
Abstract
Intermediate-risk acute myeloid leukemia (IR-AML) is a clinically heterogeneous disease, for which optimal post-remission therapy is debated. The utility of next-generation sequencing information in decision making for IR-AML has yet to be elucidated. We retrospectively studied 100 IR-AML patients, defined by European Leukemia Net classification, who had mutational information at diagnosis, received intensive chemotherapy and achieved complete remission (CR) at Cleveland Clinic (CC). The Cancer Genome Atlas (TCGA) data were used for validation. In the CC cohort, median age was 58.5 years, 64% had normal cytogenetics, and 31% required >1 induction cycles to achieve CR1. In univariable analysis, patients carrying mutations in DNMT3A, U2AF1, and EZH2 had worse overall and relapse-free survival. After adjusting for other variables, the presence of these mutations maintained an independent effect on survival in both CC and TCGA cohorts. Patients who did not have the mutations and underwent hematopoietic cell transplant (HCT) had the best outcomes. HCT improved outcomes for patients who had these mutations. RUNX1 or ASXL1 mutations did not predict survival, and performance of HCT did not confer a significant survival benefit. Our results provide evidence of clinical utility in considering mutation screening to stratify IR-AML patients after CR1 to guide therapeutic decisions.
Similar content being viewed by others


Introduction
Acute myeloid leukemia (AML) is a heterogeneous disease characterized by impaired differentiation and increased proliferation of myeloid progenitors. With the widespread use of high-throughput sequencing techniques, AML has been genetically characterized as a complex polyclonal disease with multiple somatically acquired driver mutations and disease evolution over time1. In 2013, the Cancer Genome Atlas (TCGA) Research Network profiled the genomes of 200 adult de novo AML patients and identified 23 commonly mutated genes, which were classified into 9 categories2. Subsequently, a more comprehensive study of the driver mutation landscape in AML enabled a full genomic classification with nonoverlapping subgroups3. These and multiple other studies have yielded important prognostic information and has led cytogenetically normal AML with NPM1 or biallelic CEBPA mutations in the absence of FLT3-ITD to be placed in a favorable risk category4. Moreover, patients with RUNX1, ASXL1, and TP53 mutations have recently been added to the adverse risk group in the European Leukemia Net (ELN) 2017 classification5.
Despite the increase in our knowledge of the biology of AML, treatment algorithms have not changed substantially over the last 40 years6. Regardless of their mutations and risk stratification, the majority of eligible patients receive intensive induction chemotherapy, with a primary goal of achieving complete remission (CR). This is followed by post-remission therapy tailored according to risk profile, whereby chemotherapeutic consolidation is preferred in favorable-risk AML and allogeneic hematopoietic cell transplant (HCT) is favored in poor-risk AML5. The optimal post-remission therapy for intermediate-risk AML, which comprises nearly half of all cases, is debated. Many are evaluated for HCT at the time of CR17, with reduced intensity conditioning (RIC) approaches now available for use in older populations or those with co-morbidities8,9,10,11. The prognostic role of certain mutations within intermediate-risk AML is unclear, but might aid in decision making for this clinically heterogeneous patient population.
The use of mutational data to inform clinical practice is an active area of research. In a large study of 664 AML patients treated on two phase 3 trials conducted by the German AML Cooperative Group, mutations in RUNX1, SRSF2, U2AF1, and SF3B1 were found to be independent risk factors for achievement of CR112. In addition, multivariable analysis of a smaller cohort of intermediate-risk AML patients (defined by cytogenetics alone) identified ASXL1 and FLT3-ITD as factors predicting lower chances of achieving CR113. These mutations, along with a few others (e.g., DNMT3A, TP53) are known to be associated with poor overall survival (OS)14,15. However, whether these mutations retain their predictive values after achievement of CR1, at which time the clones harboring these mutations might have already been eradicated, is unknown. Furthermore, the utility of this genetic information in decision making for intermediate-risk AML patients who have achieved CR1 has not been determined. Therefore, we set out to investigate the clinical relevance of recurrent driver gene mutations in a well-characterized cohort of intermediate-risk AML patients who were homogenously treated at a single center and achieved CR1. The analysis aims to re-visit the predictors of outcome at CR1, and identify mutations that portend poor prognosis, hence may help select patients who may benefit from HCT.
Methods
Patient cohorts and study eligibility
Cleveland clinic (CC) cohort
The diagnosis of AML was made or revised according to the 2016 WHO criteria16. Cytogenetic analysis was performed on metaphases from bone marrow aspirates taken at diagnosis, and the risk was ascribed by ELN 2010 criteria as intermediate-I or -II17. Namely, patients who had normal cytogenetics and harbored NPM1 or biallelic CEBPA mutations in the absence of FLT3-ITD were excluded. A total of 1589 AML patients treated at the Cleveland Clinic between 2002 and 2016 were screened for eligibility, of whom 825 were in ELN intermediate-risk category, and 355 received intensive induction chemotherapy. We retrospectively analyzed 100 intermediate-risk AML patients who had available pre-treatment myeloid mutational data and achieved CR1 after intensive chemotherapy. Patients who did not receive intensive therapy or achieve CR after 1 or more lines of induction (i.e., primary refractory) were excluded.
The induction regimen was 7 + 3 (i.e., 7 days of 100 mg/m2 cytarabine plus 3 days of anthracycline), and patients who had persistent disease at day 14 marrow were re-induced with 7 + 3 or 5 + 2 (i.e., 5 days of cytarabine plus 2 days of anthracycline). All patients achieved CR1 with or without count recovery. CR was defined as less than 5% bone marrow blasts with evidence of normal maturation of other marrow elements, no peripheral blast cells or extramedullary disease, peripheral blood neutrophil counts above 1 × 109/L, and platelet counts above 100 × 109/L. Complete response with inadequate count recovery (CRi) was defined as a response meeting the criteria of CR, except for residual neutropenia and/or thrombocytopenia5. Induction therapy was followed by consolidation chemotherapy (i.e., high or intermediate dose cytarabine) or allogeneic HCT. Clinical data and patient samples were collected prospectively with patient consent and approved by the Institutional Review Board in accordance with the Declaration of Helsinki.
TCGA cohort
A large publicly available database of 200 clinically annotated adult de novo AML patients with genomic and epigenomic profiling was created in 20132. The study analyzed patients from a single institution tissue banking protocol in Washington University and samples were selected to represent morphologic and cytogenetic subtypes of AML. For external validation of our findings, we studied 48 ELN intermediate-risk AML patients from this cohort, who received intensive induction chemotherapy and achieved CR1. Patients who were unfit for intensive chemotherapy, and those with primary refractory disease and acute promyelocytic leukemia were excluded.
Sample processing, DNA sequencing and mutation analysis
Multi-amplicon deep sequencing was performed as previously described18. DNA was extracted from bone marrow or peripheral blood mononuclear cells collected at the time of diagnosis. In 10 patients, subsequent serial samples were also studied. A TruSeq Custom Amplicon panel (TSCA; Illumina, San Diego, CA, USA) targeting coding exons of 62 genes with available evidence in myeloid neoplasms was used for deep sequencing (Supplementary Table). For germline confirmation, mutations were analyzed in non-clonal CD3 positive T cells whenever DNA was available. Bidirectional sequencing was performed by standard techniques using an ABI 3730xl DNA analyzer (Applied Biosystems, Foster City, CA, USA). GATK3.3 pipeline was used to extract putative variants, following recommended best practices for variant discovery. Variants with at least 10 positive reads and variant allelic frequency of 5% were prioritized for further processing and annotation. Generated VCF files were used as an input for Annovar and were annotated with multiple databases (dbSNP138, COSMIC, ExacDb). Variants found in ExacDb with allelic frequency >0.0001 were excluded. Variant allelic frequencies (VAFs) of mutations were adjusted according to the zygosity and copy number confirmed by single nucleotide polymorphism (SNP)-array. In addition, mutations in NPM1, FLT3, and CEBPA were also tested using standard methods. The sequencing method for patients in TCGA database is described previously2.
Statistical analysis
Data were presented with percentage proportions for categorical variables and medians for continuous variables. Comparison of the distribution of categorical variables was carried out with either χ2-test or Fisher’s exact test. Comparison of numerical variables between groups was performed using Wilcoxon rank sum test. Whisker plot boxes denote median and 25th and 75th percentiles, and ends of the whiskers display minimum and maximum values. Cox proportional hazards regression was used for univariable and multivariable analysis to identify the impact of clinical and genomic variables on survival outcomes. Data were presented with hazard ratios (HR) and 95% confidence intervals (CI). Overall survival (OS) was defined as the time from diagnosis until death or the last follow-up. Patients who were alive were censored at the last follow-up date. Relapse-free survival (RFS) was calculated from the time point of CR1 until the time of relapse or death or the last follow-up. Patients without relapse or death at last follow-up were censored. The survival analysis was based on Kaplan–Meier method and the log-rank test was used to compare the curves. P values were two-tailed and considered significant when <0.05. All analyses were performed using JMP software v.12.2.0 (SAS Inc. Cary, NC, USA).
Results
Patient characteristics
Clinical characteristics of patients are summarized in Table 1. In the CC cohort, the median age was 58.5 years (range, 24 to 75 years). Forty-eight percent were female, 76% had de novo AML, 19% had secondary AML after an antecedent hematologic disorder (sAML), and 5% had treatment-related AML (tAML). Since all patients in the TCGA cohort had de novo AML, there were more patients with ELN intermediate-I risk category (i.e., normal cytogenetics) as compared to the CC cohort (81 vs 64%, p = 0.007). Concomitantly, patients in the TCGA cohort had a higher WBC count and bone marrow blast percentage at diagnosis. In the CC cohort, initial 7 + 3 induction chemotherapy resulted in CR in 58%, CRi in 11%, and persistent disease in 31% of patients based on day 14 bone marrow biopsy. Of the 31 patients with persistent disease, 20 were re-induced with 5 + 2, and 11 received 7 + 3. Best response achieved was CR in 23 patients and CRi in 8 patients. The number of patients who received HCT and the time of HCT were not significantly different between cohorts. However, there were more patients who underwent haploidentical HCT in our cohort, and 13% of the transplanted patients in the TCGA cohort received autologous HCT.
The median OS and RFS of the CC cohort were 24 (range, 2 to 108 months) and 14 months (range, 1 to 70 months), respectively. Sixty patients remained in remission after a median follow-up of 14.5 months (range, 2 to 108 months). The survival outcomes of the TCGA cohort were similar to ours with a median OS of 24.4 (range, 2.2 to 118.1 months) and median RFS of 13.4 months (range, 1.7 to 77.3 months).
Spectrum of driver mutations
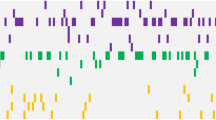
The mutational spectrum of our cohort is illustrated in Fig. 1. DNMT3A and FLT3-ITD mutations were the most common (19%), followed by RUNX1 (15%), TET2 (12%), IDH2 (12%), NPM1 (12%), ASXL1 (11%), and U2AF1 (11%) (Fig. 1a). At least one driver mutation was identified in 85% of patients. Of the remaining 15 patients, 6 had cytogenetic abnormalities. Thus, ≥1 molecularly defined lesion was detected in 91% of cases. The median number of driver gene mutations per patient was 2 (range, 0–12), and the median VAF of mutations was 33.3% (range, 5–94.3%). The spectrum of VAFs observed for genes mutated in ≥5 patients are illustrated in Fig. 1b. Of note, three genes with the lowest median VAFs (FLT3, NRAS, and CBL) are involved in growth factor signaling, supporting the previous observations that alterations in these pathways are acquired later during the evolution of the leukemic clone12,19.

a Heatmap depicting gene mutations in 100 adult AML patients in the Cleveland Clinic cohort. b Analysis of variant allelic frequencies (VAFs) of driver gene mutations found in ≥5 patients. c Number of driver gene mutations in patients with de novo AML (dnAML) compared with secondary AML (sAML) and treatment-related AML (tAML)
We studied the association of driver gene mutations with clinical and cytogenetic characteristics. The median number of mutations was higher in patients aged ≥ 60 years as compared to younger patients (3 vs 2, p = 0.03), and these patients more commonly carried mutations in SRSF2 (16% vs 2%, p = 0.02) and NRAS (16% vs 4%, p = 0.03). Mutations in ASXL1 and SRSF2 were more common in men compared to women (19% vs 2%, p = 0.005 and 13% vs 2%, p = 0.03, respectively). The number of mutated genes was not significantly different between de novo AML, sAML, and tAML (Fig. 1c). However, FLT3-ITD and NPM1 mutations were seen exclusively in de novo AML (p = 0.003 and p = 0.03, respectively). Patients with normal cytogenetics had higher rates of DNMT3A (25 vs 8%, p = 0.03) and FLT3-ITD (24 vs 8%, p = 0.04) mutations than patients who had abnormal karyotype. Finally, the frequency of persistent disease after initial induction chemotherapy was higher in patients with mutations in DNMT3A (47 vs 27%, p = 0.03) and U2AF1 (55 vs 28%, p = 0.01), while CRi was more common in patients with BCOR mutation (50% vs 8.5%, p = 0.02). Of note, all of the 6 patients with FLT3-TKD achieved CR after first induction (100% vs 55.3%, p = 0.03).
Clinical and genetic predictive features for outcomes after CR1 in intermediate-risk AML
We analyzed the impact of several prognostic variables on OS and RFS after CR1 in our cohort of intermediate-risk AML patients. In univariable analysis (Table 2), patients who achieved CRi after induction chemotherapy had a higher risk of death (HR, 2.4; 95% CI, 1.22–4.46; p = 0.01) and relapse (HR, 2.04; 95% CI, 1.1–3.6; p = 0.02) as compared to patients who achieved CR. Furthermore, patients who underwent HCT achieved better OS and RFS, represented by the unadjusted risk of 0.4 (95% CI, 0.22–0.72; p = 0.003) for death, and 0.34 (95% CI, 0.18–0.6; p = 0.0002) for relapse. Other clinical factors analyzed, such as age, sex, de novo vs. sAML/tAML, WBC count, extramedullary disease, cytogenetics, and the number of induction courses to achieve CR1 were not significant predictors for survival after CR1. Additionally, we could not demonstrate a difference in outcome when various donor sources and the intensity of conditioning regimen for HCT were compared.
Univariable analyses of gene mutations demonstrated that survival was significantly shorter for patients carrying mutations in DNMT3A, U2AF1, and EZH2 (Fig. 2). The median OS and RFS for DNMT3A mutated patients were 12 and 9 months respectively, as compared to 35 and 16 months in patients without mutation (p = 0.002 and 0.045; Fig. 2a, b). The presence of a DNMT3A mutation conferred an unadjusted risk of 2.46 (95% CI, 1.16–4.86; p = 0.02) for death, and 1.9 (95% CI, 0.95–3.37; p = 0.06) for relapse (Table 2). Patients with U2AF1 mutation had significantly worse OS (median, 9 vs 36 months, p = 0.0003) and RFS (median, 7 vs 16 months, p = 0.013) than patients without mutation (Fig. 2c, d), with an unadjusted risk of 3.61 (95% CI, 1.61–7.31; p = 0.003) for death, and 2.37 (95% CI, 1.08–4.64; p = 0.03) for relapse. Finally, the EZH2 mutation was associated with significantly shorter OS (median, 13 vs 26 months, p = 0.02) and RFS (median, 5 vs 15 months, p = 0.038) (Fig. 2e, f), with unadjusted risk of 3.75 (95% CI, 0.88–10.98; p = 0.06) and 3.26 (95% CI, 0.77–9.46; p = 0.09), respectively.

Kaplan–Meier overall survival (OS) and relapse-free survival (RFS) curves of intermediate-risk AML patients stratified based on their mutations in DNMT3A a, b, U2AF1 c, d, and EZH2 e, f in the Cleveland Clinic cohort
To verify whether the presence of these three mutations could improve the prognostic stratification of patients with intermediate-risk AML at CR1, we compared CC patients who harbored any of the three mutations with those who did not (Fig. 3). The risk stratification for OS was improved with the combined model as compared to the use of single mutations (Fig. 3a). The median OS for patients with mutations was 13 months, as compared to 41 months in patients without them (p < 0.0001). The median RFS was 9 vs 16 months (p = 0.003) (Fig. 3b). Similarly, when the mutation-based stratification was applied to intermediate-risk patients in the TCGA cohort, mutated patients had significantly poorer OS (median, 9.8 vs 46.8 months, p = 0.0002) and RFS (median, 9.3 vs 17 months, p = 0.02) (Fig. 3c, d). Moreover, after adjusting for the type of response (CR/CRi) and HCT status in multivariable analysis, the presence of the mutations maintained an independent effect on OS and RFS (HR, 3.22; 95% CI, 1.66–6.16; p = 0.0007 and HR, 2.17; 95%CI, 1.78–3.87; p = 0.01, respectively) (Table 3).

Kaplan–Meier overall survival (OS) and relapse-free survival (RFS) curves of intermediate-risk AML patients stratified based on their mutations in DNMT3A, U2AF1, and EZH2 in the Cleveland Clinic (CC) a, b and the Cancer Genome Atlas (TCGA) cohorts c, d
Outcomes of patients with DNMT3A, U2AF1, and EZH2 mutations based on the performance of HCT
Based on the finding that mutations and HCT were the major predictors of outcome after CR1, we analyzed the outcomes of patients harboring mutations in relation to HCT (Fig. 4). In both CC and TCGA cohorts, patients who did not have the mutations and underwent HCT had the best outcomes. In the CC cohort, the median OS and RFS for these patients were 45 months and not reached, respectively, as compared to 22 and 13 months for patients who did not undergo HCT (p = 0.05 for OS and p = 0.002 for RFS). In the TCGA cohort, performance of HCT did not have a significant impact on OS and RFS in patients without these mutations. However, HCT improved outcomes for patients who had these mutations in both cohorts. In the CC cohort, median OS and RFS for these patients who had HCT were 14 and 11 months, respectively, as compared to 7.5 and 5 months in patients who did not undergo HCT (p = 0.04 for OS and p = 0.002 for RFS). In the TCGA cohort, patients who harbored mutations and did not undergo HCT had significantly worse OS (median, 6.3 vs 24.6 months, p = 0.001) and RFS (median, 7.8 vs 12.5 months, p = 0.003), while there were no statistically significant differences between survival of patients with mutations who underwent HCT and patients who did not have the mutations.

Kaplan–Meier overall survival (OS) and relapse-free survival (RFS) curves of intermediate-risk AML patients, stratified based on the presence of mutations in DNMT3A, U2AF1, and EZH2, and performance of hematopoietic cell transplant (HCT) in the Cleveland Clinic (CC) a, b and the Cancer Genome Atlas (TCGA) cohorts c, d
In view of the recently proposed ELN 2017 criteria that place intermediate-risk patients with RUNX1 or ASXL1 mutations into the adverse risk category, which leads to the recommendation of HCT for this group, we explored outcomes of these patients at the time of CR1 (Supplementary Figure 1). Presence of these mutations did not predict OS and RFS in our cohort, and performance of HCT in mutated patients did not confer a statistically significant survival benefit.
Driver mutation dynamics and clinical course
In light of the above results, we next evaluated the serially collected samples from 10 patients (Fig. 5). The serial sequencing data reveal interesting and informative aspects about the clonal evolution of AML. The majority of patients harbored >1 clone at diagnosis, which were eradicated with intensive therapy. For example, patient 1 had DNMT3A and IDH2 mutations at VAF > 40%, which disappeared after 7 + 3. The patient underwent HCT in CR1 and was disease-free at 25 months follow-up. Patients 2 and 3 had U2AF1 mutated clones, which were absent following HCT in CR1. However, both of them relapsed shortly after HCT and died at 12 and 14 months follow-up, respectively. Additionally, patient 4 harbored three different mutations, which did not predict survival in this study, and they were eradicated with induction chemotherapy. However, the patient had evidence of an emerging DNMT3A clone, and relapsed 9 months after achieving CR1. Of interest, patient 5 had clones with FLT3-ITD, DNMT3A, RUNX1, and WT1 mutations, which persisted after chemotherapy and increased in size at the time of relapse. This patient died 2 months after the second sampling. Finally, patient 6 had a dominant BCORL1 clone, which disappeared with therapy, and relapsed with the emergence of new ASXL1 and TET2 mutations. The patient achieved CR3 after relapse therapy and was disease-free at 40 months follow-up. The evidence of clonal evolution for additional three patients is summarized in Supplementary Figure 2. We could not demonstrate driver mutations at diagnosis and during follow-up in one patient.

Dynamics of major driver mutations revealed by targeted sequencing and graphed with time charts showing changes in their variant allelic frequencies (VAFs) during disease progression in six representative patients
Discussion
The optimal post-remission consolidation therapy for intermediate-risk AML has been debated for years due to the clinical heterogeneity in outcomes of this group. These patients were sub-categorized based on normal vs abnormal cytogenetics (e.g., trisomy 8, MLLT3-MLL rearrangement) as intermediate-I and -II, respectively, but subsequent studies demonstrated no survival difference in between, which has led to the lumping of two groups in the recently proposed classification5,20. Considering the insufficiency of cytogenetic data to distinguish the differences in outcomes of this group, we hypothesized that driver mutations may have an effect in predicting outcomes of intermediate-risk AML and aid in clinical decision making with a particular focus on performance of HCT. Since achieving CR after intensive induction is a prerequisite for long-term survival in AML and 1-year mortality rate is >75% for primary refractory cases21, the present study focused on patients who achieved CR with 1 or more induction courses. This strategy enabled a more accurate assessment of the impact of HCT vs consolidation with chemotherapy on outcomes of patients stratified by mutational status. In two independent cohorts, we demonstrated that mutations in DNMT3A, U2AF1, and EZH2 genes were independent predictors of relapse and OS in patients who achieved CR, and performance of HCT in this group translated into a significant survival benefit.
One major difference between the CC and TCGA cohorts was that all patients in the latter had de novo AML, which accounted for higher WBC count, bone marrow blast percentage, and percentage of patients with normal cytogenetics in TCGA cohort. Donor sources were also slightly different for transplanted patients. However, none of these differences were found to predict survival in our univariable analysis. The cohorts were otherwise similar in terms of demographics, numbers of transplanted patients, and median survival durations.
The prognostic significance of DNMT3A and U2AF1 mutations in AML has been shown in a few other studies12,22,23. DNMT3A mutations in patients <60 years and U2AF1 mutations were independently associated with a lower CR rate after induction therapy12. In a large cohort of AML patients reported by Ley, et al.15, DNMT3A mutation was an independent predictor of poor survival in intermediate-risk cytogenetics group. Additionally, EZH2 inactivation was associated with poor prognosis in myelodysplastic syndromes, while its role in AML remains unclear24. In this study, patients with mutated EZH2 had significantly shorter OS and RFS, and a model integrating these three mutations could stratify intermediate-risk AML patients into two risk groups both in the CC and TCGA cohorts. Furthermore, eradication of these clones with induction chemotherapy, followed by HCT led to long-term survival in patients with available serial sequencing data. On the contrary, emergence or persistence of these mutations was associated with worse outcomes. Therefore, assessments of the mutations at diagnosis and during follow-up might offer an opportunity to improve prognostication and management of intermediate-risk AML. While patients with mutations who underwent HCT had improved OS and RFS, the median survival was still shorter than transplanted patients without mutations, indicating the importance of developing further therapies targeting these clones.
Integration of the accumulating information on gene mutations, cytogenetics, and other markers into risk stratification and management algorithms is a critical yet challenging task. Since the demonstration of favorable prognosis associated with NPM1 and biallelic CEBPA mutations, they have been incorporated into prognostic models and routinely tested in all patients at the time of diagnosis4,17. HCT is no longer recommended at CR1 for patients with these mutations, who were once classified in intermediate-risk category and transplanted without additional survival benefit. With rigorous efforts towards better risk-stratification in intermediate-risk group, several studies reported adverse outcomes in patients with RUNX1 and ASXL1 mutations25,26,27,28,29,30. These reports convinced the ELN panel and led to the re-classification of intermediate-risk patients harboring these two mutations in the poor-risk group5. However, when we stratified our cohort based on RUNX1/ASXL1 mutational status, no survival difference was appreciated. More importantly, performance of HCT did not extend survival at a significant level. These results might be attributed to two factors: First, our cohort was selected for patients who achieved CR and mutations in RUNX1/ASXL1 are known to be associated with refractory disease. Therefore, the poor prognostic impact of these mutations might disappear with the achievement of CR. The lack of survival difference, as well as no survival benefit in transplanted patients, might be due to a relatively lower number of patients enrolled. However, an important biological characteristic reported by multiple studies is that the mutations in RUNX1 and ASXL1 tend to co-occur, but are mutually exclusive with mutations in NPM1 and CEBPA2,3,12. While previous reports have shown independent prognostic value of RUNX1 and ASXL1, some focused only on cytogenetically normal AML without adjusting for NPM1 and biallelic CEBPA in multivariable models26,29, while in other reports, the impact on OS was lost when adjusted for these mutations25,27,30. Therefore, the survival differences in these studies are skewed, as RUNX1/ASXL1 mutated intermediate-risk patients were compared with those who had favorable NPM1 and CEBPA mutations. Based on the results of present study, and highlighted issues in previous reports, the utility of RUNX1 and ASXL1 mutations in stratifying intermediate-risk AML is debated and further investigation of HCT with these mutations is warranted.
There are potential limitations in our work, mainly related to the retrospective nature of this study. These include missing sequencing data and samples in a proportion of patients, different types of transplantation and conditioning regimens. Furthermore, in the absence of a matched control sample, distinguishing somatic and germline variants is challenging. Despite these limitations, clinical and molecular data were available in the majority of original patient population, and the results were successfully validated in an external cohort. In addition, the landscape of truly somatic mutations in analyzed genes has been well defined from large-scale genomic studies, which allowed us to make confident predictions. Finally, comparisons of HCT should be treated with caution, since no statistical method can adjust for the unmeasured selection factors involved in a retrospective analysis.
Collectively, our results provide evidence of clinical utility in considering mutation screening to stratify intermediate-risk AML patients after CR1 to guide therapeutic decisions. Mutations in DMT3A, U2AF1, and EZH2 might be useful to select patients who would benefit from HCT. On the contrary, RUNX1 and ASXL1 mutations were not as useful to predict patients with poor prognosis. A prospective validation of our findings is needed and we believe that our results may contribute to improving prognostication of patients with AML and the design of clinical trials.
References
Döhner, H. W. D. & Bloomfield, C. D. Acute myeloid leukemia. N. Engl. J. Med. 373, 1136–1152 (2015).
Network CGAR. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 368, 2059–2074 (2013).
Papaemmanuil, E. G. M. et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 374, 2209–2221 (2016).
Schlenk, R. F. et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N. Engl. J. Med. 358, 1909–1918 (2008).
Döhner, H. E. E. et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 129, 424–447 (2017).
Saygin, C. & Carraway, H. E. Emerging therapies for acute myeloid leukemia. J. Hematol. Oncol. 10, 93 (2017).
Cornelissen, J. J. & Blaise, D. Hematopoietic stem cell transplantation for patients with AML in first complete remission. Blood 127, 62–70 (2016).
Ferrara, F. Renaissance of autologous stem cell transplantation for AML?. Lancet Oncol. 13, 121–123 (2012).
Sengsayadeth, S. et al. Reduced intensity conditioning allogeneic hematopoietic cell transplantation for adult acute myeloid leukemia in complete remission - a review from the Acute Leukemia Working Party of the EBMT. Haematologica 100, 859–869 (2015).
Versluis, J. et al. Prediction of non-relapse mortality in recipients of reduced intensity conditioning allogeneic stem cell transplantation with AML in first complete remission. Leukemia 29, 51–57 (2015).
Cornelissen, J. J. et al. Comparative therapeutic value of post-remission approaches in patients with acute myeloid leukemia aged 40-60 years. Leukemia 29, 1041–1050 (2015).
Metzeler, K. H. et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood 128, 686–698 (2016).
Wang, B. et al. Mutational spectrum and risk stratification of intermediate-risk acute myeloid leukemia patients based on next-generation sequencing. Oncotarget 7, 32065–32078 (2016).
Patel, J. P. et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 366, 1079–1089 (2012).
Ley, T. J. et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 363, 2424–2433 (2010).
Daniel, A. et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127, 2391–2405 (2016).
Döhner, H. et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 115, 453–474 (2010).
Nazha, A. et al. Incorporation of molecular data into the Revised International Prognostic Scoring System in treated patients with myelodysplastic syndromes. Leukemia 30, 2214–2220 (2016).
Ding, L. et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481, 506–510 (2012).
Mrózek, K. et al. Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J. Clin. Oncol. 30, 4515–4523 (2012).
Schlenk, R. F. et al. Risk-adapted postremission therapy in acute myeloid leukemia: results of the German multicenter AML HD93 treatment trial. Leukemia 17, 1521–1528 (2003).
Hou, H. A. et al. Splicing factor mutations predict poor prognosis in patients with de novo acute myeloid leukemia. Oncotarget 7, 9084–9101 (2016).
Hamilton, B. K. et al. Impact of allogeneic hematopoietic cell transplant in patients with myeloid neoplasms carrying spliceosomal mutations. Am. J. Hematol. 91, 406–409 (2016).
Ernst, T. et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat. Genet. 42, 722–726 (2010).
Gaidzik, V. I. et al. RUNX1 mutations in acute myeloid leukemia: results from a comprehensive genetic and clinical analysis from the AML study group. J. Clin. Oncol. 29, 1364–1372 (2011).
Mendler, J. H. et al. RUNX1 mutations are associated with poor outcome in younger and older patients with cytogenetically normal acute myeloid leukemia and with distinct gene and MicroRNA expression signatures. J. Clin. Oncol. 30, 3109–3118 (2012).
Gaidzik, V. I. et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features. Leukemia 30, 2282 (2016).
Metzeler, K. H. et al. ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN Favorable genetic category. Blood 118, 6920–6929 (2011).
Schnittger, S. E. C. et al. ASXL1 exon 12 mutations are frequent in AML with intermediate risk karyotype and are independently associated with an adverse outcome. Leukemia 27, 82–91 (2013).
Paschka, P. et al. ASXL1 mutations in younger adult patients with acute myeloid leukemia: a study by the German-Austrian Acute Myeloid Leukemia Study Group. Haematologica 100, 324–330 (2015).
Acknowledgements
This work was supported by grants NIH/NHLBI, R01 HL118281, NIH/NHLBI, R01 HL123904, and NIH/NHLBI HL132071.
Authors’ contributions
C.S.: conceptualization, methodology, validation, formal analysis, investigation, visualization, writing-original draft and editing. C.H., B.P.: conceptualization, methodology, investigation, writing-review and editing. M.A.S., B.K.H., M.K., H.E.C., A.T.G., S.M., A.N., R.S.: conceptualization, methodology, supervision, writing-review and editing. C.G., D.A.: conceptualization, methodology, A.S.A.: conceptualization, methodology, validation, visualization, supervision, writing-review and editing, and project administration.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Saygin, C., Hirsch, C., Przychodzen, B. et al. Mutations in DNMT3A, U2AF1, and EZH2 identify intermediate-risk acute myeloid leukemia patients with poor outcome after CR1. Blood Cancer Journal 8, 4 (2018). https://doi.org/10.1038/s41408-017-0040-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-017-0040-9
This article is cited by
-
Epigenetic regulation in hematopoiesis and its implications in the targeted therapy of hematologic malignancies
Signal Transduction and Targeted Therapy (2023)
-
Clinical and prognostic profile of SRSF2 and related spliceosome mutations in patients with acute myeloid leukemia
Molecular Biology Reports (2023)
-
Estrogens revert neutrophil hyperplasia by inhibiting Hif1α-cMyb pathway in zebrafish myelodysplastic syndromes models
Cell Death Discovery (2022)
-
Mutational spectrum and prognosis in NRAS-mutated acute myeloid leukemia
Scientific Reports (2020)
-
CEBPE expression is an independent prognostic factor for acute myeloid leukemia
Journal of Translational Medicine (2019)
