PRKN pathway.
Chaperone-mediated autophagy (CMA)
CMA involves a selective pathway to directly degrade cytoplasmic proteins in lysosomes [97]. The process of CMA starts with heat shock protein family A (Hsp70) member 8 (HSPA8/HSC70) binding to proteins with a KFERQ-like motif, and then delivering the targeted proteins to the lysosome-associated membrane protein type 2A (LAMP2A) on the lysosomes for degradation [98]. Heat shock protein 90 (HSP90) directly interacts with LAMP2A at the lysosomal membrane and preserves the stability of LAMP2A for CMA activation [99]. Interestingly, GPX4 (a protein containing KEFRQ-like motif) is degraded during erastin-induced ferroptosis by HSP90-mediated CMA in vitro [100]. The knockdown of HSP90 subunits (e.g., HSP90AA1 or HSP90AB1) or administration of the HSP90 inhibitor 2-amino-5-chloro-N,3-dimethylbenzamide (CDDO) reduces glutamate- or erastin-induced ferroptosis in human cancer cells or neural cells in vitro [100]. CDDO inhibits the interaction between HSP90 and LAMP2A, thereby preventing CMA-mediated degradation of GPX4 during erastin-induced ferroptosis in vitro [100]. CDDO also prevents RSL3-induced degradation of GPX4 in vitro, although it cannot inhibit RSL3-induced ferroptosis [100]. Therefore, CMA-mediated degradation of GPX4 by HSP90 may play a selective role in mediating ferroptosis. However, certain HSPs (e.g., heat shock protein family B [small] member 1 [HSPB1] and heat shock protein family A [hsp70] member 5 [HSPA5]) protect against ferroptosis through different mechanisms. For example, the combination of HSPA5 (an ER-associated HSP) and GPX4 prevents the degradation of GPX4 caused by erastin in human pancreatic cancer cells in vitro [101]. The development of precise cancer medicines may be facilitated by the in-depth comprehension of CMA targets relevant to ferroptosis.
Autophagy regulator in ferroptosis
Positive autophagy regulators
BECN1
In addition to promoting ferroptosis through its classic function in autophagy, beclin 1 (BECN1) also promotes ferroptosis by directly interacting with SLC7A11, thereby inhibiting system xc− activity [102]. The loss of BECN1 inhibits ferroptosis induced by SLC7A11 inhibitors (e.g., erastin, sulfasalazine, and sorafenib), but not by GPX4 inhibitors (e.g., RSL3 and FIN56) in vitro [102]. Moreover, AMP-activated protein kinase (AMPK)-mediated phosphorylation of BECN1 at Ser90/93/96 is required for the formation of the BECN1-SLC7A11 complex and subsequent ferroptosis induction in vitro [102]. AMPK activation also has the ability to inhibit ferroptosis by blocking fatty acid synthesis via the phosphorylation of acetyl-CoA carboxylase [103]. How the energy sensor AMPK selectively phosphorylates different substrates to regulate ferroptosis remains unclear. In vivo, the administration of BECN1 activator peptide (namely Tat-beclin 1) increases the anticancer activity of erastin by inducing autophagy-dependent ferroptosis in mouse xenograft models, supporting a previous finding that Tat-beclin 1 is an inducer of autosis (a type of autophagy-dependent cell death) [102]. In addition to human cancer cells, the RNA-binding protein ELAVL1/HuR promotes sorafenib-induced ferroptosis by increasing BECN1 mRNA stability and subsequent ferritinophagy in human hepatic stellate cells in vitro [79]. Thus, BECN1 may play an autophagy-dependent and -independent role in promoting ferroptosis. Elucidating the complex BECN1 network in autophagy and other types of cell death (e.g., apoptosis and ferroptosis) likely depends on the identification of additional proteins within the fluctuating BECN1 interactome [104, 105].
STING1
Stimulator of interferon response cGAMP interactor 1 (STING1/STING/TMEM173) plays a key role in immune, coagulation, and inflammatory responses through its ability to trigger type I interferon and cytokine releases as well as inflammasome activation [106,107,108]. Moreover, the STING1-containing ER-Golgi intermediate compartment can serve as a membrane source for the initiation of autophagosomes in response to host DNA damage, thereby playing a positive role in autophagy [109]. In addition to promoting various types of cell death (e.g., apoptosis, necroptosis, and pyroptosis) [110], STING1 is also involved in the induction of ferroptosis [46]. In particular, zalcitabine, an antiviral drug for the treatment of human immunodeficiency virus infection, induces autophagy-dependent ferroptotic human cancer cell death by activating mitochondrial DNA (mtDNA) stress-mediated STING1 activation in preclinical models [46]. Zalcitabine inhibits DNA polymerase gamma, catalytic subunit (POLG, the mtDNA replication and repair regulator) and then induces the proteasomal degradation of transcription factor A, mitochondrial by the mitochondrial protease Lon peptidase 1 mitochondrial, leading to mtDNA stress and STING1-dependent autophagy in human pancreatic cancer cells in vitro [46]. Finally, ALOX5 functions as a mediator of lipid peroxidation for autophagy-dependent ferroptosis after STING1 activation [46]. Although these findings indicate a molecular link between mtDNA damage, autophagy, and ferroptosis, it remains to be seen to which extent STING1 contributes to ferroptotic damage-mediated macrophage polarization during tumor formation [111].
CTSB
Lysosomes seem to be the crossroads of various types of cell death closely linked to autophagy. Lysosomal inhibitors, including bafilomycin A1 (a vacuolar ATPase inhibitor), ammonium chloride (an endosome-lysosome system acidification inhibitor) and pepstatin A (a lysosomal aspartic protease inhibitor), block erastin-induced ROS production and ferroptosis in vitro [19, 20]. Cathepsins are the principal proteases in the lysosome and play an active role in the degradation of autophagic substrates. Among them, the increased expression of cathepsin B (CTSB) is required for ferroptosis [112]. Erastin induces CTSB expression by activating signal transducer and activator of transcription 3 (STAT3), which is a transcription factor for lysosomal cell death [19]. The translocation of CTSB from the lysosome to the nucleus causes DNA damage and subsequent STING1-dependent ferroptosis in vitro [112]. Therefore, CTSB mediates a lysosome-nuclear pathway of communication that promotes DNA damage-induced ferroptosis.
HMGB1
High mobility group box 1 (HMGB1) plays a location-dependent role in promoting autophagy. Nuclear HMGB1-mediated HSPB1 expression promotes cytoskeleton reorganization, which is required for mitophagy [113]. Cytosolic HMGB1 can bind BECN1 to induce autophagosome formation [114]. Extracellular HMGB1 induces autophagy by binding its receptor advanced glycosylation end-product specific receptor to mediate PtdIns3K complex activation [115]. Extracellular HMGB1 is a damage-associated molecular pattern that triggers inflammation and immunity response during ferroptosis induced by RSL3 and erastin in vitro [115], whereas intracellular HMGB1 promotes erastin-induced ferroptosis in human leukemia cells by activating mitogen-activated protein kinase (MAPK)-dependent transferrin receptor (TFRC) expression [116]. HMGB1-mediated autophagy (e.g., ferritinophagy) may enhance iron accumulation during ferroptotic damage. Therefore, neutralization of HMGB1 provides an opportunity to treat ischemia-reperfusion injury, which is a process driven by cell death-mediated sterile inflammation [117, 118].
Negative autophagy regulators
MTOR
Mechanistic target of rapamycin kinase (MTOR) is a key kinase that functions as a repressor of the induction of autophagy by regulating ULK1 or PtdIns3K complex activity [9]. MTOR-specific inhibitors, such as rapamycin and its derivatives, are classic autophagy inducers and are used to treat specific forms of cancer [119]. The interaction between MTOR and GPX4 signals can fine-tune autophagy-dependent ferroptosis. Both rapamycin (high-dose) and RSL3 cause autophagy-dependent cell death and GPX4 protein degradation in human pancreatic cancer cells [120]. In mouse models, rapamycin-induced tumor suppression is reversed by liproxstatin-1 and hydroxychloroquine [120], further suggesting that the inhibition of MTOR triggers autophagy-dependent ferroptosis. Because MTOR can also directly inhibit the CMA pathway [121], the degradation of GPX4 induced by MTOR inhibitors may be due to the activation of CMA. The function of SLC7A11 can be repressed by MTOR-mediated phosphorylation [122]. Moreover, the activation of MTOR limits ferroptosis in human cancer cells by maintaining stearoyl-CoA desaturase (SCD/SCD1)-mediated MUFA production [123]. The protective effect of MTOR on ferroptosis has also been observed in non-cancer cells, including cardiomyocytes [124]. Collectively, these findings suggest that MTOR, a central regulator of cell responses to growth stimuli, inhibits autophagy-dependent ferroptosis.
PEBP1
Phosphatidylethanolamine binding protein 1 (PEBP1/RKIP) is a conserved protein that was first isolated based on its ability to bind phospholipids [125]. PEBP1 acts as an endogenous inhibitor of the RAF-MEK-ERK pathway by directly blocking the activation of Raf-1 proto-oncogene, serine/threonine kinase (RAF1) [126]. The binding of ALOX15 and PEBP1 enhances the substrate competence of ALOX15 to generate hydroperoxy-phosphatidylethanolamine (PE), which promotes RSL3-induced ferroptosis in asthmatic human airway epithelial cells in vitro [47]. PEBP1 also inhibits autophagy by directly interacting with MAP1LC3 through its WXXL motif [127]. During RSL3-induced ferroptosis, an increase in the interaction between ALOX15 and PEBP1 limits the formation of a PEBP1-MAP1LC3 complex in vitro, thereby increasing autophagy [128]. Of note, the loss of MAP1LC3 has been shown to inhibit or promote erastin- or RSL3-induced ferroptosis in vitro [78, 128]. The inhibition of a single MAP1LC3 gene may lead to a compensatory increase in the expression of its family members [129], thereby activating MAP1LC3-independent autophagosome activation during ferroptosis. This hypothesis needs to be addressed in the future.
DUSP1
Dual specificity phosphatases (DUSPs), also known as MAPK phosphatases, specifically dephosphorylates threonine and tyrosine residues in the MAPK P loop, play a unique role in regulating the functions of the MAPK family in cancer cells. Among the ten members of the DUSP family, dual specificity phosphatase 1 (DUSP1) can have its expression and activity increased in human pancreatic cancer cells in vitro by erastin and RSL3 in an ERK-dependent manner [130]. The increased DUSP1 forms a feedback mechanism to inhibit autophagy-dependent ferroptosis in vitro and in vivo [130]. DUSP1 may limit the formation of autophagosomes by blocking the phosphorylation of ULK1 or BECN1. Small molecules that inhibit DUSP1 may offer new possibilities for the therapeutic induction of ferroptosis.
Conclusion and perspectives
Among the ever-expanding number of molecular mechanisms affecting ferroptosis, degradation pathways occupy a significant role. The specificity of each degradation pathway is reflected in the selectivity of its substrate. This means that a degradation pathway may play a dual role in the regulation of ferroptosis. Simply put, if the substrate is an anti-ferroptosis modulator, the corresponding degradation pathway promotes ferroptosis. In contrast, if the substrate is a pro-ferroptosis modulator, the corresponding degradation pathway logically inhibits ferroptosis. In this review, we discussed progress in mechanistically understanding how degradation pathways affect ferroptosis, hoping that targeting them in a specific fashion may ultimately lead to novel therapeutic interventions on malignant, metabolic, and neurodegenerative diseases. In view of the complexity of the ferroptosis process [131], we suggest that the following issues need special consideration in future research on degradation systems in ferroptosis. First, we need to determine the key effector protein(s) that execute(s) ferroptotic cell death downstream of lipid peroxidation. Second, we must identify the E3 ubiquitin ligases or autophagy receptors for individual targets in ferroptosis. Third, we need to understand the interaction between UPS and autophagy in ferroptosis. Fourth, the development of new small molecules targeting specific degradation pathways may greatly accelerate the therapeutic manipulation of ferroptosis.
References
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25:486–541.
Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29:347–64.
Tang D, Kroemer G. Ferroptosis. Curr Biol. 2020;30:R1292–7.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of non-apoptotic cell death. Cell. 2012;149:1060–72.
Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2020. https://doi.org/10.1038/s41422-020-00441-1.
Stockwell BR, Angeli JPF, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–85.
Rousseau A, Bertolotti A. Regulation of proteasome assembly and activity in health and disease. Nat Rev Mol Cell Biol. 2018;19:697–712.
Levine B, Kroemer G. Biological functions of autophagy genes: a disease perspective. Cell. 2019;176:11–42.
Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–64.
Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–22.
Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40:280–93.
Denton D, Kumar S. Autophagy-dependent cell death. Cell Death Differ. 2019;26:605–16.
Chen X, Li JB, Kang R, Klionsky DJ, Tang DL. Ferroptosis: machinery and regulation. Autophagy. 2020. https://doi.org/10.1080/15548627.2020.1810918.
Kuang F, Liu J, Tang D, Kang R. Oxidative damage and antioxidant defense in ferroptosis. Front Cell Dev Biol. 2020;8:586578.
Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell. 2019;73:354–363 e353.
Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3:e02523.
Alborzinia H, Ignashkova TI, Dejure FR, Gendarme M, Theobald J, Wolfl S, et al. Golgi stress mediates redox imbalance and ferroptosis in human cells. Commun Biol. 2018;1:210.
Song X, Xie Y, Kang R, Hou W, Sun X, Epperly MW, et al. FANCD2 protects against bone marrow injury from ferroptosis. Biochem Biophys Res Commun. 2016;480:443–9.
Gao H, Bai Y, Jia Y, Zhao Y, Kang R, Tang D, et al. Ferroptosis is a lysosomal cell death process. Biochem Biophys Res Commun. 2018;503:1550–6.
Torii S, Shintoku R, Kubota C, Yaegashi M, Torii R, Sasaki M, et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem J. 2016;473:769–77.
Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. 2020;585:603–8.
Tang D, Kroemer G. Peroxisome: the new player in ferroptosis. Signal Transduct Target Ther. 2020;5:273.
Louandre C, Ezzoukhry Z, Godin C, Barbare JC, Maziere JC, Chauffert B, et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer. 2013;133:1732–42.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31.
Eaton JK, Furst L, Ruberto RA, Moosmayer D, Hilpmann A, Ryan MJ, et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat Chem Biol. 2020;16:497–506.
Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12:497–503.
Yang L, Chen X, Yang Q, Chen J, Huang Q, Yao L, et al. Broad spectrum deubiquitinase inhibition induces both apoptosis and ferroptosis in cancer cells. Front Oncol. 2020;10:949.
Sato H, Shiiya A, Kimata M, Maebara K, Tamba M, Sakakura Y, et al. Redox imbalance in cystine/glutamate transporter-deficient mice. J Biol Chem. 2005;280:37423–9.
Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8:237–48.
Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 2020;368:85–89.
Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, et al. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe. 2018;24:97–108.e4.
Canli O, Alankus YB, Grootjans S, Vegi N, Hultner L, Hoppe PS, et al. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood. 2016;127:139–48.
Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–8.
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature.2019;575:688–92.
Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Muller C, Zandkarimi F, et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. 2020;6:41–53.
Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol. 2020;16:1351–60.
Dai E, Meng L, Kang R, Wang X, Tang D. ESCRT-III-dependent membrane repair blocks ferroptosis. Biochem Biophys Res Commun. 2020;522:415–21.
Dai E, Zhang W, Cong D, Kang R, Wang J, Tang D. AIFM2 blocks ferroptosis independent of ubiquinol metabolism. Biochem Biophys Res Commun. 2020;523:966–71.
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA. 2016;113:E4966–4975.
Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol. 2019;26:420–432 e429.
Tesfay L, Paul BT, Konstorum A, Deng Z, Cox AO, Lee J, et al. Stearoyl-CoA desaturase 1 protects ovarian cancer cells from ferroptotic cell death. Cancer Res. 2019;79:5355–66.
Yuan H, Li X, Zhang X, Kang R, Tang D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478:1338–43.
Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90.
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–98.
Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol. 2019;21:579–91.
Li C, Zhang Y, Liu J, Kang R, Klionsky DJ, Tang D. Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy. 2020. https://doi.org/10.1080/15548627.2020.1739447.
Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell. 2017;171:628–641 e626.
Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16:302–9.
Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017;20:1692–704.
Yang WH, Huang Z, Wu J, Ding CC, Murphy SK, Chi JT. A TAZ-ANGPTL4-NOX2 axis regulates ferroptotic cell death and chemoresistance in epithelial ovarian cancer. Mol Cancer Res. 2020;18:79–90.
Yang WH, Ding CC, Sun T, Rupprecht G, Lin CC, Hsu D, et al. The Hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Rep. 2019;28:2501–2508 e2504.
Chen X, Yu C, Kang R, Tang D. Iron metabolism in ferroptosis. Front Cell Dev Biol. 2020;8:590226.
Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428.
Liu T, Jiang L, Tavana O, Gu W. The deubiquitylase OTUB1 mediates ferroptosis via stabilization of SLC7A11. Cancer Res. 2019;79:1913–24.
Dai C, Chen X, Li J, Comish P, Kang R, Tang D. Transcription factors in ferroptotic cell death. Cancer Gene Ther. 2020;27:645–56.
Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–23.
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173–84.
Anandhan A, Dodson M, Schmidlin CJ, Liu P, Zhang DD. Breakdown of an ironclad defense system: the critical role of NRF2 in mediating ferroptosis. Cell Chem Biol. 2020;27:436–47.
Sun X, Niu X, Chen R, He W, Chen D, Kang R, et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology. 2016;64:488–500.
Cao JY, Poddar A, Magtanong L, Lumb JH, Mileur TR, Reid MA, et al. A genome-wide haploid genetic screen identifies regulators of glutathione abundance and ferroptosis sensitivity. Cell Rep. 2019;26:1544–1556 e1548.
Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447:864–8.
Yang Y, Luo M, Zhang K, Zhang J, Gao T, Connell DO, et al. Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat Commun. 2020;11:433.
Wang Y, Liu Y, Liu J, Kang R, Tang D. NEDD4L-mediated LTF protein degradation limits ferroptosis. Biochem Biophys Res Commun. 2020;531:581–7.
Gonzalez FJ, Xie C, Jiang C. The role of hypoxia-inducible factors in metabolic diseases. Nat Rev Endocrinol. 2018;15:21–32.
Yang M, Chen P, Liu J, Zhu S, Kroemer G, Klionsky DJ, et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv. 2019;5:eaaw2238.
Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun. 2019;10:1617.
Miess H, Dankworth B, Gouw AM, Rosenfeldt M, Schmitz W, Jiang M, et al. The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene. 2018;37:5435–50.
Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. 2017;551:639–43.
Xie Y, Kang R, Sun X, Zhong M, Huang J, Klionsky DJ, et al. Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy. 2015;11:28–45.
Schutter M, Giavalisco P, Brodesser S, Graef M. Local fatty acid channeling into phospholipid synthesis drives phagophore expansion during autophagy. Cell. 2020;180:135–149 e114.
Marino G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell. 2014;53:710–25.
Xie Y, Li J, Kang R, Tang D. Interplay between lipid metabolism and autophagy. Front Cell Dev Biol. 2020;8:431.
Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018;20:233–42.
Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509:105–9.
Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol. 2014;16:1069–79.
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–8.
Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021–32.
Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019;10:822.
Zhang Z, Yao Z, Wang L, Ding H, Shao J, Chen A, et al. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy. 2018;14:2083–103.
Masaldan S, Clatworthy SAS, Gamell C, Meggyesy PM, Rigopoulos AT, Haupt S, et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2018;14:100–15.
Yoshida M, Minagawa S, Araya J, Sakamoto T, Hara H, Tsubouchi K, et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat Commun. 2019;10:3145.
Du J, Wang T, Li Y, Zhou Y, Wang X, Yu X, et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic Biol Med. 2019;131:356–69.
Sui S, Zhang J, Xu S, Wang Q, Wang P, Pang D. Ferritinophagy is required for the induction of ferroptosis by the bromodomain protein BRD4 inhibitor (+)-JQ1 in cancer cells. Cell Death Dis. 2019;10:331.
Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature 2009;458:1131–5.
Schroeder B, Schulze RJ, Weller SG, Sletten AC, Casey CA, McNiven MA. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology. 2015;61:1896–907.
Bailey AP, Koster G, Guillermier C, Hirst EM, MacRae JI, Lechene CP, et al. Antioxidant role for lipid droplets in a stem cell niche of Drosophila. Cell. 2015;163:340–53.
Bai Y, Meng L, Han L, Jia Y, Zhao Y, Gao H, et al. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun. 2019;508:997–1003.
Gossan NC, Zhang F, Guo B, Jin D, Yoshitane H, Yao A, et al. The E3 ubiquitin ligase UBE3A is an integral component of the molecular circadian clock through regulating the BMAL1 transcription factor. Nucleic Acids Res. 2014;42:5765–75.
Chen S, Yang J, Zhang Y, Duan C, Liu Q, Huang Z, et al. Ubiquitin-conjugating enzyme UBE2O regulates cellular clock function by promoting the degradation of the transcription factor BMAL1. J Biol Chem. 2018;293:11296–309.
Ullah K, Chen S, Lu J, Wang X, Liu Q, Zhang Y, et al. The E3 ubiquitin ligase STUB1 attenuates cell senescence by promoting the ubiquitination and degradation of the core circadian regulator BMAL1. J Biol Chem. 2020;295:4696–708.
Liu Y, Wang Y, Liu J, Kang R, Tang D. The circadian clock protects against ferroptosis-induced sterile inflammation. Biochem Biophys Res Commun. 2020;525:620–5.
Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298.
Li C, Zhang Y, Cheng X, Yuan H, Zhu S, Liu J, et al. PINK1 and PARK2 suppress pancreatic tumorigenesis through control of mitochondrial iron-mediated immunometabolism. Dev Cell. 2018;46:441–455 e448.
Basit F, van Oppen LM, Schockel L, Bossenbroek HM, van Emst-de Vries SE, Hermeling JC, et al. Mitochondrial complex I inhibition triggers a mitophagy-dependent ROS increase leading to necroptosis and ferroptosis in melanoma cells. Cell Death Dis. 2017;8:e2716.
Chang LC, Chiang SK, Chen SE, Yu YL, Chou RH, Chang WC. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018;416:124–37.
Gaschler MM, Hu F, Feng H, Linkermann A, Min W, Stockwell BR. Determination of the subcellular localization and mechanism of action of ferrostatins in suppressing ferroptosis. ACS Chem Biol. 2018;13:1013–20.
Dice JF. Chaperone-mediated autophagy. Autophagy. 2007;3:295–9.
Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 2018;19:365–81.
Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol. 2008;28:5747–63.
Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci USA. 2019;116:2996–3005.
Zhu S, Zhang Q, Sun X, Zeh HJ 3rd, Lotze MT, Kang R, et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. 2017;77:2064–77.
Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc(−) activity. Curr Biol. 2018;28:2388–2399 e2385.
Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 2020;22:225–34.
Maiuri MC, Criollo A, Kroemer G. Crosstalk between apoptosis and autophagy within the Beclin 1 interactome. EMBO J. 2010;29:515–6.
Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18:571–80.
Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol. 2020;21:501–21.
Zhang H, Zeng L, Xie M, Liu J, Zhou B, Wu R, et al. TMEM173 drives lethal coagulation in sepsis. Cell Host Microbe. 2020;27:556–570.e556.
Zeng L, Kang R, Zhu S, Wang X, Cao L, Wang H, et al. ALK is a therapeutic target for lethal sepsis. Sci Transl Med. 2017;9.eaan5689.
Gui X, Yang H, Li T, Tan X, Shi P, Li M, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature. 2019;567:262–6.
Murthy AMV, Robinson N, Kumar S. Crosstalk between cGAS-STING signaling and cell death. Cell Death Differ. 2020;27:2989–3003.
Dai E, Han L, Liu J, Xie Y, Zeh H, Kang R, et al. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat Commun. 2020;11:6339.
Kuang F, Liu J, Li C, Kang R, Tang D. Cathepsin B is a mediator of organelle-specific initiation of ferroptosis. Biochem Biophys Res Commun. 2020;533:1464–9.
Tang D, Kang R, Livesey KM, Kroemer G, Billiar TR, Van Houten B, et al. High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab. 2011;13:701–11.
Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, et al. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190:881–92.
Wen Q, Liu J, Kang R, Zhou B, Tang D. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun. 2019;510:278–83.
Ye F, Chai W, Xie M, Yang M, Yu Y, Cao L, et al. HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRAS(Q61L) cells. Am J Cancer Res. 2019;9:730–9.
Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–62.
Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, et al. HMGB1 in health and disease. Mol Asp Med. 2014;40:1–116.
Vignot S, Faivre S, Aguirre D, Raymond E. mTOR-targeted therapy of cancer with rapamycin derivatives. Ann Oncol. 2005;16:525–37.
Liu Y, Wang Y, Liu J, Kang R, Tang D. Interplay between MTOR and GPX4 signaling modulates autophagy-dependent ferroptotic cancer cell death. Cancer Gene Ther. 2020. https://doi.org/10.1038/s41417-020-0182-y.
Arias E, Koga H, Diaz A, Mocholi E, Patel B, Cuervo AM. Lysosomal mTORC2/PHLPP1/Akt regulate chaperone-mediated autophagy. Mol Cell. 2015;59:270–84.
Gu Y, Albuquerque CP, Braas D, Zhang W, Villa GR, Bi J, et al. mTORC2 regulates amino acid metabolism in cancer by phosphorylation of the cystine-glutamate antiporter xCT. Mol Cell. 2017;67:128–138 e127.
Yi J, Zhu J, Wu J, Thompson CB, Jiang X. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci USA. 2020;117:31189–97.
Baba Y, Higa JK, Shimada BK, Horiuchi KM, Suhara T, Kobayashi M, et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2018;314:H659–H668.
Bernier I, Tresca JP, Jolles P. Ligand-binding studies with a 23 kDa protein purified from bovine brain cytosol. Biochim Biophys Acta. 1986;871:19–23.
Yeung K, Seitz T, Li S, Janosch P, McFerran B, Kaiser C, et al. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature 1999;401:173–7.
Noh HS, Hah YS, Zada S, Ha JH, Sim G, Hwang JS, et al. PEBP1, a RAF kinase inhibitory protein, negatively regulates starvation-induced autophagy by direct interaction with LC3. Autophagy. 2016;12:2183–96.
Zhao J, Dar HH, Deng Y, St Croix CM, Li Z, Minami Y, et al. PEBP1 acts as a rheostat between prosurvival autophagy and ferroptotic death in asthmatic epithelial cells. Proc Natl Acad Sci USA. 2020;117:14376–85.
Schaaf MB, Keulers TG, Vooijs MA, Rouschop KM. LC3/GABARAP family proteins: autophagy-(un)related functions. FASEB J. 2016;30:3961–78.
Xie Y, Kuang F, Liu J, Tang D, Kang R. DUSP1 blocks autophagy-dependent ferroptosis in pancreatic cancer. Journal of Pancreatology. 2020;3:154–60.
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–79.
Acknowledgements
We thank Dave Primm (Department of Surgery, University of Texas Southwestern Medical Center) for his critical reading of the manuscript.
Funding
GK is supported by the Ligue contre le Cancer (équipe labellisée); Agence National de la Recherche (ANR)—Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer; Cancéropôle Ile-de-France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale; a donation by Elior; European Research Area Network on Cardiovascular Diseases (ERA-CVD, MINOTAUR); Gustave Roussy Odyssea, the European Union Horizon 2020 Project Oncobiome; Fondation Carrefour; High-end Foreign Expert Program in China (GDW20171100085 and GDW20181100051), Institut National du Cancer; Inserm (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière; the Seerave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); and the SIRIC Cancer Research and Personalized Medicine program (CARPEM).
Author information
Authors and Affiliations
Contributions
All authors wrote the manuscript. GK and DT edited, reviewed, and approved the manuscript before submission.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethics statement
The review was written in accordance with ethical standards.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by G. Melino
Rights and permissions
About this article

Cite this article
Chen, X., Yu, C., Kang, R. et al. Cellular degradation systems in ferroptosis. Cell Death Differ 28, 1135–1148 (2021). https://doi.org/10.1038/s41418-020-00728-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-020-00728-1
This article is cited by
-
Melatonin: a ferroptosis inhibitor with potential therapeutic efficacy for the post-COVID-19 trajectory of accelerated brain aging and neurodegeneration
Molecular Neurodegeneration (2024)
-
Glutathione supplementation improves fat graft survival by inhibiting ferroptosis via the SLC7A11/GPX4 axis
Stem Cell Research & Therapy (2024)
-
S670, an amide derivative of 3-O-acetyl-11-keto-β-boswellic acid, induces ferroptosis in human glioblastoma cells by generating ROS and inhibiting STX17-mediated fusion of autophagosome and lysosome
Acta Pharmacologica Sinica (2024)
-
Nrf2-mediated redox balance alleviates LPS-induced vascular endothelial cell inflammation by inhibiting endothelial cell ferroptosis
Scientific Reports (2024)
-
Ferroptosis exacerbates hyperlipidemic acute pancreatitis by enhancing lipid peroxidation and modulating the immune microenvironment
Cell Death Discovery (2024)
