
Abstract
3,3′,4′,5,7-Pentahydroxyflavone-3-rhamnoglucoside (rutin) is a flavonoid with a wide range of pharmacological activities. Dietary rutin is hardly absorbed because the microflora in the large intestine metabolize rutin into a variety of compounds including quercetin and phenol derivatives such as 3,4-dihydroxyphenolacetic acid (DHPAA), 3,4-dihydroxytoluene (DHT), 3,4-hydroxyphenylacetic acid (HPAA) and homovanillic acid (HVA). We examined the potential of rutin and its metabolites as novel histone acetyltransferase (HAT) inhibitors. DHPAA, HPAA and DHT at the concentration of 25 μM significantly inhibited in vitro HAT activity with DHT having the strongest inhibitory activity. Furthermore, DHT was shown to be a highly efficient inhibitor of p300 HAT activity, which corresponded with its high degree of inhibition on intracellular lipid accumulation in HepG2 cells. Docking simulation revealed that DHT was bound to the p300 catalytic pocket, bromodomain. Drug affinity responsive target stability (DARTS) analysis further supported the possibility of direct binding between DHT and p300. In HepG2 cells, DHT concentration-dependently abrogated p300-histone binding and induced hypoacetylation of histone subunits H3K9, H3K36, H4K8 and H4K16, eventually leading to the downregulation of lipogenesis-related genes and attenuating lipid accumulation. In ob/ob mice, administration of DHT (10, 20 mg/kg, iv, every other day for 6 weeks) dose-dependently improved the NAFLD pathogenic features including body weight, liver mass, fat mass, lipid accumulation in the liver, and biochemical blood parameters, accompanied by the decreased mRNA expression of lipogenic genes in the liver. Our results demonstrate that DHT, a novel p300 histone acetyltransferase inhibitor, may be a potential preventive or therapeutic agent for NAFLD.
Similar content being viewed by others

Introduction
Nonalcoholic fatty liver disease (NAFLD), ranging from steatosis to cirrhosis to hepatocarcinoma, is characterized by an abnormal accumulation of triglycerides (TG) in the liver without alcohol consumption [1]. NAFLD has been well established to not only be confined to liver-related morbidity and mortality but is also a multisystemic disease affecting several extrahepatic organs and regulatory pathways, such as the kidney, colon, bone, endocrine and cardiovascular systems [2,3,4]. Liver steatosis, marked as the first stage of NAFLD, is closely associated with obesity, type 2 diabetes mellitus, insulin resistance, and drug-induced liver injury [5].
Recently, several examples of dynamic changes in epigenetic markers due to environmental stimuli, such as stress and nutritional interventions, have been reported, including histone acetylation as a gene expression marker [6,7,8]. Few studies have provided evidence that the dysregulation of hepatic function is orchestrated by epigenetic mechanisms in NAFLD [9]. Among the phenomena of epigenetic modulation, histone acetyltransferase (HAT)-mediated acetylation of histone tail lysine residues is usually associated with the activation of gene transcription [10]. Indeed, altered expression and activity of certain histone acetylation modifying enzymes influence gene expression in NAFLD [11]. This may lead to the alteration of hepatic metabolism and cellular transformation through the regulation of various signaling pathways, such as hepatic lipid metabolism, insulin resistance, mitochondrial damage, oxidative stress, and inflammation. These pathways are implicated in the development and progression of NAFLD [12,13,14,15,16]. Recently, beneficial effects of natural dietary compounds on the prevention and treatment of NAFLD have been reported, but little research has been conducted to analyze the molecular mechanisms related to epigenetic modulation.
3,3′,4′,5,7-Pentahydroxyflavone-3-rhamnoglucoside (rutin) is a flavonoid under the flavonol subgroup that has a wide range of pharmacological properties, such as antioxidative, antimicrobial, antifungal, and anti-allergic properties [17]. In addition, current research has shown that its multispectrum pharmacological effects benefit the treatment of various chronic diseases, such as cancer, diabetes mellitus, hypertension, and hypercholesterolemia [18]. Dietary rutin is hardly absorbed because the microflora in the large intestine metabolize rutin into a variety of compounds, including quercetin and phenol derivatives such as 3,4-dihydroxyphenolacetic acid (DHPAA), 3,4-dihydroxytoluene (DHT), 3,4-hydroxyphenylacetic acid (HPAA), and 4-hydroxy-3-methoxyphenylacetic acid (homovanillic acid, HVA) [19, 20]. Recent studies have shown the suppressive effects of rutin metabolites on oxidative stress, inflammation, and advanced glycation end product (AGE) formation [20,21,22]; however, to date, no in-depth explanations have been published regarding the molecular mechanisms of rutin metabolites in NAFLD. This study is the first attempt to demonstrate the efficacy of rutin metabolites in preventing NAFLD and to explore their roles in epigenetic modulation.
In the present study, we investigated the potential of rutin metabolites as HAT inhibitors (HATis) and demonstrated that DHT is a potent HATi that successfully ameliorated NAFLD in both in vitro and in vivo NAFLD models. DHT suppressed HAT activity, notably p300, lipid accumulation, and lipogenic gene expression induced by a mixture of oleic and palmitic acids in HepG2 cells. In silico docking simulation and the DARTS assay revealed that DHT directly binds to the bromodomain of p300. In an obese mouse model, administration of DHT reduced body weight, liver mass, plasma TG, plasma LDL cholesterol, and lipogenic-related gene expression in the liver.
Materials and methods
Chemicals and reagents
Rutin (Cat. No. R5143), DHPAA (Cat. No. 850217), DHT (Cat. No. M34200), HPAA (Cat. No. H49901), and HVA (Cat. No. H1252) in powder form were purchased from Sigma-Aldrich (St. Louis, MO, USA) and dissolved in DMSO for the experiments. Oleic acid, palmitic acid, and oil red O staining solution were also purchased from Sigma-Aldrich. Water-soluble tetrazolium salt (WST-1) used to measure cytotoxicity was obtained from Enzo Life Sciences, Inc. (Farmingdale, NY, USA).
Cell culture
HepG2 cells were purchased from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in a humidified atmosphere with 5% CO2 at 37 °C in high-glucose Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and antibiotics (Coring, NY, USA). A nonfat bovine serum albumin-conjugated combination of oleic acid (Sigma-Aldrich, St. Louis, MO, USA) and palmitic acid (Sigma) at a ratio of 4:1 (OPA) was used to establish an NAFLD model using HepG2 cells. To evaluate the effect of rutin and its metabolites on hepatic lipid accumulation, HepG2 cells were concurrently treated with the indicated concentrations of OPA for 18 h.
Cytotoxicity
HepG2 cells were seeded in 24-well plates at a density of 5 × 104 cells/well. Next, at ~70% confluency, cells were treated with the indicated concentrations of rutin metabolites in each experiment in the presence or absence of OPA. After a 24-h incubation, WST-1 solution (Enzo Life Sciences) was added to the wells, and the cells were incubated for 3 h. Thereafter, the absorbance values were measured at 450 nm (Molecular Devices, Sunnyvale, CA, USA).
Lipid content
HepG2 cells were seeded in 24-well plates at a density of 5 × 104 cells/well. After reaching ~70% confluency, the cells were treated with OPA in the presence or absence of rutin metabolites. After a 24-h incubation, the cells were fixed with 4% paraformaldehyde for 15 min at room temperature and were then incubated with 60% isopropanol for 5 min, followed by staining with 0.1% oil red O staining solution (Sigma-Aldrich) for 15 min. The cells were then washed with water, and images were captured under a light microscope (Olympus IX51; Olympus Corporation, Central Valley, PA, USA). For lipid quantification, isopropanol was added to each well to dissolve the red dye. After 10 min, the absorbance values were measured at 510 nm (Molecular Devices).
Histone extraction
HepG2 cells were seeded at a concentration of 5 × 106 in 100-mm dishes. After the cells reached 70% confluency (~2 × 108 cells), the cells were treated for 18 h with the indicated concentrations of OPA, with or without metabolites of rutin. The histone proteins from HepG2 cells were extracted using a Histone Extraction Kit (Abcam, Cambridge, MA, USA) according to the manufacturer’s protocol.
In vitro HAT and histone deacetylase (HDAC) assays
HAT and HDAC activities in the HeLa nuclear extracts (BioVision, Milpitas, CA, USA) were assessed using a commercially available kit according to the manufacturer’s protocol (BioVision). To examine the inhibitory effect of DHT on p300, 100 ng of p300 recombinant protein (Enzo Life Science) was used as the enzyme source instead of the nuclear extracts. For autoradiography-based in vitro HAT activity assays, either the HeLa cell nuclear extract or the p300 recombinant protein was incubated with HAT assay buffer (50 mM HEPES, pH 8.0; 10% glycerol; 1 mM dithiothreitol [DTT]; 1 mM phenylmethylsulfonyl fluoride; and 10 mM sodium butyrate), 1 µL of [3H] acetyl-CoA, and 5 µg of biotinylated-H4 peptide (Millipore, Billerica, MA, USA) along with metabolites of rutin at the indicated concentrations at 30 °C for 1 h. The reactions were stopped by adding 5× sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS–PAGE) sample buffer. Proteins from each sample were then separated on 15% SDS–PAGE gels and visualized via autoradiographic analysis.
Quantitative reverse-transcription polymerase chain reaction (qRT-PCR)
Cells were seeded in 24-well plates at 5 × 104 cells/well. At ~70% confluency, the cells were treated with the indicated concentrations of DHT in the presence or absence of OPA. After an 18-h incubation, total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). qRT-PCR was performed using an I Cycler iQ system (Bio-Rad Laboratories, Hercules, CA, USA) with SYBR Green PCR master mix (Thermo Fisher Scientific, Waltham, MA, USA). PCR amplification was carried out in triplicate using the primers listed in Supplementary Table 1. mRNA levels were normalized to those of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA, and relative expression levels were calculated using the comparative ΔΔCT method.
Western blot analysis
Cells were grown and treated in a similar fashion as those used for qRT-PCR. Cell extracts were then prepared using lysis buffer (Cell Signaling, Danvers, MA, USA) containing protease and phosphatase inhibitors (Roche, Basel, Switzerland) and were incubated on ice for 30 min. The lysates were centrifuged at 20,000 × g for 20 min at 4 °C. The cell lysates were separated on SDS–PAGE gels and transferred to cellulose membranes (Whatman, Dassel, Germany). The membranes were blocked with 5% (w/v) skimmed milk (BD Biosciences, Sparks, MD, USA) solution in 1× phosphate buffered saline (PBS) containing Tween-20 (PBST) for 1 h. The blocked membranes were incubated overnight with the indicated primary antibodies at 4 °C (Supplementary Table S2). The membranes were then washed with 1× PBST, incubated with the appropriate secondary anti-rabbit or anti-mouse horseradish peroxidase-conjugated antibody (Thermo Scientific, Rockford, IL, USA) for 1 h, and visualized using Fusion Solo 6 S (Vilber Lourmat, Collegien, France) with an enhanced chemiluminescence detection reagent (Thermo Scientific).
Histone tail binding assay
In the biotinylated histone peptide-binding assay, 1 µg of biotin-labeled Lys acetylated histone H3 (AcH3) tail peptide (Anaspec, Fermont, CA, USA) was bound to NeutrAvidin (Pierce); pull-down assays were performed with flag-tagged recombinant p300 (1041–1161 amino acid) containing the bromodomain (Active Motif, Carlsbad, CA, USA). The binding assay was performed at 4 °C for 2 h in binding buffer [20 mM Tris-HCl (pH 7.1), 120 mM KCl, 1 mM DTT, 1 mM ethylenediaminetetraacetic acid, 0.1% Nonidet P-40 (NP-40), 10% glycerol, and 1 mM PMSF]. The histone tail and protein complexes were separated via SDS–PAGE and detected using a flag antibody (Sigma). The biotinylated AcH3 tail was stained with Ponceau S solution (Sigma) to demonstrate the loading control.
Drug affinity responsive target stability (DARTS) assay
The DARTS assay was performed according to a previously described protocol [23]. HepG2 cells were incubated in 10-cm dishes until 80% confluency, followed by lysis in M-PER buffer (Pierce, Rockford, IL, USA), which contains 30 mM NaCl, protease, and phosphatase inhibitors. After centrifugation (14,000 × g, 10 min, 4 °C), 10× TNC buffer [500 mM Tris-HCl (pH 8.0), 500 mM NaCl, 100 mM CaCl2] was added to the lysates prior to the determination of protein concentration using a DC protein assay kit (Bio-Rad Laboratories), according to the manufacturer’s instructions. The proteins in the supernatant (2 μg/μL) were digested with pronase (1:1,000,000 protein to pronase ratio) for 30 min. Digestion was stopped by adding 5× Laemmli sample buffer, followed by denaturation at 95 °C for 5 min.
Cell and liver tissue fractions
Cells were seeded at a concentration of 5 × 106 in 100-mm dishes. After reaching ~70% confluency (~2 × 108 cells), the cells were treated with the indicated concentrations of OPA, with or without the metabolites of rutin, for 18 h. Harvested cells were lysed in lysis buffer [10 mM Tris (pH 7.4), 10 mM NaCl, 3 mM MgCl2, 0.5% NP-40, and protease inhibitor cocktail (Roche)] at 3000 r/min and 4 °C for 3 min, and the resulting supernatants were used as cytosol fractions. Nuclear fractions from HepG2 cells were obtained by following the manufacturer’s protocol (Abcam).
Liver tissues obtained at the end of the animal experiments were washed with ice-cold PBS and lysed according to the manufacturer’s protocol (Abcam) to obtain the cytosolic and nuclear fractions. Briefly, tissues were weighed and cut into small pieces. To obtain the cytosolic fractions, tissue pieces were homogenized in pre-extraction buffer containing DTT using a glass tissue homogenizer (Thomas Scientific, NJ, USA). Extraction buffer was added to the pellets, and the resulting suspensions were incubated on ice for 15 min and were then further homogenized using a Teflon pestle (Thomas Scientific). The suspensions were then centrifuged at 14,000 r/min at 4 °C for 10 min, and the supernatants were used as nuclear fractions. Each nuclear fraction was used for the HAT assays.
Animal experiments
Seven-week-old male ob/ob mice were purchased from Orient Bio (Seongnam, Gyeonggi, Korea) and housed at the College of Medicine of Ulsan University. All animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Ulsan University Guide for the Animal Care and Use Committee (2017-02-148). A total of 20 male ob/ob mice were divided into three groups (2 or 3 mice/cage) and housed in a temperature- and humidity-controlled room with a 12-h light/dark cycle, with free access to food and water. After a 1-week acclimation period, saline (control group), 10 mg, or 20 mg/kg DHT was injected into the lateral tail vein of each mouse once every alternate day. Body weights were measured at the beginning of the experiment and subsequently every week for 6 weeks. At the end of the experiment, mice were sacrificed by cervical dislocation under anesthesia (100 mg/kg ketamine + 5 mg/kg xylazine), and blood samples were collected for serum isolation via abdominal heart puncture. After laparotomy, the liver, retroperitoneal fat, and epididymal fat were weighed, and a portion of the liver from each mouse was fixed in 4% formalin solution for hematoxylin and eosin (H&E) staining. The remaining livers were used for the experiments as indicated.
H&E staining
Liver specimens were fixed in 4% buffered formalin, embedded in paraffin, and cut into 4–5-μm-thick sections, which were stained with H&E. Lipid accumulation in each liver specimen was assessed through microscopic observation using an Eclipse 80i microscope (Nikon Instruments, Inc., Melville, NY, USA).
Measurement of triglycerides (TGs), total cholesterol, and low-density lipoprotein cholesterol (LDL-C)
The serum levels of TGs, total cholesterol, and LDL-C were measured enzymatically using a commercial kit (Asan Pharm, Seoul, Korea).
Protein–ligand docking simulations for the E1A binding protein p300 (EP300)
To identify the active site in EP300, the three-dimensional X-ray crystallographic structure of EP300 was downloaded from the Protein Data Bank (http://www.rcsb.org/pdb/home/home.do) (PDB ID: 4BHW) [24]. The three-dimensional structure of DHT used as a ligand for docking was downloaded from the PubChem site (CID: 9958) (https://pubchem.ncbi.nlm.nih.gov) [25]. The specific simulation methods were described in detail in a previous study by Chung et al. [11].
Statistical analysis
Data were analyzed using Student’s t-test for comparing two groups or one-way analysis of variance (ANOVA) with Tukey’s multiple comparison test for comparing multiple groups, and values are expressed as the means ± standard deviations (SD) or means ± SEM. Statistical analyses were conducted using SPSS ver. 20 (SPSS Inc., Chicago, IL, USA). Differences were considered statistically significant at P < 0.05, P < 0.01, and P < 0.001.
Results
DHT has the most significant inhibitory effect on HAT activity
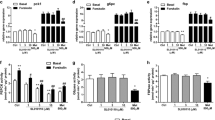
Initially, to test whether rutin and its metabolites (Fig. 1a) can modulate HAT activity, we measured HAT activity using a cell-free system. Rutin (100 µM) or one of its metabolites, including DHPAA, HPAA, DHT, and HVA, was incubated with HeLa nuclear extract. The results indicated that DHT had the most significant inhibitory effect on HAT activity compared to that of the other metabolites (Fig. 1b). To confirm the anti-HAT activity of DHT, an autoradiography assay was performed using radiolabeled H4 tail peptides. Consistent with the results of the in vitro HAT activity assay, DHT exhibited the most powerful HATi effect (Fig. 1c), although the effects of DHPAA and HPAA in the cell-free system and the autoradiography assay were slightly different from the in vitro HAT activity. To ensure that the activities of rutin and its metabolites, including DHT, were specifically directed against HATs, we assessed their effects on HDAC activities. Trichostatin A (TSA), used as a positive control, efficiently inhibited nuclear HDAC activities, whereas rutin and its metabolites did not affect HDAC activity (Fig. 1d). Together, these data showed that DHT, among the rutin metabolites, most efficiently abrogated HAT activity without affecting HDAC activity.

a The chemical structures of rutin, 4-dihydroxyphenolacetic acid (DHPAA), DHT, 3,4-hydroxyphenylacetic acid (HPAA), and 4-hydroxy-3-methoxyphenylacetic acid (homovanillic acid, HVA). b Histone acetyltransferase (HAT) activity of rutin metabolites in the in vitro cell-free system. A colorimetric HAT assay was conducted according to the manufacturer’s guidelines on a cell-free system subjected to 25 μM rutin and its metabolites. The results are shown as percentages relative to the control (in red), and the values presented are the means ± standard deviation (SD) of three independent experiments. Student’s t-test; *P < 0.05, **P < 0.01, and ***P < 0.001. c In vitro HAT assay using autoradiography to evaluate the efficacy of the HAT inhibitory effect of rutin and its metabolites on the acetylation of the synthetic H4 tail peptide by HeLa nuclear extract (NE). Rutin and its metabolites were incubated with HeLa NE (HAT enzyme source), acetylated-3H (acetyl donor), and biotinylated-H4 peptide (acetyl recipient) under the conditions mentioned in the Materials and Methods section. The relative band intensity of the biotinylated H4 peptide was normalized to that of Coomassie brilliant blue (CBB) staining. d Histone deacetylase (HDAC) activity of rutin and its metabolites in the cell-fee system in vitro. A colorimetric HDAC assay was conducted according to the manufacturer’s guidelines on a cell-free system subjected to 25 μM rutin and its metabolites. The results are shown as percentages relative to the control (in red), and the values presented are the means ± SD of three independent experiments. Student’s t-test; ***P < 0.001. Trichostatin A (TSA), an HDAC inhibitor, was used as a negative control (in blue).
DHT selectively inhibits p300 acetyltransferase activity
To further confirm the effect of DHT as a HATi, the changes in both HAT and HDAC activities were measured with DHT at various concentrations in a cell-free system. DHT diminished HAT activity in a dose-dependent manner (Fig. 2a), whereas HDAC activity was not affected by DHT (Fig. 2b). Next, we examined the enzyme specificity of DHT. The HAT activities of p300, CREB-binding protein (CBP), and p300/CBP-associated factor (pCAF) were measured either in the absence or presence of increasing concentrations of DHT. DHT was shown to be a highly efficient inhibitor of p300 acetyltransferase activity (Fig. 2c). The p300 inhibitory effect of DHT was supported by the results of an autoradiography assay using a radiolabeled H4 tail peptide (Fig. 2d). Collectively, these results showed that DHT specifically inhibited p300 acetyltransferase activity.

a Histone acetyltransferase (HAT) activity of DHT in an in vitro cell-free system. A colorimetric HAT assay was conducted using a cell-free system, with the indicated concentrations in accordance with the instrument’s guide. The results are shown as percentages relative to the control (in red), and the values presented are the means ± standard deviation (SD) of three independent experiments. ***P <0.001 (one-way analysis of variance [ANOVA] followed by Tukey’s multiple test). b Histone deacetylase (HDAC) activity of DHT in an in vitro cell-free system. A colorimetric histone deacetylase (HDAC) assay was conducted using a cell-free system, with the indicated concentrations in accordance with the instrument’s guide. The results are shown as percentages relative to the control (in red), and the values presented are the means ± SD of three independent experiments. ***P < 0.001 (Student’s t-test). Trichostatin A (TSA), an HDAC inhibitor, was used as a negative control (in blue). c Inhibitory effect of DHT on global HAT in an in vitro cell-free system. The enzyme-specific HAT inhibitory capacity of DHT against p300, CBP, and pCAF was measured using purified enzymes. A colorimetric histone acetyltransferase (HAT) assay was conducted using a cell-free system, with the indicated concentrations in accordance with the instrument’s guide. The results are shown as percentages relative to the control (in red), and the values presented are the means ± SD of three independent experiments. ***P < 0.001 (one-way analysis of variance [ANOVA] followed by Tukey’s multiple test). d In vitro HAT assay using autoradiography to evaluate the efficacy of the p300 inhibitory effect of DHT on the acetylation of the synthetic H4 tail peptide by the p300 recombinant protein. DHT was incubated with p300 recombinant protein (HAT enzyme source), acetylated-3H (acetyl donor), and biotinylated-H4 peptide (acetyl recipient) under the indicated conditions in the Materials and Methods section. The relative band intensity of the biotinylated H4 peptide was normalized to that of CBB staining.
The antilipogenic effect of DHT is associated with its HAT inhibitory capacity
We previously demonstrated that HAT activity increases in NAFLD in vitro and in vivo [11]. To determine whether the rutin metabolites attenuated the increased HAT activity found in HepG2 cells undergoing lipid accumulation, we measured the change in HAT activity after exposing an OPA-induced NAFLD model to rutin metabolites. HepG2 cells with or without various rutin metabolites were treated with OPA for 24 h. Nuclear extracts were then used to evaluate HAT enzyme activity. OPA-induced HAT activity was significantly inhibited by DHPAA, HPAA, DHT, and HVA (Fig. 3a). Interestingly, similar to the previous result (Fig. 1b), DHT showed the strongest HATi effect. To observe the correlation between HATi activity and intracellular lipid accumulation in HepG2 cells, oil red O staining was performed under the same conditions as above. The inhibition of lipid accumulation by rutin metabolites was also similar to that of the observed HATi activity (Fig. 3b). The results showed that the metabolites were not cytotoxic at the concentration used (Fig. 3c). To check the effect of different concentrations of DHT on HAT activity and lipid accumulation, we treated cells with 25–50 μM DHT and remeasured their corresponding HATi effects and degrees of inhibition of lipid accumulation. DHT showed both HATi and antilipogenic effects in a dose-dependent manner (Fig. 3d, e). Next, RT-qPCR was conducted to determine whether DHT was implicated in the expression of acetyl-CoA carboxylase (ACC), ATP-citrate lyase (ACLY), fatty acid synthase (FASN), and sterol regulatory element-binding protein 1c (SREBP1c). As expected, there was a significant increase in the mRNA expression of these genes in the OPA-treated group; mRNA expression significantly decreased in OPA-treated groups that were exposed to DHT (Fig. 3f). These data suggest that DHT decreased lipid accumulation in hepatic cells by abrogating the transcription of lipogenesis-related genes via its HATi capacity.

a Histone acetyltransferase (HAT) activity in the nuclear extract (NE) from HepG2 cells. HepG2 cells were treated with a combination of oleic and palmitic acid (OPA), with or without (w/wo) exposure to rutin and its metabolites, for 24 h. The cells were harvested, and nuclear proteins were extracted, which were then used as an enzyme source for the following HAT assay. A colorimetric HAT assay was conducted according to the manufacturer’s guidelines on a cell-free system subjected to 25 μM rutin and its metabolites. The results are shown as percentages relative to the control (in red), and the values presented are the means ± standard deviation (SD) of three independent experiments. Student’s t-test; ###P < 0.001 (control vs. OPA-treated group), ***P < 0.001 (vs. OPA-treated group) (left panel). To ensure cell fractionation, Western blotting was performed. HDAC1 and tubulin were used as nuclear and cytosolic markers, respectively (right panel). b Effect of rutin and its metabolites on OPA-induced lipid accumulation. HepG2 cells were treated for 24 h with OPA, w/wo 25 μM rutin or its metabolites, and were then stained with oil red O solution. The stained cells were destained with isopropanol; for lipid quantification, the stained cells were dissolved, and their absorbance values were then measured at 510 nm. The results are shown as percentages relative to the control (in red), and the values presented are the means ± SD of three independent experiments. Student’s t-test; ###P < 0.001 (control vs. OPA-treated group), **P < 0.01 and ***P < 0.001 (vs. OPA-treated group). c The effect of rutin and its metabolites on cellular viability. HepG2 cells were treated with OPA, w/wo 25 μM rutin or its metabolites, for 24 h, and cytotoxicity was measured using the WST assay. d The HAT inhibitory effect of DHT in HepG2 cells. HAT activity was measured in the NE from HepG2 cells. HepG2 cells were treated with OPA at the indicated concentrations, w/wo DHT, for 24 h. The subsequent process was the same as that in Fig. 3a. The results are shown as percentages relative to the control (in red), and the values presented are the means ± SD of three independent experiments. ###P < 0.001 (Student’s t-test); ***P < 0.001 (one-way analysis of variance [ANOVA] followed by Tukey’s multiple test) (left panel). To ensure cell fractionation, Western blotting was performed. HDAC1 and tubulin were used as nuclear and cytosolic markers, respectively (right panel). e DHT efficacy in attenuating OPA-induced lipid accumulation in HepG2 cells. HepG2 cells were treated with OPA, w/wo 25 μM DHT, for 24 h and were then stained with an oil red O solution. Representative photographs (right panel); staining was quantified the same way as in Fig. 3b. The results are shown as percentages relative to the control (in red), and the values presented are the means ± SD of three independent experiments. ###P < 0.001 (Student’s t-test); ***P < 0.001 (one-way analysis of variance [ANOVA] followed by Tukey’s multiple test). f Effect of DHT on the mRNA expression of lipogenesis-related genes. HepG2 cells were incubated with OPA in the presence or absence of DHT for 18 h. RNA was extracted from the cells; the corresponding cDNA was synthesized and used as a template for qRT-PCR. The mRNA expression of ACC1, ACLY, FASN, and SREBP1c was measured. The results are shown as percentages relative to the control (in red), and the values presented are the means ± SD of three independent experiments. ###P < 0.001 (Student’s t-test); ***P < 0.001 (one-way analysis of variance [ANOVA] followed by Tukey’s multiple test).
DHT binds to the bromodomain of p300 and inhibits p300–histone complex formation
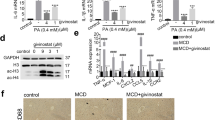
Previously, we demonstrated the selective p300 inhibitory effect of DHT. We then examined the binding affinity of DHT and p300 through docking simulation. The most likely binding site on p300 was the catalytic pocket for the bromodomain. We identified the top five docking structures with free energy values between −7.181 kcal/mol and −6.547 kcal/mol (Fig. 4a). In particular, the most likely binding residues were PRO1074, TYR1089, MET1124, ASN1127, ALA1128, and TYR1131 (Fig. 4b). To confirm DHT-p300 binding at the molecular level, we conducted a DARTS assay using HepG2 cells. The binding of chemicals to specific proteins increases protein stability and resistance to proteases. The cells were treated with DHT, and the lysates were then digested with pronase, a mixture of proteases, as indicated. DARTS analysis revealed that DHT directly bound to p300, and the resistance of p300 to pronase increased proportionately to the DHT concentration (Fig. 4c). It is well established that the bromodomain of p300 is responsible for its histone binding [26]. Based on this, we hypothesized that docking of DHT in the p300 bromodomain would inhibit histone binding with p300 and would consequently suppress histone acetylation. To support our hypothesis, we performed a histone tail binding assay. Biotinylated AcH3 and flag-tagged p300 recombinant protein containing the bromodomain were incubated with or without DHT (Fig. 4d). Then, biotinylated AcH3 was immunoprecipitated with avidin beads, and the bound p300 protein was detected using an anti-flag antibody. Remarkably, our results demonstrated that the binding of p300 and histone H3 decreased as the concentration of DHT increased (Fig. 4d). To observe whether a decrease in the binding of p300 and histone protein would actually lead to a reduction in histone acetylation, we detected the histone acetylation status following DHT treatment. DHT effectively obstructed the OPA-induced hyperacetylation of the histone subunits H3K8, H3K36, H4K8, and H4K16 (Fig. 4e). Together, these data indicate that DHT interrupted p300-histone protein binding by docking at the histone binding site of p300 and consequentially inhibiting hyperacetylation of histone proteins caused by OPA-induced lipid accumulation.

a Top five docking results of DHT with p300. DHT was expected to bind to the p300 catalytic pocket bromodomain. Gray ribbon, p300; the structure is presented in cylinder mode. Free energy (lowest energy+(-kTlnN), k: Boltzmann constant, T: Kelvin temperature, N: the number of structures in each cluster) was used to select a number of ligands with the lowest energy in the cluster. b The most likely DHT-p300 docking is presented. Gray ribbon, p300; the structure is presented in cylinder mode (upper left). The catalytic pocket surface containing the bromodomain of p300 is shown in orange (upper right). The most likely binding residues for DHT docking in the p300 bromodomain were predicted. Gray ribbon, p300; the structure is presented in cylinder mode. c DHT directly binds to p300 and increases p300 stability in HepG2 cells. To assess the binding of DHT to p300, the DARTS assay was conducted. For the DARTS assay, HepG2 cell lysates were incubated with DHT at the indicated concentrations for 1 h. The lysates were digested with pronase and subjected to Western blotting (left and middle panels). The bands were quantified by normalizing the intensity values using the control (in red) using ImageJ software (right panel). d DHT inhibited the binding of p300 to histone protein. Biotinylated AcH3 and flag-tagged p300 recombinant protein containing the bromodomain were incubated with or without DHT. Biotinylated-AcH3 is immunoprecipitated with avidin beads, and then the histone bound-p300 is detected using an anti-flag antibody by Western blotting (upper panel). The biotinylated AcH3 was visualized using Ponceaus S staining (lower panel). The relative band intensity of histone-bound p300 was normalized to that of biotinylated AcH3. e DHT suppresses OPA-induced hyperacetylation of histones. DHT-treated HepG2 cells were treated with the indicated concentrations of OPA for 24 h. Core histones were extracted from the cells, and the acetylation status of the indicated lysine residues of histones H3 and H4 was detected by Western blot assays (left panel). To confirm histone extraction, CBB staining was performed (right panel). The relative band intensity of histone acetylation marks (H3K9ac, H3K36ac, H4K8ac, and H4K16ac) was normalized to that of their corresponding histone (H3 or H4) in CBB staining.
DHT administration improves the NAFLD pathogenic features in vivo
To strengthen the previously gathered data, we examined the effect of DHT on significant NAFLD features. A dose of 10 or 20 mg/kg DHT was injected into the lateral tail vein once every alternate day for 6 weeks in ob/ob mice (Fig. 5a). The DHT-injected mouse groups demonstrated a lower average body weight than the control mouse group (Fig. 5b). The average weight gain was also significantly lower in the DHT-injected groups than in the control group (Fig. 5c), and there were no significant differences in daily food intake between the DHT-injected mice and the control mice (P < 0.05) (Fig. 5d). Enlarged tissue sizes of the liver and retroperitoneal and epididymal fat and increased tissue mass were observed in the control group but not in the DHT-injected group (Fig. 5e, f). In H&E-stained liver tissue, less lipid accumulation was detected in DHT-injected mice than in control mice (Fig. 5g). In the blood test, TG, LDL-C, alkaline phosphatase (ALP), and alanine aminotransferase (ALT) levels effectively decreased following DHT treatment (Fig. 5h). Moreover, the mRNA expression of lipogenic genes, such as FASN, ACLY, and PPARγ, decreased in the DHT-injected groups (Fig. 5i). These results established that DHT administration ameliorated NAFLD peculiarities in ob/ob mice.

a A workflow of the animal experiment. The procedure of the experiment is described in detail in the Materials and methods section. b Body weight changes measured every week in the mice injected with 10 or 20 mg/kg DHT via their tail vein every other day for 6 weeks (n = 6/group). The values presented are the means ± SEM. Means with different superscript letters are significantly different, P < 0.05 (one-way analysis of variance [ANOVA]). c Body weight gain is calculated by subtracting the weights of the mice at the start from their weights at the end of the experiment. The values presented are the means ± SEM. One-way ANOVA was used for statistical analysis (P < 0.05). d Daily food intake was measured for 6 weeks and represented as kilocalories per day (kcal/daily) (n = 6/group). The values presented are the mean ± SEM. One-way ANOVA was used for statistical analysis. n.s. not significant. e Representative images of the liver, epididymal fat, and retroperitoneal fat from mice. f Average values for the liver epididymal fat and retroperitoneal fat mass after a 6-week DHT treatment are shown as the means ± SEM (n = 6/group, right panel). **P < 0.01; ***P < 0.001 (one-way analysis of variance [ANOVA] followed by Tukey’s multiple test). g After a 6-week DHT treatment, hematoxylin and eosin (H&E) staining of mouse liver specimens was carried out. Representative images are shown. h The serum levels of TG, LDL-C, ALP, and ALT were measured following the 6-week DHT treatment (n = 6/group). The values presented are the means ± SEM. *P < 0.05, **P < 0.01 (one-way analysis of variance [ANOVA] followed by Tukey’s multiple test). i DHT inhibited the mRNA overexpression of lipogenic genes in mouse livers. After a 6-week DHT injection, the mRNA expression levels of FASN, ACLY, and PPARγ2 were measured by qRT-PCR. The values presented are the means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (one-way analysis of variance [ANOVA] followed by Tukey’s multiple test).
Discussion
NAFLD is one of the major health concerns worldwide. Its development and progression are affected by various factors, such as genetics, epigenetics, and environmental factors. A large number of studies have provided evidence to support that the critical components implicated in the progression of NAFLD pathogenesis are epigenetically regulated [27,28,29]. In addition, recent studies have shown that epigenetic modifications are reprogrammed for NAFLD [30, 31]. For this reason, there is increasing interest in developing new preventive or therapeutic applications that target NAFLD based on epigenetics owing to its reversible properties. In particular, epigenetic regulation using natural compounds with favorable safety profiles is expected to be a novel preventive or therapeutic strategy for metabolic syndromes, including NAFLD [7, 32, 33]. In a preliminary study, we observed that sprouts containing a large amount of rutin, which has known beneficial effects against metabolic diseases, show anti-HAT capacity. Based on our preliminary observations, in this study, we elucidated the in-depth epigenetic mechanism underlying NAFLD control by rutin and its metabolites at the molecular level.
Initially, we investigated the anti-HAT efficacy of rutin and its metabolites, namely, DHPAA, HPAA, DHT, and HVA. Our data showed that DHPAA, HPAA, and DHT showed potential in effectively inhibiting HAT activity. In particular, DHT, a phenol derivative of rutin, demonstrated the most potent HAT inhibitory effect among all the rutin metabolites. Several small molecules targeting HAT that have been derived from natural products, such as anacardic acid, curcumin, and garcinol [34, 35], contain phenolic structures that are prone to oxidation; thus, it is occasionally difficult to distinguish whether their efficacy in disease models is due to the antioxidant effect or the anti-HAT effect. Chen et al. reported that DHT had the most significant inhibitory effect on LPS-induced NO production and inflammatory cytokine production among rutin metabolites [20]. Recently, a study indicated that DHT is a plausible rutin colonic metabolite that suppresses oxidative stress by enhancing peroxiredoxin-6 protein expression, a protein responsible for the cellular antioxidant defense system [21]. These studies allow us to indirectly or directly infer that the beneficial effects of DHT are mediated by its antioxidant effect. However, to the best of our knowledge, no studies have shown the efficacy of DHT as a HAT inhibitor. Our data clearly demonstrate that HAT activity is regulated in a DHT dose-dependent manner. Based on the above facts, we believe that it is justifiable to identify the bioactive effects of DHT based on its HAT inhibitory capacity.
The acetylation modification of proteins in the liver is associated with a broad range of cellular activities. This is supported by a study showing that ~1000 proteins in human liver tissue are subjected to lysine acetylation modification [36]. The development and progression of NAFLD is no exception. A pregnant woman’s high-fat diet was shown to trigger the development of NAFLD in the fetal liver; statistically significant hyperacetylations and gross alterations of acetylation at the H3K14, H3K9, and H3K18 subunits have also been observed [37]. Global histone H3 acetylation also increases in a serotonin reuptake inhibitor-induced NAFLD model [38]. We recently reported that increased HAT activity following treatment with a mixture of oleic and palmitic acids (OPA) induces histone hyperacetylation and promotes NAFLD phenotypes such as steatosis and lipogenic gene expression in HepG2 cells [39]. Our data showed that histones H3 and H4 were hypoacetylated in an OPA-induced NAFLD model in HepG2 cells. This result was attributed to the increased HAT activity in the model. Interestingly, DHT effectively blocked the phenomena through its anti-HAT activity, especially by selectively inhibiting p300. According to our data, the effect of HATs appeared to be tissue-specific; in the differentiation of preadipocytes (3T3-L1) to adipocytes, pCAF activity was more significant than p300 activity (data not shown). P300 dynamically upregulates many transcription factors, including SREBP1c and PPARγ, which are responsible for lipogenesis [40, 41], and consequently enhances the expression of lipogenic genes in NAFLD [42]. Thus, regardless of its molecular mechanism, these results show that inhibition of p300 activity can serve as a key to prevent the development and progression of NAFLD.
In the present study, we provided a plausible mechanism underlying p300 inhibition caused by the derivative of a natural compound. To explain this, we proved the mode of action of DHT as a p300 inhibitor through the DARTS assay and docking simulations. The results demonstrated that DHT bound to p300, and the docking site was predicted to be a bromodomain located in the p300 catalytic pocket. The domain not only increases p300 activity by providing a docking surface for acetyl-CoA, the intracellular acetyl donor [43], but also plays an important role in activating the target level of gene transcription through binding with histone H3 [44]. The bromodomain has emerged as a novel therapeutic target for cancer, cardiovascular disease, and type 2 diabetes, and its inhibitors, such as apabetalone and JQ-1, disrupt the interaction of the domain with acetylated histones [45]. We elucidated that DHT directly interrupted the access of the p300 bromodomain to histone H3 using the histone tail binding assay. Therefore, the results shown in our study suggested that DHT inhibited p300 activity by obstructing the binding of both acetyl-CoA and the histone tail to the p300 bromodomain. Physically, acetylation of the histone tail changes the positive charge of lysine residues to negative, thereby weakening its electrostatic affinity to DNA, resulting in conformational changes in euchromatin and enhancing gene transcription [46, 47]. Thus, from an epigenetic point of view, our hypothesis that DHT prevents the onset and progression of NAFLD through its p300 inhibitory capacity is firmly supported by the compelling results obtained in this study. Our data showed that DHT induced inhibition of the hyperacetylation of histone proteins, including H3K9, H3K36, H4K8, and H4K16, and decreased expression of lipogenic genes, such as ACC1, ACLY, FASN, and SREBP1c. Consistent with our results, we recently reported that tannic acid, a plant-derived polyphenol, suppresses HAT, notably p300 activity, hyperacetylation of histone marks, and lipogenic gene expression and ameliorates NAFLD [11]. Based on the present findings and the previous literature, it can be postulated that the beneficial effects of DHT on the development and progression of NAFLD are closely associated with DHT-dependent epigenetic regulation of the lipid metabolic pathway.
In conclusion, we have not only provided powerful evidence that DHT is a new p300 inhibitor but also elucidated a plausible scenario describing the underlying molecular mechanism controlling NAFLD through DHT (Fig. 6). However, we did not observe whether DHT affected the occupancy of p300 on the promoter region of the lipogenic genes. Considering that this study provides strong evidence to support DHT-mediated epigenetic regulation in controlling NAFLD, further relevant in-depth studies are needed.

DHT showed the most powerful histone acetyltransferase (HAT) inhibitory effect among the rutin metabolites. In particular, DHT is a potent, novel p300 HAT inhibitor and can potentially prevent the development of NAFLD through its anti-p300 HAT activity. DHT attenuated p300 activity by docking onto the bromodomain of a p300 catalytic pocket and obstructed the binding of p300 to the histone tail, which consequentially reduced histone acetylation. Finally, histone hypoacetylation decreased the mRNA expression of lipogenic genes and improved the significant NAFLD features. Thus, DHT is a promising candidate that may be used for the prevention of NAFLD.
References
Shankar E, Kanwal R, Candamo M, Gupta S. Dietary phytochemicals as epigenetic modifiers in cancer: promise and challenges. Semin Cancer Biol. 2016;40–41:82–99.
Fotbolcu H, Zorlu E. Nonalcoholic fatty liver disease as a multi-systemic disease. World J Gastroenterol. 2016;22:4079–90.
Sun C, Fan JG, Qiao L. Potential epigenetic mechanism in non-alcoholic fatty liver disease. Int J Mol Sci. 2015;16:5161–79.
Tariq A, Mussarat S, Adnan M. Review on ethnomedicinal, phytochemical and pharmacological evidence of Himalayan anticancer plants. J Ethnopharmacol. 2015;164:96–119.
Choi WJ, Kim SK, Park HK, Sohn UD, Kim W. Anti-inflammatory and anti-superbacterial properties of sulforaphane from shepherd’s purse. Korean J Physiol Pharmacol. 2014;18:33–9.
Lee Y-H, Kwak J, Choi H-K, Choi K-C, Kim S, Lee J, et al. EGCG suppresses prostate cancer cell growth modulating acetylation of androgen receptor by anti-histone acetyltransferase activity. Int J Mol Med. 2012;30:69–74.
Shankar S, Kumar D, Srivastava RK. Epigenetic modifications by dietary phytochemicals: implications for personalized nutrition. Pharmacol Ther. 2013;138:1–17.
Wang G-L, Salisbury E, Shi X, Timchenko L, Medrano EE, Timchenko NA. HDAC1 cooperates with C/EBPα in the inhibition of liver proliferation in old mice. J Biol Chem. 2008;283:26169–78.
Bose M, Lambert JD, Ju J, Reuhl KR, Shapses SA, Yang CS. The major green tea polyphenol,(-)-epigallocatechin-3-gallate, inhibits obesity, metabolic syndrome, and fatty liver disease in high-fat–fed mice. J Nutr. 2008;138:1677–83.
Chen YK, Cheung C, Reuhl KR, Liu AB, Lee MJ, Lu YP, et al. Effects of green tea polyphenol (−)-epigallocatechin-3-gallate on newly developed high-fat/western-style diet-induced obesity and metabolic syndrome in mice. J Agric Food Chem. 2011;59:11862–71.
Chung MY, Song JH, Lee J, Shin EJ, Park JH, Lee SH, et al. Tannic acid, a novel histone acetyltransferase inhibitor, prevents non-alcoholic fatty liver disease both in vivo and in vitro model. Mol Metab. 2019;19:34–48.
Asrih M, Jornayvaz FR. Inflammation as a potential link between nonalcoholic fatty liver disease and insulin resistance. J Endocrinol. 2013;218:R25–36.
Kitade H, Chen G, Ni Y, Ota T. Nonalcoholic fatty liver disease and insulin resistance: new insights and potential new treatments. Nutrients. 2017;9:387.
Li Z, Li Y, Zhang HX, Guo JR, Lam CWK, Wang CY, et al. Mitochondria‐mediated pathogenesis and therapeutics for non‐alcoholic fatty liver disease. Mol Nutr Food Res. 2019;63:1900043.
Masarone M, Rosato V, Dallio M, Gravina AG, Aglitti A, Loguercio C, et al. Role of oxidative stress in pathophysiology of nonalcoholic fatty liver disease. Oxid Med Cell Longev. 2018;2018:7864316.
Simões IC, Fontes A, Pinton P, Zischka H, Wieckowski MR. Mitochondria in non-alcoholic fatty liver disease. Int J Biochem Cell Biol. 2018;95:93–9.
Al-Dhabi NA, Arasu MV, Park CH, Park SU. An up-to-date review of rutin and its biological and pharmacological activities. EXCLI J. 2015;14:59–63.
Sharma S, Ali A, Ali J, Sahni JK, Baboota S. Rutin: therapeutic potential and recent advances in drug delivery. Expert Opin Investig Drugs. 2013;22:1063–79.
Jaganath IB, Jaganath IB, Mullen W, Edwards CA, Crozier A. The relative contribution of the small and large intestine to the absorption and metabolism of rutin in man. Free Radic Res. 2006;40:1035–46.
Su KY, Yu CY, Chen YP, Hua KF, Chen YLS. 3, 4-Dihydroxytoluene, a metabolite of rutin, inhibits inflammatory responses in lipopolysaccharide-activated macrophages by reducing the activation of NF-κB signaling. BMC Complement Alter Med. 2014;14:21.
Morales AM, Mukai R, Murota K, Terao J. Inhibitory effect of catecholic colonic metabolites of rutin on fatty acid hydroperoxide and hemoglobin dependent lipid peroxidation in Caco-2 cells. J Clin Biochem Nutr. 2018;63:175–80.
Giménez‐Bastida JA, Zielinski H, Piskula M, Zielinska D, Szawara‐Nowak D. Buckwheat bioactive compounds, their derived phenolic metabolites and their health benefits. Mol Nutr Food Res. 2017;61:1600475.
Lomenick B, Hao R, Jonai N, Chin RM, Aghajan M, Warburton S, et al. Target identification using drug affinity responsive target stability (DARTS). Proc Natl Acad Sci U S A. 2009;106:21984–9.
Sharma R, Zhou MM. Partners in crime: The role of tandem modules in gene transcription. Protein Sci. 2015;24:1347–59.
Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Bryant SH. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009;37:W623–33.
Manning ET, Ikehara T, Ito T, Kadonaga JT, Kraus WL. p300 forms a stable, template-committed complex with chromatin: role for the bromodomain. Mol Cell Biol. 2001;21:3876–87.
da Silva RP, Kelly KB, Al Rajabi A, Jacobs RL. Novel insights on interactions between folate and lipid metabolism. Biofactors. 2014;40:277–83.
Lu SC, Alvarez L, Huang Z-Z, Chen L, An W, Corrales FJ, et al. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proc Natl Acad Sci U S A. 2001;98:5560–5.
Sinton MC, Hay DC, Drake AJ. Metabolic control of gene transcription in non-alcoholic fatty liver disease: the role of the epigenome. Clin Epigenet. 2019;11:104.
Feng D, Liu T, Sun Z, Bugge A, Mullican SE, Alenghat T, et al. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science. 2011;331:1315–9.
Ferreira DM, Simão AL, Rodrigues CM, Castro RE. Revisiting the metabolic syndrome and paving the way for micro RNA s in non‐alcoholic fatty liver disease. FEBS J. 2014;281:2503–24.
Legeay S, Rodier M, Fillon L, Faure S, Clere N. Epigallocatechin gallate: a review of its beneficial properties to prevent metabolic syndrome. Nutrients. 2015;7:5443–68.
Shanak S, Saad B, Zaid H. Metabolic and epigenetic action mechanisms of antidiabetic medicinal plants. Evid Based Complement Alternat Med. 2019;2019:3583067.
Balasubramanyam K, Swaminathan V, Ranganathan A, Kundu TK. Small molecule modulators of histone acetyltransferase p300. J Biol Chem. 2003;278:19134–40.
Marcu MG, Jung YJ, Lee S, Chung EJ, Lee MJ, Trepel J, et al. Curcumin is an inhibitor of p300 histone acetylatransferase. Med Chem. 2006;2:169–74.
Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–4.
Aagaard-Tillery KM, Grove K, Bishop J, Ke X, Fu Q, McKnight R, et al. Developmental origins of disease and determinants of chromatin structure: maternal diet modifies the primate fetal epigenome. J Mol Endocrinol. 2008;41:91–102.
De Long NE, Hardy DB, Ma N, Holloway AC. Increased incidence of non‐alcoholic fatty liver disease in male rat offspring exposed to fluoxetine during fetal and neonatal life involves the NLRP3 inflammasome and augmented de novo hepatic lipogenesis. J Appl Toxicol. 2017;37:1507–16.
Chung S, Hwang JT, Park JH, Choi HK. Free fatty acid-induced histone acetyltransferase activity accelerates lipid accumulation in HepG2 cells. Nutr Res Pr. 2019;13:196–204.
Gelman L, Zhou G, Fajas L, Raspé E, Fruchart JC, Auwerx J. p300 interacts with the N-and C-terminal part of PPARγ2 in a ligand-independent and-dependent manner, respectively. J Biol Chem. 1999;274:7681–8.
Giandomenico V, Simonsson M, Grönroos E, Ericsson J. Coactivator-dependent acetylation stabilizes members of the SREBP family of transcription factors. Mol Cell Biol. 2003;23:2587–99.
Tian Y, Wong VWS, Chan HLY, Cheng ASL. Epigenetic regulation of hepatocellular carcinoma in non-alcoholic fatty liver disease. Semin Cancer Biol 2013;23:471–82.
Maksimoska J, Segura-Peña D, Cole PA, Marmorstein R. Structure of the p300 histone acetyltransferase bound to acetyl-coenzyme A and its analogues. Biochemistry. 2014;53:3415–22.
Delvecchio M, Gaucher J, Aguilar-Gurrieri C, Ortega E, Panne D. Structure of the p300 catalytic core and implications for chromatin targeting and HAT regulation. Nat Struct Mol Biol. 2013;20:1040–6.
Cochran AG, Conery AR, Sims RJ. Bromodomains: a new target class for drug development. Nat Rev Drug Discov. 2019;18:609–28.
Chang L, Takada S. Histone acetylation dependent energy landscapes in tri-nucleosome revealed by residue-resolved molecular simulations. Sci Rep. 2016;6:34441.
Zhang F, Huang Q, Yan J, Chen Z. Histone acetylation induced transformation of B-DNA to Z-DNA in cells probed through FT-IR spectroscopy. Anal Chem. 2016;88:4179–82.
Acknowledgements
This study was supported by the Main Research Program (E-0150301) of the Korea Food Research Institute (KFRI), funded by the Ministry of Science, ICT & Future Planning.
Author information
Authors and Affiliations
Contributions
HKC and JTH designed the research. JL, JHS, MYC, and HKC performed the research. JL, JHL, TGN, and JHP contributed to the data analysis. JL, HKC, and JTH wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Lee, J., Song, JH., Chung, MY. et al. 3,4-dihydroxytoluene, a metabolite of rutin, suppresses the progression of nonalcoholic fatty liver disease in mice by inhibiting p300 histone acetyltransferase activity. Acta Pharmacol Sin 42, 1449–1460 (2021). https://doi.org/10.1038/s41401-020-00571-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00571-7
Keywords
This article is cited by
-
The influence of Mg(II) and Ca(II) ions on the autoxidation of 4-methylcatechol in weakly alkaline aqueous solutions
Reaction Kinetics, Mechanisms and Catalysis (2022)
