
Abstract
G protein-coupled receptor kinases (GRKs) constitute seven subtypes of serine/threonine protein kinases that specifically recognize and phosphorylate agonist-activated G protein-coupled receptors (GPCRs), thereby terminating the GPCRs-mediated signal transduction pathway. Recent research shows that GRKs also interact with non-GPCRs and participate in signal transduction in non-phosphorylated manner. Besides, GRKs activity can be regulated by multiple factors. Changes in GRKs expression have featured prominently in various tumor pathologies, and they are associated with angiogenesis, proliferation, migration, and invasion of malignant tumors. As a result, GRKs have been intensively studied as potential therapeutic targets. Herein, we review evolving understanding of the function of GRKs, the regulation of GRKs activity and the role of GRKs in human malignant tumor pathophysiology.
Similar content being viewed by others

Introduction
The superfamily of G protein-coupled receptors (GPCRs) is the largest group of receptor membrane proteins in mammals and mediates a variety of ligand signals, including neurotransmitters, hormones, and other small molecules, and it is also recognized as the target of a variety of drugs [1, 2]. G protein-coupled receptor kinases (GRKs) are key modulators of GPCRs signaling. They constitute a family of seven subtypes that phosphorylate agonist-bound receptor, thereby terminating the GPCRs-mediated signal transduction. There are more than 800 subtypes of GPCRs, whereas there are only seven GRK subtypes [3]. The significant difference between GRKs and GPCRs suggests that one GRK subtype may interact with multiple receptors, which is an explanation for GRKs involvement in a variety of physiological and pathological effects in humans [4, 5]. In addition to their classical role in promoting GPCRs desensitization and internalization, emerging data also suggest novel functions of GRKs at the receptor interface, including regulating non-GPCRs and non-receptor substrates, or participating in cellular responses in a phosphorylation-independent manner [6]. The classical and novel roles of GRKs involved in signaling pathways are being increasingly elucidated, and they are associated with angiogenesis, proliferation, migration, and invasion of malignant tumors [7, 8]. This review summarizes our current knowledge of the function of GRKs, the regulation of GRKs activity and the role of GRKs in human tumors.
Distribution and structure specificity of GRKs
GRKs consist of a family of seven mammalian serine/threonine protein kinases, named GRK1 ~ GRK7 according to the time they were found. Based on sequence homology, the members of the GRK family can be subdivided into three main groups: the visual GRK subfamily (GRK1 and GRK7); the GRK2/GRK3 subfamily, which consists of GRK2 (β-ARK1) and GRK3 (β-ARK2); and the GRK4 subfamily (GRK4, GRK5, and GRK6) [1]. GRK1 and GRK7 are mainly distributed in the retina, whereas GRK2, GRK3, GRK5, and GRK6 are distributed widely in the heart, brain, lung, kidney, and other tissues. GRK4 is primarily in the testes and has weak expression in some brain regions and the kidney [9]. Moreover, the distributional differences of GRKs present high specificity in their receptor preference.
The seven isoforms of GRKs share a number of structural and functional similarities. The basic structure is mainly composed of three parts, including the N-terminal domain, catalytic domain, and C-terminal domain. The N-terminal domain of GRKs is a region that may be important in receptor recognition. The C-terminal domain of GRKs contributes to their subcellular localization and agonist-dependent translocation [10]. In addition to the visual GRKs, other subtypes share a regulator of G-protein signaling (RGS) domain within the N-terminus region, which may be involved in GRK regulated signal transduction via a phosphorylation-independent mechanism [11]. In addition, signal transduction of GRK4-6 is highly correlated with catalytic activity. The C-terminal of GRK2 and GRK3 is longer than that of the GRK4 subfamily and contains a 125-amino acid pleckstrin homology (PH) domain. This domain plays an important role in the targeting and translocation of cytosolic GRKs to membranes after GPCRs activation [12].
Function and kinase activity modulation of GRKs
GRKs were originally identified as inhibitors of GPCRs signaling. Activated GPCRs are the targets of GRKs, which phosphorylate the specific intracellular domains of receptors. This event promotes the recruitment of β-arrestins, thus leading to uncoupling from G proteins and GPCRs internalization [13]. This rapid desensitization process helps uncouple phosphorylated receptors from the G proteins, thus abrogating further signal transduction. Moreover, β-arrestins also serve to mediate the internalization of certain GPCRs, which may regulate cellular activity both by mediating long-term desensitization through the degradation of receptors and by recycling desensitized receptors back to the cell surface to initiate other signaling [14].
Emerging evidence also suggests novel functions of GRKs at the receptor interface, including regulating non-GPCRs and non-receptor substrates or participating in cellular responses in a phosphorylation-independent manner [15]. GRKs can also regulate signaling mediated by membrane receptors that do not belong to the GPCRs, such as tyrosine kinase receptors of the insulin-like growth factor (IGF), platelet-derived growth factor (PDGF), and epidermal growth factor (EGF) families [16,17,18,19]. For example, activated EGFR attracts the recruitment of GRK2, leading to its phosphorylation at tyrosine residues, thereby increasing its catalytic activity, which terminates opioid receptor transregulation. In addition, GRKs also identify a number of non-receptor substrates, including tubulin, Smads, ezrin, p38 MAPK, and phosducin, which can be phosphorylated by GRK2 [20, 21]. Other substrates such as histone deacetylase 5 (HDAC5) can be identified and phosphorylated by GRK5 [22]. Some phosphorylation events led to an inhibitory effect, but other phosphorylation events may cause stimulatory effects. For example, GRK2 inhibits transforming growth factor β (TGF-β)-mediated cell growth inhibition and apoptosis by inducing R-Smads phosphorylation, whereas GRK5 phosphorylates HDAC5, a repressor of myocyte enhancer factor-2 (MEF2), enhancing the activation of MEF2, which induces MEF2-mediated gene transcription [15, 20, 22]. In addition to the phosphorylation-dependent manner discussed above, GRKs also modulate cellular responses in a phosphorylation-independent manner. GRKs were reported to associate with Gαq, Gβγ, PI3K, caveolin, MEK, Akt, Raf kinase inhibitor protein (RKIP), RalA GTPase, and the APC protein [10, 23,24,25,26,27]. For example, upon agonist stimulation, GRK2 interacts with PI3Kγ, favoring PI3K recruitment to the membrane and contributing to receptor endocytosis and desensitization [26].
GRKs are capable of interacting with many cellular components, which are involved in the direct modulation of kinase activity and subcellular targeting. GRK2 and GRK3 contain a PH domain (residues 561–655) that partially overlaps with a Gβγ-binding region [12]. By binding Gβγ, GRK2 activity is increased by promoting the GPCR-mediated allosteric activation of this kinase. Cellular calcium levels appear to regulate GRKs activity via the interaction of calcium-sensing proteins [28]. Calmodulin, a universal mediator of calcium signals, can inhibit the activity of GRK2-6 with different potencies. Calmodulin interacts with GRK2 at sites located at both the N- and C-terminal domains of the kinase (residues 18–37 and 593–689), leading to direct inhibition of GRK2 activity [29]. In addition, some other kinases emerge as important for modulating GRKs activity and protein stability, such PKA, PKC, c-Src, and MAPK [30,31,32,33]. These protein kinases regulate the activity of GRKs not only by directly phosphorylating GRKs but also by mechanisms of combination. Moreover, caveolin and RKIP also play an important role in the regulation of GRKs activity [34, 35]. Therefore, GRKs phosphorylate GPCRs and modulate the non-GPCRs, and their own activity can be regulated by many factors as well.
Roles of GRKs in tumors
Emerging evidence indicates that GRKs act as oncomodulators [36], which contribute to cancer progression via different mechanisms depending on the specific tumor and cell type (Table 1).
GRKs in central nervous system cancers
The C-X-C chemokine receptor type 4 (CXCR4) is highly expressed and has prognostic significance in brain tumors. Woerner et al. found that the alteration in CXCR4 function is correlated with increased ERK-dependent phosphorylation and inhibition of GRK2. The data suggest that changes in GRK expression or activity may promote oncogenic GPCRs function [37]. On this basis, further investigation identified that GRK3 expression is frequently decreased in glioblastoma multiforme (GBM), which possesses signature amplification or mutational activation of the EGF receptor [37]. The results indicate that GRK3 is a negative regulator of cell growth in GBM, whereas GRK5, another member of the GRK family, is significantly upregulated in GBM compared to low-grade glioma. Moreover, aggressive recurrent GBM, which is often resistant to chemotherapy and radiation, expresses even higher levels of GRK5, and knockdown of GRK5 with shRNA results in a significant decrease in glioblastoma-derived stem cell (GSC) proliferation [38].
Metastasis in medulloblastoma (MB) is a reason for poor survival. Previous studies reported that overexpression of the PDGF receptor (PDGFR) promotes migration through ERK-dependent activation in MB. Relative to other GRK isoforms, the percentage of GRK6 expression is lower in MB tumors with metastasis (22%) compared to those without metastasis (43%). These data lead to the conclusion that overexpression of GRK6 suppresses growth factor receptor/PDGFR-Src-mediated CXCR4 signaling by inactivating the phosphorylation of ERK, which promotes MB cell migration [39]. Therefore, targeting GRK6 may represent a potent new therapeutic strategy for MB.
GRKs in thyroid carcinoma
The thyrotropin receptor (TSHR) plays an important role in the proliferation and differentiation of thyroid cells, which is mediated by cAMP via an adenylylcyclase-activating Gs protein. TSHR belongs to the GPCR family, which can be regulated by GRK2 and GRK5. Métayé et al. [40] demonstrate that GRK5 protein expression is significantly decreased in differentiated thyroid carcinoma (DTC), whereas GRK3 and GRK6 proteins were not detected in human thyroid tissue. Moreover, this study indicates that the decrease in GRK5 expression may cause a reduction in the desensitization of TSHR in DTC, contributing to increased cAMP levels in these tumors. However, another study shows an increased expression of GRK2 in non-medullary thyroid cancers compared with adjacent normal tissues. Surprisingly, GRK2 reduced cell proliferation instead of stimulating it, revealing a new role for this kinase in the growth of thyroid cancers [41]. The mechanisms remain to be further explored.
GRKs in breast cancer
Recent data from Nogués et al. [42] indicate a relevant oncomodulator role of GRK2 in breast tumorigenesis. Elevated GRK2 protein levels are observed in breast cancer cell lines and in a significant proportion of invasive ductal carcinoma patients, whereas endothelial GRK2 expression is decreased in human breast cancer vessels [43]. Increased GRK2 functionality promotes the phosphorylation and activation of histone deacetylase 6 (HDAC6), leading to de-acetylation of the prolyl isomerase Pin1, a central modulator of tumor progression. The activation of GRK2-HDAC6-Pin1 signaling helps trigger a self-perpetuating positive feedback loop of growth factor transduction cascades, thus driving cell survival and proliferation [42]. In contrast, down-regulation of GRK2 in endothelial cells is relevant in triggering the tumor angiogenic switch by altering the response to TGF-β and other angiogenic stimuli [43]. These data raise new questions regarding the integrated impact of potential GRK2 inhibitors in breast cancer development. Exploring the potential role of GRK2 in the modulation of crosstalk among other pathways and GPCRs in breast cancer cells is an attractive area for future research.
Triple negative breast cancer (TNBC) accounts for approximately 15–20% of all breast cancers and is a heterogeneous disease that has a poor prognosis and limited treatment options [44, 45]. Most TNBC is associated with high CXCR4 expression and increased metastatic potential [46]. Analysis of public human breast cancer microarray data shows that GRK3 is expressed at statistically significant lower levels in tumor cells compared to normal breast tissue, and there is an inverse correlation between GRK3 and CXCR4 expression. High CXCR4 levels in human TNBC cell lines, combined with decreased intracellular regulatory GRK3, lead to impaired receptor internalization and more active C-X-C motif chemokine 12 (CXCL12)-mediated migration of breast cancer cells [47]. Silencing GRK3 expression does not appear to influence cell proliferation or detachment-induced death. Furthermore, GRK3 deficiency enhanced mammary tumor formation and lung and liver metastasis in an in vivo syngeneic mouse model [47].
GRK4 is highly expressed in human breast cancer, but not in normal epithelia. In addition, GRK4 overexpression activates MAPK through ERK and JNK phosphorylation and enhances the proliferation of breast cancer-derived cell lines, whereas GRK4 silencing promotes the opposite effect. Notably, silencing of β-arrestin 1 and 2 blocked the effects of GRK4 overexpression, suggesting that GRK4-mediated activation of MAPK module and breast tumor cell growth by favoring β-arrestin pathways [48]. These data suggest that GRK4 may be implicated in breast cancer carcinogenesis.
GRKs in ovarian neoplasm
Granulosa cell tumors (GCT) are serious ovarian neoplasms that can occur in women of all ages. Researchers examined GRKs expression in nonmalignant human granulosa cells and in the human GCT cell line KGN. GRK4 is expressed as multiple splice variants. The variants for GRK4 are designated as α, β, γ, and δ. The results show that KGN tumor cells express significantly less GRK4 α/β protein and higher levels of GRK2 and GRK4 γ/δ protein compared to nonmalignant human granulosa cells. In human GCT samples, GRK4 α/β protein was detected in 3 of 13 tumor samples, whereas GRK4 γ/δ and GRK2 protein expression was detected in all samples [49]. In addition, the differential expression of either GRK2 or GRK4 isoforms may result in impaired follicle-stimulating hormone (FSH) receptor uncoupling and desensitization. However, further confirmation is needed to determine the mechanisms of action.
GRKs in prostate cancer
Prostate cancer accounts for approximately one-third of all male cancer cases in the United States [50]. Immunohistochemistry (IHC) on a tissue microarray containing 651 cores of primary prostate cancer samples and benign prostatic hyperplasias (BPH) from 175 patients shows that a subgroup of patients with high Gleason scores (Gs7–9) is characterized by a complete loss of the GRK2 protein [51]. Down-regulation of GRK2 may constitute a mechanism to trigger androgen-independent tumor growth.
However, another group shows that GRK3 is overexpressed in human prostate cancers, particularly in metastatic tumors. GRK3 is not only necessary for the survival and proliferation of metastatic cells in vitro and in vivo, but is also sufficient to promote primary prostate tumor growth and metastasis in mouse xenograft models. Furthermore, GRK3 stimulates angiogenesis, at least in part, through the down-regulation of thrombospondin-1 and plasminogen activator inhibitor type 2 [52, 53]. These findings support a role for GRK3 in human prostate cancer progression, and GRK3 may serve as a novel biomarker or part of a panel of biomarkers for prostate cancer progression.
GRK5 was discovered with high expression in the aggressive prostate cancer PC3 cell line, and its siRNA knockdown attenuates cell proliferation. A study showed that GRK5 knockdown leads to G2/M arrest in the cell cycle, suggesting great significance of GRK5 in tumor growth. Kim et al. [54] provide support that GRK5 activity is critical in regulating Rb-mediated E2F transcriptional activity, cell cycle progression, and tumor growth. Moreover, knockdown of endogenous GRK5 expression attenuates the migration and invasion of prostate cancer cells and increases cell attachment and spreading. In the mechanism involved, GRK5 forms a complex with moesin and phosphorylates moesin principally on T66 residue [55]. Recent research demonstrates the enrichment of GRK5 and GRK6 in exosomes from prostate cancer cells, both of which regulate Src and IGF-I receptor signaling and are implicated in cancer [17]. Therefore, these results indicate that GRK5 may potentially be exploited for therapeutic targeting in prostate cancer.
GRKs in digestive system cancers
Colorectal cancer remains the fourth most commonly diagnosed cancer in the world. In colorectal cancer, studies found that the EP4 receptor/β-arrestin/c-Src complex plays an important role in prostaglandin E2 (PGE2)-mediated Akt signaling, suggesting that GRKs/β-arrestins may be involved in regulating colorectal cancer migration and metastasis. Thus, novel approaches targeting the GRKs/β-arrestins pathway may provide new therapeutic advances for the treatment of colorectal cancer [56]. Additionally, Wu et al. provide a new role for the tazarotene-induced gene 1 (TIG1) in colon cancer. TIG1 is a retinoid-inducible type II tumor suppressor gene. Knockdown of GRK5 expression relieves TIG1A-induced growth inhibition of the colon cancer cell line HCT116, suggesting that GRK5 mediates the suppression of cancer cell growth by TIG1A [57]. Together, these data suggest a functional role for GRKs as mediators of metastasis and cell growth in colorectal cancer, while the regulation of GRKs likely contributes to control the progression of colorectal cancer.
Hepatocellular carcinoma (HCC) is a complicated human disease in terms of etiology and molecular carcinogenic mechanisms. An increasing number of researchers are focused on developing new strategies for HCC treatment. By inducing the overexpression of GRK2 through recombinant adenovirus transduction in human HCC cells, cell proliferation and apoptosis are investigated. These data indicate that the anti-proliferative function of elevated GRK2 is associated with delayed cell cycle progression through G2/M phase arrest and in a GRK2 activity-dependent manner [58]. Based on this study, Wei et al. conducted further research on the role of GRK2 in HCC. The effects of a decreased protein level of GRK2 on the insulin-like growth factor 1 receptor (IGF-1R) signal pathway in HepG2 cells are investigated. Knockdown of GRK2 increases the phosphorylation of Akt, indicating a stronger activation of the IGF-1R signaling pathway [59]. Recent data from our laboratory also indicate an inhibitory role of GRK2 in HCC tumorigenesis. IHC results demonstrate that GRK2 expression in human HCC tissues is clearly decreased compared with tumor-adjacent and normal liver tissues. Down-regulation of GRK2 expression in HCC cells by transfection with GRK2 siRNA can significantly promote cell proliferation, migration, and invasion [60]. Furthermore, reduced levels of GRK2 induce a small cell cycle arrest at the G2/M phase by enhancing the expression of cyclin A, B1, and E. GRK2 overexpression inhibits the IGF1-induced proliferation and migration of human HCC cells by downregulating early growth response protein 1 (EGR1) [61]. These results indicate that enforced GRK2 may offer a potential therapeutic approach against HCC.
Contrary to HCC, GRK2 may play a protumoral role in pancreatic cancer. Using a high-content screening approach, the results demonstrate that the ADRBK1/GRK2 gene is not only significantly overexpressed in human pancreatic cancer, but also mediates pro-proliferative effects in pancreatic cancer cells. Recombinant overexpression of the kinase in cell lines without endogenous expression accelerates cell growth, whereas RNAi-mediated knockdown of endogenous ADRBK1 expression significantly inhibits growth via induction of a G1/S cell cycle arrest [62]. Analysis of tissue microarrays confirms that the ADRBK1 protein is undetectable in all non-neoplastic tissues (chronic pancreatitis and normal pancreas), whereas it is readily detectable in 33 of 65 (51%) pancreatic ductal adenocarcinoma samples [62]. Consistent with the results, another study by Zhou et al. [63] also found that GRK2 is overexpressed in pancreatic cancer compared to corresponding non-tumor tissues and may serve as a potential indicator of an unfavorable prognosis. Therefore, GRK2 may function as a proto-oncogene and serve as a prognostic indicator for pancreatic cancer.
GRKs in lung cancer
Using murine models of human lung cancer, researchers investigated the role of GRK6 in tumorigenesis. Mice deficient in GRK6 exhibit a significant increase in Lewis lung cancer growth and metastasis relative to control littermates (GRK6+/+). GRK6 deletion has no effect on the expression of vascular endothelial growth factor (VEGF) but upregulates matrix metalloproteinase MMP-2 and MMP-9 release, thereby promoting tumor angiogenesis [64]. Studies in lung adenocarcinoma (LADC) show that GRK6 expression is significantly down-regulated in LADC patients but high in normal lung tissue. Additionally, protein expression of GRK6 is significantly associated with staging and the pathology grade of LADC [65]. Thus, decreased expression of GRK6 may serve as an independent predictor of overall survival in LADC patients.
GRKs and other tumors
The melanocortin 1 receptor (MC1R), a GPCR, is a key regulator of epidermal melanocyte proliferation and differentiation and is a determinant of human skin phototype and skin cancer risk. GRK6 can inhibit MC1R agonist-independent constitutive signaling, whereas expression of a dominant negative GRK2 mutant in melanoma cells increases their cAMP response to agonists [66]. Moreover, stable transfection with GRK6 initiates a decrease in agonist-stimulated cAMP production in melanoma cells. The results suggest that GRK2 and GRK6 play a key role in modulating the MC1R signaling pathway and may be determinants of skin cancer.
In oral squamous cell carcinomas, overexpressed genes of the β-adrenergic receptor complex were identified by cDNA array analysis. It was found that malignant cells and surrounding tissue overexpress the GRK3 (ADRBK2) gene. The β-adrenergic receptor is closely related to cell proliferation, apoptosis, chemotaxis and metastasis [67]. The study reveals that activation of the β-adrenergic receptor by GRK3 in oral squamous cell carcinomas may impart greater potential for tumor malignancy and invasion.
In myeloma cells, GRK6 inhibition is selectively lethal to KMS11, INA6, and JJN3 myeloma cells. Furthermore, GRK6 is ubiquitously expressed in lymphoid tissues and myeloma. Heat shock protein HSP90 regulates GRK6 expression in myeloma, and GRK6 knockdown causes a comprehensive loss of signal transducer and activator of transcription 3 (STAT3) tyrosine phosphorylation and promotes cytotoxicity [68]. The results indicate that inhibition of GRK6 represents a uniquely targeted novel therapeutic strategy in human multiple myeloma.
In Kaposi’s sarcoma, down-modulation of GRK2 was reported, leading to enhanced migration and invasion of endothelial cells via activation of CXCR2/Akt signaling [69]. Kaposi’s sarcoma-associated herpesvirus (KSHV) may be involved in the pathogenesis of Kaposi’s sarcoma and is consistently present in the tissues of patients with Kaposi’s sarcoma. KSHV contains a gene that encodes a G protein-coupled receptor (KSHV-GPCR). Research by another group showed that GRK5, but not GRK2, inhibits KSHV-GPCR–stimulated proliferation of rodent fibroblasts [70].
Chen et al. identified p53, a key tumor suppressor, as a novel GRKs substrate in vivo, revealing a previously unknown function of GRKs in the regulation of genome stability. GRK5 phosphorylates p53 at the Thr-55 residue and promotes its degradation, thus inhibiting p53-mediated apoptosis in cultured human osteosarcoma cells in vitro. Moreover, GRK5 knock-out mice show an elevated p53 level in multiple tissue types [71]. The results suggest that GRK5 may play an essential role in the regulation of genome integrity and tumorigenesis.
Perspectives and conclusions
In the present review, we summarized recent evidence that deconstructs the traditional concept of GRKs as merely GPCRs-desensitizing kinases. Growing evidence indicates that GRKs play an essential role in regulating non-GPCRs and non-receptor substrates or participate in cellular responses in a phosphorylation-independent manner (Fig. 1). The interaction of GRKs with cellular proteins contributes directly or indirectly to the modulation of kinase activity and subcellular targeting. Alterations in GRKs activity and expression were found in the pathologies of various diseases, including tumors. In recent years, a variety of malignant tumors were found to associate closely with the abnormal expression of different GRK subtypes and present specific regulation in the progression of malignant tumor. For example, GRK3 serves as a negative regulatory factor in modulating glioblastoma tumor growth, whereas in oral squamous cell carcinoma, GRK3 interestingly promotes tumor development. Another finding is that GRK5 helps to promote the proliferation of prostate carcinoma PC3 cells, whereas high expression of GRK5 inhibits tumor cell growth in colon cancer [54, 57]. Increasing data showed that signaling pathways involved in the classical and novel role of GRKs, such as c-Src, MEK/ERK, and PI3K/Akt, are also important to tumor progression. However, further studies are needed to understand the complex mechanism. Better knowledge of the regulation of GRKs in tumor-relevant signaling pathways and its functional consequences will help us better understand the progression of tumors, as well as explore potentially therapeutic targeting in malignant tumors.

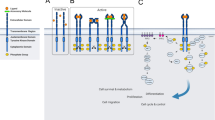
Function and GRKs-mediated signal pathways. GRKs phosphorylate agonist-activated GPCRs, which allows the recruitment of β-arrestins, thereby terminating or inducing the GPCRs-mediated signal transduction pathways. In addition, GRKs can also interact with non-GPCRs or non-receptor substrates, including participating in signal transduction in both a phosphorylated manner and non-phosphorylated manner. This associated signal regulation by GRKs promotes or inhibits different cellular functions, such as cell growth, proliferation, migration, metastasis, and survival
References
Pitcher JA, Freedman NJ, Lefkowitz RJ. G protein-coupled receptor kinases. Annu Rev Biochem. 1998;67:653–92.
Manglik A, Kruse AC. Structural basis for G protein-coupled receptor activation. Biochem-Us. 2017;56:5628–34.
Liu Y, An S, Ward R, Yang Y, Guo XX, Li W, et al. G protein-coupled receptors as promising cancer targets. Cancer Lett. 2016;376:226–39.
Metaye T, Gibelin H, Perdrisot R, Kraimps JL. Pathophysiological roles of G-protein-coupled receptor kinases. Cell Signal. 2005;17:917–28.
Komolov KE, Benovic JL. G protein-coupled receptor kinases: past, present and future. Cell Signal. 2018;41:17–24.
Penela P, Murga C, Ribas C, Lafarga V, Mayor FJ. The complex G protein-coupled receptor kinase 2 (GRK2) interactome unveils new physiopathological targets. Br J Pharmacol. 2010;160:821–32.
Lymperopoulos A, Bathgate A. Pharmacogenomics of the heptahelical receptor regulators G-protein-coupled receptor kinases and arrestins: the known and the unknown. Pharmacogenomics. 2012;13:323–41.
Nogues L, Palacios-Garcia J, Reglero C, Rivas V, Neves M, Ribas C, et al. G protein-coupled receptor kinases (GRKs) in tumorigenesis and cancer progression: GPCR regulators and signaling hubs. Semin Cancer Biol. 2018;48:78–90.
Harris RC. Abnormalities in renal dopamine signaling and hypertension: the role of GRK4. Curr Opin Nephrol Hypertens. 2012;21:61–5.
Ribas C, Penela P, Murga C, Salcedo A, Garcia-Hoz C, Jurado-Pueyo M, et al. The G protein-coupled receptor kinase (GRK) interactome: role of GRKs in GPCR regulation and signaling. Biochim Biophys Acta. 2007;1768:913–22.
Siderovski DP, Strockbine B, Behe CI. Whither goest the RGS proteins? Crit Rev Biochem Mol Biol. 1999;34:215–51.
Clapham DE, Neer EJ. G protein beta gamma subunits. Annu Rev Pharmacol Toxicol. 1997;37:167–203.
Claing A, Laporte SA, Caron MG, Lefkowitz RJ. Endocytosis of G protein-coupled receptors: roles of G protein-coupled receptor kinases and beta-arrestin proteins. Prog Neurobiol. 2002;66:61–79.
Kohout TA, Lefkowitz RJ. Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol Pharmacol. 2003;63:9–18.
Murga C, Penela P, Ribas C, Mayor F Jr. G protein-coupled receptor kinases: specific phosphorylation of 7TM receptors and beyond. Drug Discov Today Technol. 2010;7:e1–94.
Cipolletta E, Campanile A, Santulli G, Sanzari E, Leosco D, Campiglia P, et al. The G protein coupled receptor kinase 2 plays an essential role in beta-adrenergic receptor-induced insulin resistance. Cardiovasc Res. 2009;84:407–15.
DeRita RM, Zerlanko B, Singh A, Lu H, Iozzo RV, Benovic JL, et al. c-Src, insulin-like growth factor I receptor, G-protein-coupled receptor kinases and focal adhesion kinase are enriched into prostate cancer cell exosomes. J Cell Biochem. 2017;118:66–73.
Hildreth KL, Wu JH, Barak LS, Exum ST, Kim LK, Peppel K, et al. Phosphorylation of the platelet-derived growth factor receptor-beta by G protein-coupled receptor kinase-2 reduces receptor signaling and interaction with the Na(+)/H(+) exchanger regulatory factor. J Biol Chem. 2004;279:41775–82.
Chen Y, Long H, Wu Z, Jiang X, Ma L. EGF transregulates opioid receptors through EGFR-mediated GRK2 phosphorylation and activation. Mol Biol Cell. 2008;19:2973–83.
Ho J, Cocolakis E, Dumas VM, Posner BI, Laporte SA, Lebrun JJ. The G protein-coupled receptor kinase-2 is a TGFbeta-inducible antagonist of TGFbeta signal transduction. EMBO J. 2005;24:3247–58.
Peregrin S, Jurado-Pueyo M, Campos PM, Sanz-Moreno V, Ruiz-Gomez A, Crespo P, et al. Phosphorylation of p38 by GRK2 at the docking groove unveils a novel mechanism for inactivating p38MAPK. Curr Biol. 2006;16:2042–7.
Martini JS, Raake P, Vinge LE, DeGeorge BJ, Chuprun JK, Harris DM, et al. Uncovering G protein-coupled receptor kinase-5 as a histone deacetylase kinase in the nucleus of cardiomyocytes. Proc Natl Acad Sci USA. 2008;105:12457–62.
Ferguson SS. Phosphorylation-independent attenuation of GPCR signalling. Trends Pharmacol Sci. 2007;28:173–9.
Aziziyeh AI, Li TT, Pape C, Pampillo M, Chidiac P, Possmayer F, et al. Dual regulation of lysophosphatidic acid (LPA1) receptor signalling by Ral and GRK. Cell Signal. 2009;21:1207–17.
Wang L, Gesty-Palmer D, Fields TA, Spurney RF. Inhibition of WNT signaling by G protein-coupled receptor (GPCR) kinase 2 (GRK2). Mol Endocrinol. 2009;23:1455–65.
Naga PS, Laporte SA, Chamberlain D, Caron MG, Barak L, Rockman HA. Phosphoinositide 3-kinase regulates beta2-adrenergic receptor endocytosis by AP-2 recruitment to the receptor/beta-arrestin complex. J Cell Biol. 2002;158:563–75.
Jimenez-Sainz MC, Murga C, Kavelaars A, Jurado-Pueyo M, Krakstad BF, Heijnen CJ, et al. G protein-coupled receptor kinase 2 negatively regulates chemokine signaling at a level downstream from G protein subunits. Mol Biol Cell. 2006;17:25–31.
Sallese M, Iacovelli L, Cumashi A, Capobianco L, Cuomo L, De Blasi A. Regulation of G protein-coupled receptor kinase subtypes by calcium sensor proteins. Biochim Biophys Acta. 2000;1498:112–21.
Pronin AN, Satpaev DK, Slepak VZ, Benovic JL. Regulation of G protein-coupled receptor kinases by calmodulin and localization of the calmodulin binding domain. J Biol Chem. 1997;272:18273–80.
Cong M, Perry SJ, Lin FT, Fraser ID, Hu LA, Chen W, et al. Regulation of membrane targeting of the G protein-coupled receptor kinase 2 by protein kinase A and its anchoring protein AKAP79. J Biol Chem. 2001;276:15192–9.
Mahavadi S, Huang J, Sriwai W, Rao KR, Murthy KS. Cross-regulation of VPAC2 receptor internalization by m2 receptors via c-Src-mediated phosphorylation of GRK2. Regul Pept. 2007;139:109–14.
Elorza A, Sarnago S, Mayor FJ. Agonist-dependent modulation of G protein-coupled receptor kinase 2 by mitogen-activated protein kinases. Mol Pharmacol. 2000;57:778–83.
Sarnago S, Elorza A, Mayor FJ. Agonist-dependent phosphorylation of the G protein-coupled receptor kinase 2 (GRK2) by Src tyrosine kinase. J Biol Chem. 1999;274:34411–6.
Carman CV, Lisanti MP, Benovic JL. Regulation of G protein-coupled receptor kinases by caveolin. J Biol Chem. 1999;274:8858–64.
Deiss K, Kisker C, Lohse MJ, Lorenz K. Raf kinase inhibitor protein (RKIP) dimer formation controls its target switch from Raf1 to G protein-coupled receptor kinase (GRK) 2. J Biol Chem. 2012;287:23407–17.
Nogues L, Reglero C, Rivas V, Neves M, Penela P, Mayor FJ. G-protein-coupled receptor kinase 2 as a potential modulator of the hallmarks of cancer. Mol Pharmacol. 2017;91:220–8.
Woerner BM, Luo J, Brown KR, Jackson E, Dahiya SM, Mischel P, et al. Suppression of G-protein-coupled receptor kinase 3 expression is a feature of classical GBM that is required for maximal growth. Mol Cancer Res. 2012;10:156–66.
Kaur G, Kim J, Kaur R, Tan I, Bloch O, Sun MZ, et al. G-protein coupled receptor kinase (GRK)-5 regulates proliferation of glioblastoma-derived stem cells. J Clin Neurosci. 2013;20:1014–8.
Yuan L, Zhang H, Liu J, Rubin JB, Cho YJ, Shu HK, et al. Growth factor receptor-Src-mediated suppression of GRK6 dysregulates CXCR4 signaling and promotes medulloblastoma migration. Mol Cancer. 2013;12:18.
Metaye T, Menet E, Guilhot J, Kraimps JL. Expression and activity of G protein-coupled receptor kinases in differentiated thyroid carcinoma. J Clin Endocrinol Metab. 2002;87:3279–86.
Metaye T, Levillain P, Kraimps JL, Perdrisot R. Immunohistochemical detection, regulation and antiproliferative function of G-protein-coupled receptor kinase 2 in thyroid carcinomas. J Endocrinol. 2008;198:101–10.
Nogues L, Reglero C, Rivas V, Salcedo A, Lafarga V, Neves M, et al. G protein-coupled receptor kinase 2 (GRK2) promotes breast tumorigenesis through a HDAC6-Pin1 axis. EBioMedicine. 2016;13:132–45.
Rivas V, Carmona R, Munoz-Chapuli R, Mendiola M, Nogues L, Reglero C, et al. Developmental and tumoral vascularization is regulated by G protein-coupled receptor kinase 2. J Clin Invest. 2013;123:4714–30.
Chalakur-Ramireddy N, Pakala SB. Combined drug therapeutic strategies for the effective treatment of triple negative breast cancer. Biosci Rep. 2018;38: pii: BSR20171357. https://doi.org/10.1042/BSR20171357.
Wang C, Kar S, Lai X, Cai W, Arfuso F, Sethi G, et al. Triple negative breast cancer in Asia: An insider’s view. Cancer Treat Rev. 2018;62:29–38.
Chu QD, Panu L, Holm NT, Li BD, Johnson LW, Zhang S. High chemokine receptor CXCR4 level in triple negative breast cancer specimens predicts poor clinical outcome. J Surg Res. 2010;159:689–95.
Billard MJ, Fitzhugh DJ, Parker JS, Brozowski JM, McGinnis MW, Timoshchenko RG, et al. G protein coupled receptor kinase 3 regulates breast cancer migration, invasion, and metastasis. PLoS ONE. 2016;11:e152856.
Matsubayashi J, Takanashi M, Oikawa K, Fujita K, Tanaka M, Xu M, et al. Expression of G protein-coupled receptor kinase 4 is associated with breast cancer tumourigenesis. J Pathol. 2008;216:317–27.
King DW, Steinmetz R, Wagoner HA, Hannon TS, Chen LY, Eugster EA, et al. Differential expression of GRK isoforms in nonmalignant and malignant human granulosa cells. Endocrine. 2003;22:135–42.
Catalona WJ. Prostate cancer screening. Med Clin North Am. 2018;102:199–214.
Prowatke I, Devens F, Benner A, Grone EF, Mertens D, Grone HJ, et al. Expression analysis of imbalanced genes in prostate carcinoma using tissue microarrays. Br J Cancer. 2007;96:82–8.
Li W, Ai N, Wang S, Bhattacharya N, Vrbanac V, Collins M, et al. GRK3 is essential for metastatic cells and promotes prostate tumor progression. Proc Natl Acad Sci USA. 2014;111:1521–6.
Sang M, Hulsurkar M, Zhang X, Song H, Zheng D, Zhang Y, et al. GRK3 is a direct target of CREB activation and regulates neuroendocrine differentiation of prostate cancer cells. Oncotarget. 2016;7:45171–85.
Kim JI, Chakraborty P, Wang Z, Daaka Y. G-protein coupled receptor kinase 5 regulates prostate tumor growth. J Urol. 2012;187:322–9.
Chakraborty PK, Zhang Y, Coomes AS, Kim WJ, Stupay R, Lynch LD, et al. G protein-coupled receptor kinase GRK5 phosphorylates moesin and regulates metastasis in prostate cancer. Cancer Res. 2014;74:3489–500.
Buchanan FG, Gorden DL, Matta P, Shi Q, Matrisian LM, DuBois RN. Role of beta-arrestin 1 in the metastatic progression of colorectal cancer. Proc Natl Acad Sci USA. 2006;103:1492–7.
Wu CC, Tsai FM, Shyu RY, Tsai YM, Wang CH, Jiang SY. G protein-coupled receptor kinase 5 mediates Tazarotene-induced gene 1-induced growth suppression of human colon cancer cells. BMC Cancer. 2011;11:175.
Wei Z, Hurtt R, Ciccarelli M, Koch WJ, Doria C. Growth inhibition of human hepatocellular carcinoma cells by overexpression of G-protein-coupled receptor kinase 2. J Cell Physiol. 2012;227:2371–7.
Wei Z, Hurtt R, Gu T, Bodzin AS, Koch WJ, Doria C. GRK2 negatively regulates IGF-1R signaling pathway and cyclins’ expression in HepG2 cells. J Cell Physiol. 2013;228:1897–901.
Xu ZW, Yan SX, Wu HX, Chen JY, Zhang Y, Li Y, et al. The influence of TNF-alpha and Ang II on the proliferation, migration and invasion of HepG2 cells by regulating the expression of GRK2. Cancer Chemother Pharmacol. 2017;79:747–58.
Ma Y, Han CC, Huang Q, Sun WY, Wei W. GRK2 overexpression inhibits IGF1-induced proliferation and migration of human hepatocellular carcinoma cells by downregulating EGR1. Oncol Rep. 2016;35:3068–74.
Buchholz M, Honstein T, Kirchhoff S, Kreider R, Schmidt H, Sipos B, et al. A multistep high-content screening approach to identify novel functionally relevant target genes in pancreatic cancer. PLoS ONE. 2015;10:e122946.
Zhou L, Wang MY, Liang ZY, Zhou WX, You L, Pan BJ, et al. G-protein-coupled receptor kinase 2 in pancreatic cancer: clinicopathologic and prognostic significance. Hum Pathol. 2016;56:171–7.
Raghuwanshi SK, Smith N, Rivers EJ, Thomas AJ, Sutton N, Hu Y, et al. G protein-coupled receptor kinase 6 deficiency promotes angiogenesis, tumor progression, and metastasis. J Immunol. 2013;190:5329–36.
Yao S, Zhong L, Liu J, Feng J, Bian T, Zhang Q, et al. Prognostic value of decreased GRK6 expression in lung adenocarcinoma. J Cancer Res Clin Oncol. 2016;142:2541–9.
Sanchez-Mas J, Guillo LA, Zanna P, Jimenez-Cervantes C, Garcia-Borron JC. Role of G protein-coupled receptor kinases in the homologous desensitization of the human and mouse melanocortin 1 receptors. Mol Endocrinol. 2005;19:1035–48.
Perez-Sayans M, Somoza-Martin JM, Barros-Angueira F, Gayoso-Diz P, Otero-Rey EM, Gandra-Rey JM, et al. Activity of beta2-adrenergic receptor in oral squamous cell carcinoma is mediated by overexpression of the ADRBK2 gene: a pilot study. Biotech Histochem. 2012;87:179–86.
Tiedemann RE, Zhu YX, Schmidt J, Yin H, Shi CX, Que Q, et al. Kinome-wide RNAi studies in human multiple myeloma identify vulnerable kinase targets, including a lymphoid-restricted kinase, GRK6. Blood. 2010;115:1594–604.
Hu M, Wang C, Li W, Lu W, Bai Z, Qin D, et al. A KSHV microRNA directly targets G protein-coupled receptor kinase 2 to promote the migration and invasion of endothelial cells by inducing CXCR2 and activating AKT signaling. PLoS Pathog. 2015;11:e1005171.
Geras-Raaka E, Arvanitakis L, Bais C, Cesarman E, Mesri EA, Gershengorn MC. Inhibition of constitutive signaling of Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor by protein kinases in mammalian cells in culture. J Exp Med. 1998;187:801–6.
Chen X, Zhu H, Yuan M, Fu J, Zhou Y, Ma L. G-protein-coupled receptor kinase 5 phosphorylates p53 and inhibits DNA damage-induced apoptosis. J Biol Chem. 2010;285:12823–30.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81770605, 81300332), the Natural Science Foundation of the Higher Education Institutions of Anhui Province (No. gxyqZD2018024, KJ2018A0200) and the Natural Science Foundation of Anhui Medical University (No. 2017xkj032). The authors acknowledge the help of the staff members of the Institute of Clinical Pharmacology, Anhui Medical University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Sun, Wy., Wu, Jj., Peng, Wt. et al. The role of G protein-coupled receptor kinases in the pathology of malignant tumors. Acta Pharmacol Sin 39, 1699–1705 (2018). https://doi.org/10.1038/s41401-018-0049-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-018-0049-z
Keywords
This article is cited by
-
Transcriptome signatures of host tissue infected with African swine fever virus reveal differential expression of associated oncogenes
Archives of Virology (2024)
-
Computational analysis of G-protein-coupled receptor kinase family members as potential targets for colorectal cancer therapy
Egyptian Journal of Medical Human Genetics (2022)
-
G protein-coupled receptor kinase 2 modifies the cellular reaction to cisplatin through interactions with NADPH oxidase 4
Molecular and Cellular Biochemistry (2021)
