
Abstract
Cardiovascular disease (CVD) is the leading cause of death in the world, with a high incidence and a youth-oriented tendency. RNA modification is ubiquitous and indispensable in cell, maintaining cell homeostasis and function by dynamically regulating gene expression. Accumulating evidence has revealed the role of aberrant gene expression in CVD caused by dysregulated RNA modification. In this review, we focus on nine common RNA modifications: N6-methyladenosine (m6A), N1-methyladenosine (m1A), 5-methylcytosine (m5C), N7-methylguanosine (m7G), N4-acetylcytosine (ac4C), pseudouridine (Ψ), uridylation, adenosine-to-inosine (A-to-I) RNA editing, and modifications of U34 on tRNA wobble. We summarize the key regulators of RNA modification and their effects on gene expression, such as RNA splicing, maturation, transport, stability, and translation. Then, based on the classification of CVD, the mechanisms by which the disease occurs and progresses through RNA modifications are discussed. Potential therapeutic strategies, such as gene therapy, are reviewed based on these mechanisms. Herein, some of the CVD (such as stroke and peripheral vascular disease) are not included due to the limited availability of literature. Finally, the prospective applications and challenges of RNA modification in CVD are discussed for the purpose of facilitating clinical translation. Moreover, we look forward to more studies exploring the mechanisms and roles of RNA modification in CVD in the future, as there are substantial uncultivated areas to be explored.
Similar content being viewed by others

Introduction
Dynamic chemical modifications are prevalent in biological macromolecules, particularly RNA and protein, determining the fate of these molecules and regulating their function. For instance, demethylation of DNA can lead to chromatin reprogramming and gene transcription.1 The phosphorylation of a protein is crucial to its ubiquitination and degradation by the proteasome.2 Over the past half century, many RNA modifications have been thoroughly documented. Numerous studies have confirmed that RNA modification plays an indispensable role in physiologic processes and pathologic changes in mammals. RNA stimulates the mammalian innate immune system by activating toll-like receptors (TLRs), which is significantly inhibited by the incorporation of modified nucleosides 5-methylcytidine (m5C), N6-methyladenosine (m6A), 5-methyluridine (m5U), 2-thiouridine (s2U), or pseudouridine (denoted by psi, Ψ).3 m6A is involved in mammalian cortical neurogenesis by promoting the decay of mRNA associated with neurogenesis and neuronal differentiation.4 Ψ-mediated post-transcriptional modification of tRNA leads to translation dysregulation, which affects stem cell commitment during early embryogenesis, and is commonly seen in aggressive subtypes of human myelodysplastic syndromes.5 Recent studies indicate an association between RNA modification and therapeutic tolerance. Rapino et al.6 found that enzymes catalyzing wobble uridine 34 (U34) tRNA modification are responsible for the transformation driven by BRAF V600E oncogene and resistance to targeted therapy of melanoma.
The role of RNA modification in disease is gradually being revealed, providing potential small molecules, and new targets for drug development and intervention strategies (Supplementary Table 1). N1-methyladenosine (m1A)-modified early tRNAs improve the translation efficiency of MYC protein and promote T cell proliferation, and interventions based on this mechanism can alleviate colitis in mouse model.7 Yankova et al.8 reported a highly selective m6A “writer” inhibitor STM2457. Their study demonstrates that STM2457 acts as an anti-cancer agent by inhibiting METTL3, resulting in the selective reduction of m6A levels on the mRNA of known leukemia genes with reduced translation. Delaunay et al.9 revealed that some antibiotics could be used as adjuvants in cancer therapy to inhibit metastasis by targeting m5C in mitochondrial tRNAs (mt-tRNAs). Combined blocking of N7-methylguanosine (m7G) tRNA methyltransferase METTL1 and its downstream chemokine pathway can enhance the efficacy of anti-PD-1 in the treatment of intrahepatic cholangiocarcinoma, providing a new idea for developing effective immunotherapy.10 In the tremendous progress made in the chemical modification of biomacromolecules, we have seen several drugs move from clinical trials to approval to benefit patients.11 Drugs based on RNA modification are in clinical trials,12 and we expect new and improved results from them.
In this review, we mainly focus on nine RNA modifications (Fig. 1) and their effects on cardiovascular diseases, ranging from the development and physiological function of the cardiovascular system to the occurrence and progression of the disease, and the potential therapeutic strategies.

A schematic diagram of chemical modifications on mammal RNAs. Several RNA modifications with their chemical structures are highlighted in (a) transcripts (such as mRNA, miRNA and lncRNA) and (b) tRNAs. For tRNAs, anticodons are located at positions 34 to 36, and modification at position 34 (known as U34 modification) regulates wobble pairing, including m5C, hm5C, f5C, A-to-I editing, mcm5U, mcm5s2U, mcm5Um, τm5U, τm5s2U, and cmnm5U. m7G, 7-methylguanosine; m1A, N1-methyladenosine; m6A, N6-methyladenosine; m5C, 5-methylcytosine; ac4C, N4-acetylcytidine; Ψ, pseudouridine; A-to-I editing, adenosine-to-inosine RNA editing; mcm5U, 5-methoxycarbonylmethyluridine; mcm5s2U, 5-methoxycarbonylmethyl-2-thiouridine; mcm5Um, 5-methoxycarbonylmethyl-2’-O-methyluridine; τm5U, 5-taurinomethyluridine; τm5s2U, 5-taurinomethyl-2-thiouridine; cmnm5U, 5-carboxyaminomethyluridine
Overview of RNA modifications
The development of various scientific and technical methods (e.g., liquid chromatography/mass spectrometry and high-throughput sequencing) has promoted the progression of RNA modifications (Fig. 2; for details on technologies for mapping modified nucleotides, see the review by Bartee et al.13 and Wiener et al.14). Currently, over 100 RNA modifications have been identified and characterized on various types of RNAs, including messenger RNAs (mRNAs) and non-coding RNAs (ncRNAs), such as ribosomal RNAs (rRNAs) and transfer RNAs (tRNAs), some of them are known to have specific biochemical and physiological effects, and indispensable roles in human diseases.15

The historical milestone in RNA modification
RNA modification is a dynamic and reversible process theoretically regulated by certain proteins or complexes. The “writers” are responsible for installing modification molecules that can be removed by the “erasers”, which are usually pairs of enzymes with opposite functions, such as methyltransferase and demethylase. In addition, modification sites can be recognized and bound by several proteins, called “readers”, thus regulating RNA metabolism or function. Most “writers”, “erasers”, and “readers” are identified as m6A regulators, while regulators in other modifications are reported less frequently.
RNA modification determines or affects RNA fate and function. 5′-cap modification and the poly(A) tail are common structures found in almost every eukaryotic mRNA, which control their stability and facilitate translation. 5′-cap is the m7G modified guanine nucleotide, located in the 5′-end of eukaryotic mRNA, while poly(A) tail is ~200 adenosine residues, added to the 3′-end of mRNA in the process of transcription, and is associated with export to the cytoplasm. Moreover, some internal modifications (including but not limited to m6A, m1A, m5C, 2′-O-methylation) on mRNA have recently received more attention. These internal modifications can also be found in various ncRNAs, such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs). Regarding function, m6A modification affects the processing, structure, splicing, localization, stability, degradation, and translation of RNAs and their functions, such as RNA–protein and RNA–RNA interactions.16,17,18 The m5C can perform a similar role.19,20,21,22 Recently, m1A and ac4C have been found to promote the stability and translation of methylated mRNA.23,24,25 Ψ, a more common modification in ncRNAs, stabilizes the structure of tRNAs and rRNAs and regulates mRNAs in response to environmental signals in humans and yeast.26
tRNA is a linking molecule carrying amino acids and anticodons, with abundant modification sites for various modifications (including but not limited to: m1A, m5C, m7G, ac4C, and Ψ), which plays an indispensable role in the accurate translation of proteins. For example, the T/D arm and variable loop modifications are normally associated with structural stability of tRNA,27,28,29 while anticodon loop modifications are involved in accurate translation.30 Aberrant modifications of cytoplasmic tRNA and mt-tRNA can cause various diseases, such as neurological disorders and tumors. Suzuki summarized the aberrant modifications of tRNAs and the corresponding regulatory genes in these diseases.31
The ribosome comprises two subunits containing rRNAs and ribosomal proteins (RPs): the small subunit (SSU) identifying mRNAs and the large subunit (LSU) carrying peptidyl transferase center, translation factor binding sites, and exit tunnel. During the biogenesis of ribosomes, some rRNAs are mainly modified with methylated sugars and Ψs at the subunit inner cores and the interface. How these modifications occur and their effects on rRNA structure and function are discussed by Sharma and Lafontaine.32
In addition, with the revelation of functions of mRNAs, lncRNAs and circulating RNAs (circRNAs) in human health and diseases were previously considered junk without biological function. Their interaction with chemical modifications that regulate the transcription, stability, export, and function of these ncRNAs is also beginning to be revealed. For instance, m6A modification promotes THAP7-AS1 transcription,33 DIAPH1-AS1 stability,34 and circNSUN2 export.35 Wang et al.36 found that oxidative modification confers miR-184 to target Bcl-xL and Bcl-w, inhibiting their translation and promoting apoptosis. The addition of terminal uridine to the 3′-end of miR-26 eliminates its inhibition of IL-6.37 Moreover, these ncRNAs, in turn, can regulate chemical modification. LncRNA can serve as a decoy to inhibit histone methyltransferase,38 and a scaffold of histone modification complexes to regulate histone modifications on target genes.39,40
The mechanisms of RNA modifications
N6-methyladenosine (m6A)
Nucleotide methylation in HeLa cells, including m6A and m7G, was identified about 50 years ago,41,42 and has been studied extensively since then. To date, m6A is believed to be the most common internal modification of eukaryotic mRNA. However, due to the limitations of the technical methods at the time, much was still unknown in this field. By 2012, Dominissini et al.43 used a novel technique, m6A-seq (also called MeRIP-seq), to identify transcriptome-wide m6A modifications in humans and mice. Their results suggest that m6A, which is mainly found around the stop codons and in the longer internal exons, plays an essential regulatory role in gene expression. Using the same technique, Meyer et al.44 obtained similar results in mammals that m6A sites are enriched near the stop codons, and in 3′-untranslated regions (3'UTRs) which may be related to miRNA-binding sites. The m6A mapping and measure techniques, and their advantages and disadvantages, are comprehensively summarized elsewhere (see ref. 45).
m6A is added to specific sites of RNAs by a multi-subunit complex, methyltransferase-like 3 (METTL3)-METTL14, which is stable in cells.46 METTL3 (used to be MT-A70) has enzymatic activity,47 and its allosteric homolog METTL14 with higher methyltransferase activity48 is critical for target recognition and binding.49,50 Wilms’ tumor 1-associating protein (WTAP), without catalytic domains, is identified as a key subunit in the m6A methyltransferase complex, which can interact with METTL3 and METTL14 to affect their activity and accumulation in nuclear speckles markedly.46,48 It is worth mentioning that the effect of METTL14 and WTAP on m6A level in cells is greater than that of METTL3.46 In addition, METTL16, VIRMA (also known as KIAA1429), HAKAI (also called CBLL1), ZC3H13, RBM15, RBM15B, METTL5-TRMT112, and ZCCHC4 are identified as m6A “writers” in mammals.51,52,53,54 METTL5-TRMT112 and ZCCHC4 are responsible for m6A modification of 18S rRNA and 28S rRNA, respectively.54 Moreover, methyltransferases are sequence-specific with site preference rather than structural preference for the target RNA.46,52 METTL3 and WTAP bind to a variety of types of RNAs, but most of them are mRNA. Their binding sites are mainly located in the coding sequence (CDS) and 3′UTR.48 GGAC and GACU are the main enrichment binding motifs of METTL3-METTL14 and WTAP, respectively.46 VIRMA-mediated m6A on mRNA is concentrated in the 3′UTR and close to the stop codon, particularly with the GGACU motif.52 RRACH (R = G/A; H = A/C/U) is a consensus-specific methylated site with highly conservation.43,44,55,56,57 Demethylase converts the m6A into A in RNAs, called the m6A “eraser”. Fat mass and obesity-associated protein (FTO) and ALKBH5, members of the AlkB family, are currently reported as m6A “erasers” in mammals.58,59 In the dynamic process of m6A modification, there is a class of proteins, known as m6A “readers”, which recognize and bind the m6A sites of RNAs, participate in the molecular mechanisms, and mediate important functions. At present, proteins are identified as m6A “readers” in mammals, including but not limited to YTH domain family proteins (YTHDF1-3), YTH domain-containing proteins (YTHDC1-2), heterogeneous nuclear ribonucleoprotein C (HNRNPC), HNRNPG, HNRNPA2B1, IGF2BPs, eIF3, and cytosol METTL3. Depending on the binding mechanism, they can be divided into three categories: direct binding, indirect binding, and m6A structural switch.45 The m6A switch refers to m6A-dependent RNA structural remodeling and RNA-protein interaction.16 Functionally, m6A modification is involved in RNA processing and maturation in the nucleus, affects RNA transport to the cytoplasm, and regulates RNA stability and translation in the cytoplasm. Moreover, some m6A “readers” coordinate these processes (Fig. 3).

Biological functions of dynamic m6A modification on RNAs. m6A modification is a reversible process in which “writers” (such as METTL3 and METTL14) add m6A to RNAs and “erasers” (ALKBH5 and FTO) are responsible for removal in the nucleus. The “readers” recognize and bind to m6A-RNA, which affects RNA processing and export to the cytoplasm. In the cytoplasm, m6A finally regulates RNA degradation, stability and translation
RNA processing
Before RNAs mature, precursor mRNA (pre-mRNA), pre-miRNA and primary miRNA (pri-miRNA) experience alternative processing events, such as the splicing of RNA, the 5′-capping and 3′-polyadenylation of mRNAs, and the cleavage of miRNA by RNase III enzyme Drosha and Dicer successively. Dominissini et al.43 reported that METTL3-knockdown (KD) in HepG2 cells results in the differential expression of transcript isoforms with enriched m6A peaks, suggesting that m6A may affect splicing. Moreover, compared to METTL3 depletion, FTO depletion significantly affects m6A levels in the surrounding regions of constitutive and alternative splice sites.60 The underlying mechanism is that FTO-dependent m6A sites spatially overlap with the sites of exonic splicing enhancers (ESEs), which results in co-recruiting the splicing factors SRSF1/2. Bartosovic et al.61 also observed m6A-mediated exon-skipping events in FTO-knockout (KO) cells. Since then, multiple studies have confirmed the role and mechanism of m6A in RNA splicing and maturation.
The nuclear “writer” YTHDC1 reportedly interacts with five other splicing factors (SRSF1, SRSF3, SRSF7, SRSF9, and SRSF10). YTHDC1-SRSF3 tends to promote exon inclusion, while YTHDC1-SRSF10 promotes exon skipping.62 Interestingly, the nuclear AURKA interaction with HNRNPK and YTHDC1 inhibits RBM4 exon inclusion by m6A-YTHDC1-SRSF3 and results in RBM4 exon skipping in an m6A-YTHDC1-HNRNPK-dependent manner.63 In addition, YTHDC1 is associated with pre-mRNA 3′-end processing factors, including CPSF4, CPSF6, SRSF3, SRSF7, and FIP1L1, to inhibit alternative polyadenylation and alter the 3′-UTR length of the target transcript.64,65 Similarly, NKAP, acting as an m6A “reader”, combines with m6A and recruits splicing factor SFPQ for transcription termination site (TTS) splicing.66 Moreover, m6A also affects the processing of miRNAs. The microprocessor Drosha-DGCR8 complex plays a critical role in processing pri-miRNAs into pre-miRNAs, a first step in miRNA biogenesis. Tavazoie and colleagues67 observed that METTL3 depletion leads to a downregulation of overall mature miRNAs and a significant upregulation of pri-miRNAs. They also revealed the underlying mechanism that DGCR8 effectively recognizes and binds pri-miRNAs containing METTL3-labeled m6A markers, facilitating the processing of most pri-miRNAs into pre- and mature miRNAs. Furthermore, it is reported that m6A “readers”, such as HNRNPA2B1 and NKAP, play an important role in the METTL3-mediated miRNA maturation by recognizing and directly binding m6A sites (especially RGAC motif) to recruit the DGCR8.17,68 This mechanism does not appear to be unique to METTL3-mediated m6A, as the same phenomenon is observed in cells overexpressing METTL14 where METTL14 is proved to interact with DGCR8,69 or to benefit the interaction between DGCR8 with pri-miRNAs.70 Another mechanism is that m6A-mediated RNA structural changes regulate the RNA-structure-dependent accessibility (also known as m6A-switch) to some RBPs conducting RNA processing events, such as HNRNPC and HNRNPG. In this case, m6A results in the exposure of HNRNPC and HNRNPG preferential binding sites, which promotes splicing of nearby exons.16,71 Moreover, HNRNPG binds to the m6A-modified pre-mRNA, which prolongs the RNA polymerase II (RNAPII) occupancy near the exon-intron junction and is attributed to the recruitment of spliceosome components or splice factors.72
Although “readers” may be primary regulators of m6A-mediated alternative splicing, the role of “writers” and “erasers” cannot be ignored. METTL16 is identified as the methyltransferase of MAT2A and U6 spliceosomal small nuclear RNA (snRNA). METTL16-mediated methylation of MAT2A hairpins (hp), especially its occupancy on hp1 in MAT2A 3'UTR, is crucial in inducing alternative splicing of MAT2A,73 while METTL16-mediated m6A modification of U6 snRNA at A43 may be related to 5'-splicing site recognition on pre-mRNA.51,74 Tang et al.75 reported that about 41% of m6A sites overlap with exon skipping/inclusion or intron skipping/retention, enriched near the sites where abnormal splicing events occur (such as producing the aberrant shorter transcript isomers in spermatocytes). They found that ALKBH5-dependent m6A demethylation regulates the correct splicing of long 3′UTR transcripts. METTL3 typically mediates the m6A modification of an intron at the 5'-splicing site, which enhances its downstream splicing, suppresses upstream splicing, and may provide a basis for specifying other splicing outcomes.76 However, more evidence is needed to explore specific mechanisms.
RNA nuclear export
Mature nuclear mRNAs and most ncRNAs require transport into the cytoplasm to function. It was observed that nuclear mRNA output in ALKBH5-deficient cells is accelerated with cytoplasmic mRNA level significantly increased, which may be the result of the phosphorylation of SF2/ASF (coded by SFRS1) to switch from splicing factors to export adapter proteins in an m6A-dependent manner.59 This mechanism can also be applied to SRSF3. Roundtree and colleagues reported that YTHDC1-SRSF3 mediates the nuclear export of m6A-mRNA.77 They found that YTHDC1 facilitates the binding of the mRNA export receptor NXF1. These results explain why YTHDC1-KD accumulates m6A-mRNAs in the nuclear, while they deplete in the cytoplasm. Moreover, YTHDC1 promotes the nuclear export of m6A-circRNAs, such as circNSUN235 and circSPECC1.78 In addition, the m6A “reader” FMRP, containing nuclear localization and export sequences, is reported to preferentially bind target mRNAs with m6A modification and interact with nuclear export protein CRM1 to promote nuclear export of target mRNAs.79
RNA stability
The stability of RNA protected by a 5′-cap structure and a 3′-poly(A) tail plays a critical role in controlling RNA metabolism. It is reported that m6A significantly shortens the half-life (2.5 h shorter on average) of RNAs and increases the decay rate (to 9 from 5.4 h on average) of mRNAs.80 m6A’s preference for the last exon of transcripts, especially the longer one, may be related to mRNA stability, as it facilitates the selection of 3′-poly(A) sites which play important roles in promoting translation and preventing mRNA degradation.81 Slobodin and colleagues82 revealed that both m6A and CCR4-NOT deadenylase complex regulate poly(A) tail length and mRNA stability. Moreover, m6A “readers” are reportedly involved in deacetylation-mediated mRNA decay. For instance, YTHDF2 recruits the CCR4-NOT complex through a direct interaction between its N terminus and the superfamily homology (SH) domain of CNOT1, a scaffold subunit of the CCR4-NOT complex, thus destabilizing m6A-containing RNAs by accelerating deadenylation.83 In contrast, IGF2BP1 inhibits deadenylation-mediated mRNA degradation by competitively binding to PABPC1, a poly(A)-binding protein (PABP), to prevent CCR4-NOT complex recruitment to PABPC1, thereby enhancing m6A-modified mRNA stability and expression.84 FMR1 reportedly interacts with the PABPs to polyadenylate/deadenylate m6A-mRNAs and thus affects their stability.85
YTHDC1 reportedly regulates nonsense-mediated mRNA decay (NMD) based on m6A modification near the initiation codon,86 and destabilizes the subsets of m6A-labeled chromosome-associated regulatory RNAs (carRNAs) via the nuclear exosome targeting (NEXT) complex,87 and the m6A-labeled polyadenylated RNAs (such as MYC mRNA) via the poly(A) tail exosome targeting (PAXT) complex.88 YTHDC2 is found to have RNA-induced ATPase activity and 3′→5′ RNA helicase activity, and it interacts with 5′→3′ exoribonuclease XRN1 to regulate RNA decay.89 Furthermore, m6A “readers” recognize and bind with m6A modified RNAs, which can locate protein-RNA complex to RNA decay sites, thereby controlling the metabolism of RNAs. For example, YTHDF2 plays a dominant role in m6A-mediated RNA stability. YTHDF2-KD results in an accumulation of untranslated mRNAs, with an increased m6A/A ratio of the total mRNA, and a prolonged lifetime of the target mRNA, confirming that YTHDF2 destabilizes the m6A-containing mRNA. Specifically, the C-terminal domain of YTHDF2 selectively binds to m6A-containing mRNAs, while the N-terminal domain helps its targeted mRNAs to localize from the translatable pool to mRNA decay sites (such as processing bodies) for sustained degradation.18 YTHDF2 mediates mRNA decay, by directly interacting m6A-mRNA and argonaute RISC catalytic component 2 (AGO2),90 or by indirectly recognizing m6A-circRNA.91 Moreover, YTHDF1/3 are also involved in YTHDF2-mediated RNA degradation.90,92 Depletion of YTHDF2 or any two YTHDFs or all three of them results in increased stability of m6A-mRNA, but they may be cell-specific.93,94 Recent studies have demonstrated that YTHDF1/3 attenuates the stability of target transcripts.95,96,97,98 These results suggest that YTHDFs play an important role in m6A-mediated RNA stability, but their biological function may vary from cell to cell, where they may cooperate, compensate or compete with each other. More evidence is needed to verify this assumption. Moreover, both FMRP and MSI2 act as m6A “readers” to protect their targeted m6A-mRNAs from YTHDF2-dependent degradation,99,100 but the mechanisms have not been clarified. In contrast to YTHDFs-mediated RNA decay, m6A modified target mRNAs of IGF2BPs appear to be more stable by recruiting the RNA stabilizers such as HuR, MATR3, PABPC1, and stress granule marker TIAR,101,102,103 or by inhibiting the miRNA-dependent decay.104,105 Other studies showed that some ncRNAs (such as lncRNAs and cirRNAs) are also involved in m6A-mediated mRNA stability by interaction with YTHDF2 or IGF2BPs,106,107,108,109,110,111 but the specific mechanism remains unclarified. In addition, m6A affects the binding of RNA stabilizers to target mRNAs (such as ETS1, ZMYM1, and GATA3) and ncRNAs (such as DDIT4-AS1), thus regulating transcript stability.112,113,114,115 For example, m6A repels the HuR binding to U-rich regions at 3′UTR and the G3BP1 interaction with GG(m6A)CU-containing targets.116,117 Thus, MELLT3-KD enhances the stability of both HuR and G3BP1 target RNAs.
mRNA translation
Translation events mainly include initiation (TI), elongation, and termination. Depending on the mechanisms of TI, there are cap-dependent and independent translations. The former is mediated by cap-binding complexes such as the eukaryotic initiation factor 4F (eIF4F) and CBP80/20 complexes.118,119 The internal ribosome entry site (IRES) trans-acting factors (ITAFs, such as G3BP1) mediates translation in a cap-independent manner, also known as IRES-dependent translation.118 m6A reportedly regulates mRNA translation through various mechanisms affecting initiation and elongation.
The classic mode of translation initiation in eukaryotic cells is that eIF4E, (a subunit of eIF4F complex) binds to the 5′-cap and recruits the 40S ribosomal subunit to form a 43S preinitiation complex with the help of eIF3.118 In the cytoplasm, METTL3 reportedly acts as an m6A “reader” to regulate the target mRNA translation (such as EGFR and TAZ) in CBP80/20-dependent and eIF4E-dependent manners by interaction with eIF3,120 and “reader” METTL3 facilitates cap-dependent translation by binding with eIF3H subunit to 3′UTR specific sites near the stop codon and leading to mRNA circularization.121 Similarly, cytoplasmic METTL16 exerts a methyltransferase activity-independent function in regulating translation. Mechanistically, the Mtase domain of METTL16 binds to the 5′-cap and interacts with eIF3A/B and 18S rRNA.122 Cytoplasmic METTL16 promotes eIF4E-dependent translation by interacting with eIF4E2 and repressing eIF4E2’s competition with eIF4E for cap binding.123 YTHDF1-mediated translation relies on the interaction with eIFs (such as eIF3) and the eIF4G-dependent loop formation.124 YTHDF1 promotes cap-independent translation of mRNA with m6A in CDS by binding the elongation factor eEF2.125 Moreover, RNA helicase-containing m6A “reader” YTHDC2 is required for elongation of mRNA with m6A in CDS by resolving the mRNA structural hurdle and facilitating ribosome removement.126 It has been shown that YTHDF3 also participates in cap-independent translation by interacting with the 40S and 60S ribosomal proteins rather than eIFs.92,127,128 This mechanism also applies to the translation of circRNAs in response to stress.129 More importantly, m6A provides a new mechanism for translation in a cap-independent manner (also known as m6A specific translation), which is distinct from cap- or IRES-dependent translation. Under this condition, eIF3 directly binds m6A for cap-independent ribosome recruitment.118 Meyer and colleagues130 found that the 5′-cap and eIF4E are not indispensable for the translation of 5′UTR m6A-mRNA (such as Hsp70 mRNA in mouse embryonic fibroblasts), which directly recruits the 48S complex (composed of eIF1, 1A, 2, 3, and 40S subunit) and selectively initiates translation from the first AUG.
In addition, METTL5 and ZCCHC4 mediate methylation of 18S rRNA and 28S rRNA, which affects the 40S ribosome structure, 80S activity, and occupancy of specific codons, thereby regulating global translation.131,132,133,134 If m6A is added to the mRNA codon, it prolongs cognate tRNA decoding, which depends on the position and context of m6A in the codon.135
N1-methyladenosine (m1A)
m1A is when methyl is added to the N1 position of adenosine. Half a century ago, m1A was identified primarily in tRNAs and rRNAs of various types of fungi and bacteria. In 2016, Dominissini and colleagues23 explored m1A in mammalian mRNAs using MeRIP-seq based on the m1A antibody. Their results showed that m1A is mainly distributed in the 5′UTR and CDS regions, and methylated transcripts contain more alternative TISs, suggesting that m1A may be involved in the initiation of translation. However, Grozhik et al.136 point out that m1A is not a prevalent high stoichiometric modification in mRNAs, and there may be false positives in m1A antibody-based on mapping methods. They found that m1A is difficult to detect at the transcription-start nucleotide, and that the m1A antibody specifically interacts with the m7G-cap of mRNA in an m1A-independent manner, which may explain why m1A peaks are displayed at the 5′UTR.
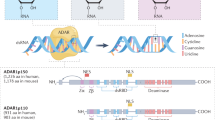
The m1A “writer” TRMT61B is the first identified methyltransferase modifying both tRNAs and rRNAs in humans,137,138 while TRMT10C is responsible for the m1A methylation of tRNAs (Fig. 4a).139 Subsequent studies reported that both TRMT61B and TRMT10C regulate m1A levels in mitochondrial mRNAs.140,141 It should be noted that m1A in mitochondrial transcripts is enriched mainly in the CDS region and the third position of a codon, affecting translation in mitochondria. Moreover, these studies demonstrated that the TRMT6/TRMT61A-dependent m1A has a high sequence preference within the cytosol, predominantly distributed in a GUUCRA motif with a strong hairpin structure, identified as the tRNA T-loop. Like m6A, m1A modification is a reversible process, which is removed by “erasers” such as ALKBH3.142 ALKBH3-mediated demethylation of CSF-1 mRNA significantly improves the stability of CSF-1 in breast and ovarian cancer cells,143 while in human retinal pigmented epithelial 1 (RPE-1) cells, ALKBH3 deletion inhibits the Aurora A mRNA stability as well as its translation.144 Also, ALKBH3-mediated demethylation of tRNA is crucial for translation initiation and promotes the generation of tRNA-derived small RNAs (tDRs).145 These results suggest that ALKBH3 can regulate transcriptional degradation and translation in an m1A-dependent manner by various mechanisms. Another “eraser” ALKBH1 is identified as a demethylase of tRNAs. The ALKBH1-mediated demethylation of tRNAs at position 58 affects their stability and translation, including translation initiation by influencing the assembly of the 80S monomers, and translation elongation by eEF1A promoting the enrichment of tRNAs in the active translation pool.146 ALKBH7 also has demethylase activity and its substrate is mitochondrial pre-tRNA.147 FTO is also a demethylase of nuclear and cytoplasmic m1A-tRNAs and is selective for stem-loop structures, which promote protein translation efficiency.148 Furthermore, serum starvation and H2O2 stimuli result in specific m1A peaks, suggesting that different stress conditions may regulate m1A in mRNAs.142 Similarly, glucose deprivation leads to reduced m1A-tRNA levels in cells, thereby inhibiting translation, which can be reversed by ALKBH1-KD.146

Dynamic m1A and m5C modification on RNAs. (a) TRMT6/TRMT61A are m1A methylases of mRNAs and tRNAs in the nucleus. TRMT10C/TRMT61B catalyze m1A on mitochondrial (mt-) tRNA, rRNA and mRNA (such as ND5 mRNA). ALKBH1/3/7 and FTO regulate their demethylation. Functionally, m1A mediates RNA stability and translation via various mechanisms. YTHDFs may function as m1A “readers”. (b) NSUN1-6 are “writers” for RNA m5C modification to regulate the nuclear export, metabolism and functions of RNA. ALYREF, YBX1, and YTHDF2 reportedly act as m5C “readers”. ALKBH1 and TNT2 are responsible for the oxidizing, rather than the removing m5C to hm5C, f5C, and ca5C
5-methylcytosine (m5C)
m5C has been once thought to be one of the DNA markers. With the development and application of detection technologies (recently reviewed in detail by Guo et al.149), m5C is widely found in eukaryotic RNAs as well, especially tRNAs and rRNAs (Fig. 4). In mammalian RNAs, m5C “writers” mainly include NOL1/NOP2/SUN (NSUN) family members and tRNA aspartic acid methyltransferase 1 (TRDMT1, also known as DNMT2). NSUN2 is mainly responsible for mRNAs (especially in CG-rich and CDS regions), tRNAs and microRNAs.20,29,150,151 NSUN3, NSUN6 and TRDMT1 are the main methyltransferases of tRNAs.150,152,153,154 NSUN1 and NSUN5 target rRNAs.155,156 NSUN4 catalyzes mRNA and mitochondrial rRNA.157,158 The Aly/REF export factor (ALYREF, an mRNA transport adapter), YTHDF2, and Y-box binding protein 1 (YBX1) are identified as m5C “readers”.19,20,159 The controversy about the m5C “erasers” still exists. It is unclear whether m5C modification is a reversible process like m6A or m1A, but some studies found that m5C can be oxidized, rather than demethylated, to 5-hydroxymethylcytosine (hm5C), 5-formylcytosine (f5C) and 5-carboxylcytosine (ca5C) by ALKBH1 and ten-eleven-translocator (TET) enzymes.9,160,161,162 Functionally, m5C, together with its oxidized derivatives, regulates RNA nuclear export, stability, processing, and mRNA translation, thus participating in the organ development and function, as well as the occurrence and progression of diseases such as cancer, mitochondrial disease, infection, and colitis.20,152,159,163,164,165
N7-methylguanosine (m7G)
As early as fifty years ago, m7G was identified as a post-translational modification widely seen in the 5′-cap structure of mammalian mRNA, typically connected with a triphosphate and 2′-O-methyl (Nm) modifications.166,167 m7G is also found in ncRNAs such as miRNA, tRNA, and rRNA. In mammals, m7G “writers” mainly include METTL1, WD repeat domain 4 (WDR4), Williams-Beuren syndrome chromosome region 22 protein (WBSCR22, coded by BUD23 gene), TRMT112, RNA guanine-7 methyltransferase (RNMT) and RNMT-activating miniprotein (RAM). RNMT-RAM primarily targets mRNA;168 WBSCR22-TRMT112 adds m7G to 18S rRNA;169 and METTL1-WDR4 are responsible for mRNA, tRNA, and miRNA.170,171,172 Functionally, m7G is thought to regulate the export and translation of mRNA and the maturation of 18S rRNA (Fig. 5a).

m7G and ac4C modification on RNAs. a In the nucleus, METTL1-WDR4 are m7G “readers” of mRNA, pre-miRNA and tRNA. RNMT-RAM mainly targets mRNA, while WBSCR22-TRMT112 is responsible for 18S rRNA. Functionally, m7G facilitates RNA nuclear export, translation, 40S ribosome biogenesis, and selective miRNA maturation. b In the nucleus, NAT10-THUMPD1 catalyzes ac4C mainly on rRNA, tRNA and mRNA, which is related to ribosome biogenesis, translation and mRNA stability, respectively
The m7G cap structure primarily determines mRNA’s stability, splicing, and translation. It is demonstrated that m7G protects mRNA from 5′→3′ exonuclease degradation and promotes splicing.173,174,175 Pabis and colleagues176 found that m7G-mediated splicing is due to the role of CBC in recruiting snRNPs and spliceosome assembly. As discussed above, cap-dependent translation initiation is mediated by the cap-binding protein eIF4E. The eIF4E affinity to the m7G cap was enhanced by the interaction of eIF4G peptides with the cap.177 The middle domain of the AGO2 protein is similar to the cap-binding domain of eIF4E and can bind specifically to the m7G cap, which impedes the recruitment of eIF4E and thus inhibits translation initiation.178 The endogenous let-7 microribonucleoproteins (miRNPs) may play a similar role in m7G cap-dependent translation.179 Moreover, METTL1-WDR4 can add m7G to the internal of target mRNAs, which facilitates translation.172 In mice, m7G is enriched in tRNAs with the RAGGU motif, affecting translation.180 METTL1-mediated m7G-tRNA promotes the use of m7G codons in mRNA translation.171,181
Moreover, the m7G cap also affects RNA nuclear export and miRNA maturation. In the nucleus, eIF4E regulates RNA export. For example, eIF4E is physically associated with cyclin D1 mRNA and promotes its transportation.182 CBC-PHAX binds to m7G-capped pre-miRNAs and facilitates their transportation via XPO1 rather than via the canonical XPO5 pathway.183,184 In the cytoplasm, only mature 3p-miRNA is produced along with the extended 5p-miRNA.183 In addition, HNRNPC may inhibit PHAX activity through interactions with CBC.185
N4-acetylcytosine (ac4C)
N4-acetylcytosine is thought to be the only acetylation in post-transcriptional modification. In the past, ac4C has been explored mainly in bacterial and fungal tRNA and rRNA. The discovery of N-acetyltransferase 10 (NAT10) promotes the exploration of RNA ac4C modification in mammals (Fig. 5b). In HeLa cells, NAT10 reportedly mediates ac4C1842 in 18S rRNA in an ATP-dependent manner. It regulates ribosome biogenesis, but the exact mechanism remains to be investigated further.186 A recent study reported that SNORD13 assists ac4C1842 in 18S rRNA, but the mechanism has not been clarified.187 In human colon cancer cells, NAT10 depletion significantly reduces tRNA acetylation.188 Moreover, the NAT10-THUMPD1 complex is responsible for acetylating human tRNA, whereas THUMPD1 primarily acts as a tRNA adapter. With the development of ac4C detection technologies, Arango and colleagues189 identified ac4C-modified mRNAs in HeLa cells in 2018. Their results show that ac4C significantly enhances mRNA stability through uncoupled exonuclease resistance, particularly in the CDS. The translation of ac4C-mRNA is significantly enhanced, possibly mediated by the enhanced mRNA stability rather than the 18S rRNA and tRNAser/leu acetylation. Moreover, if ac4C is present at the wobble site of the codon, it is conducive to cognate tRNA recognition, significantly improving the decoding efficiency. Schwartz et al.190 comprehensively mapped the ac4C modification in eukaryotic RNA subsequently. They found that the ac4C site in mRNA, rRNA, and tRNA, induced by NAT10-THUMPD1 overexpression, almost occurs at the CCG motif and that ac4C is absent or maintained at relatively low levels in endogenous eukaryotic mRNA. A recent study pointed out that acetylation in 5′UTR promotes the translation initiation in upstream sequences.191 These results suggested that ac4C in different regions of mRNA may affect translation through different mechanisms. NAT10 also mediates the acetylation of lncRNA CTC-490G23.2 in esophageal squamous cell carcinoma.192
The role of mammalian ac4C-modified RNA in the occurrence and development of diseases has been gradually revealed. NAT10-mediated ac4C positively regulates runt-related transcription factor 2 (RUNX2) mRNA stability and protein expression, thus promoting the differentiation of bone marrow-derived mesenchymal stem cells (BMSCs).193 Similarly, NAT10 is dysregulated in various cancers, which mediates ac4C modification of target mRNA (such as BCL9L, SOX4, AKT1, FSP1 and KIF23), and promotes cancer progression.194,195,196,197,198 In terms of therapeutic applications, ac4C is expected to be used in nucleic acid therapeutics, as Nance et al.199 found that ac4C significantly inhibits synthetic mRNA-mediated inflammation, which may be beneficial for repeated drug administration.
Uridylation
Uridylation is a post-transcriptional modification mainly found in the poly(A) tail, especially the short one (5–25 nt), also known as the U-tail.200,201 This poly(A)-tail length-dependent uridylation may also be associated with PABPs, because PABPC1 preferentially binds RNAs with long poly(A) tail and against terminal uridylyl transferases (TUTs). There are exceptions that the 3′-uridylation modification also exits in histone mRNAs without poly(A) tails.202,203 Uridylation is added by TUTs and terminal nucleotidyl transferases (TENTs). Currently reported mammalian uridylation “writers” include TUT2, TUT4 (ZCCHC11), TUT7 (ZCCHC6), TENT2, TENT4B, and TENT5C. A recent study indicated that TUT4, rather than TUT7, is primarily responsible for the uridylation of mature miRNAs and that TENT2 selectively modifies mature miRNAs with little effect on their abundance.204
Uridylation is primarily present in mRNA and miRNA to influence their metabolism (Fig. 6a). Firstly, the role of deadenylases (e.g., CCR4-NOT complex) and PABPs-mediated deadenylation in RNA stability is described above (see Section “RNA stability”). Similarly, uridylation and deadenylation regulators mediate RNA metabolism through antagonistic or synergistic effects. CCR4 promotes the dissociation of PABPC1 from the poly(A) tail, which is conducive to mRNA decay mediated by TUTs or decay factors.205 In addition, miR-1, which acts as a deadenylation inducer, also affects uridylation and mRNA decay.201 TUT4/7 depletion significantly prolongs the half-life of miR-1 target mRNAs. The final step in mRNA degradation is handled by 5′ → 3′ (e.g., exoribonuclease XRN1) or 3′ → 5′ (e.g., exosomes and exoribonuclease DIS3L2) decay factors. XRN1 contributes to the degradation of mRNA with U-tail added by TUT4/TUT7.201 DIS3L2 preferentially degrades uridylated RNA.206 TUT-DIS3L2 surveillance (TDS) pathway plays a crucial role in quality control and degradation of cytoplasmic RNAs in the mammal.207 PABPN1 facilitates the 3′→5′ decay of uridylated mRNA by recruiting DIS3L2.208 Uridylated miRNAs are significantly enriched in exosomes and degraded.209,210 Exosomes promote the uridylation of AGO-bound pre-miRNAs by cooperating with TUT7/4 to recognize and degrade pre-miRNAs without function for quality control. Furthermore, some RBPs, such as HuR and zinc-finger proteins, interact with adenylate-uridylate-rich elements (AREs) and regulate RNA decay.211,212,213,214 In addition, TUT7 uridylates Zc3h12a mRNA (encoding ribonuclease Regnase-1) and indirectly regulates the stability of a subset of cytokines, including IL-6.215

Uridylation and A-to-I editing on RNAs. a In the cytoplasm, TUT2/4/7 catalyzes the 3′-uridylation of precursor miRNA (pre-miRNA), mature miRNA, and mRNA, whereas TUT1 is a uridylation “writer” of snRNA in the nucleus. TENT2 is responsible for the mature miRNA. Uridylation regulates the maturation of miRNA and snRNA, the functions of miRNA, and RNA degradation. b ADAR1/2 plays as A-to-I editing “writers” of primary miRNA (pri-miRNA), mature miRNA, and mRNA, which affects the maturation and function of miRNA and the stability and export of mRNA
Uridylation is also involved in miRNA biogenesis. In the cytoplasm, ribonuclease (RNase) III Dicer cleaves pre-miRNA formed by the Drosha-DGCR8 complex. Heo et al.216 found that Lin28 recruits TUT4 specifically to bind to the GGAG motif and uridylates cytoplasmic pre-let-7, which blocks Dicer processing and significantly reduces mature let-7. This mechanism also applies to miR-1.217 Interestingly, they later reported that in the absence of Lin28, such as in HeLa cells, TUT7/4/2 mediated 3′-strand (3p) mono-uridylation in most pre-let-7 and pre-miR-105 is essential for Dicer processing.218 Also, the uridylation frequency in 3p.1 has a negative correlation with the 5p/3p.1 ratio, because the 3p uridylation alters the Dicer cleavage site selection of pre-miR-324, leading to arm switching of miR-324.219 The uridylation of mature miRNAs influences their biological function. For example, uridylated miR-26a and miR-26b repress their inhibition of target genes such as IL-6 mRNA.37,220
Adenosine-to-Inosine (A-to-I) RNA Editing
Adenosine-to-inosine (A-to-I) RNA editing refers to the conversion of adenosine to inosine in RNA by adenosine deaminases acting on RNA (ADARs), which is widely found in double-stranded RNAs (dsRNAs) with Alu and LINE (long interspersed nuclear element) elements.221,222 RNA editing leads to changes in RNA secondary structure, amino acid sequence, alternative splicing, miRNA-target regulation, and gene expression, which can affect cell phenotypes and lead to diseases.223,224,225,226,227 The editing alters the RNA secondary structure at the Dicer cleavage site and inhibits miRNA biogenesis.228 In cardiomyocytes (CMs), edited miR-34a undoes its inhibition to target mRNAs.229 Various RBPs, such as HNRNPC, Drosha, and HuR, prefer or repel edited sites, connecting RNA editing events to RBP-mediated biological processes.230,231,232 Specifically, for example, edited CTSS mRNA benefits HuR recruitment with improved stability,231 while ADAR1 occupancy on repetitive Alu elements antagonizes their interaction with STAU1 and STAU1-mediated RNA export.233 This section mainly focuses on advances in the last decade to avoid excessive duplication, since Kazuko Nishikura detailed A-to-I editing and ADARs in mammals (see ref. 221).
ADAR1 and ADAR2 (ADARB1) have enzyme activity for A-to-I RNA editing (Fig. 6b). They are not mutually compensatory in vital functions nor have unique substrate preferences.234 ADAR1 acts as an oncogene in cancer, while ADAR2 has an inhibitory effect.235,236 They also play a similar role in circRNA regulation. ADAR1 inhibits or promotes circRNAs equally, but ADAR2 mainly acts as an inhibitor.232 Moreover, ADAR1 is dominant in editing. In human umbilical arterial fibroblasts (HUAFs), ADAR1 almost participates in the editing of all pri-microRNAs and mature microRNAs, whereas ADAR2 is only responsible for pri-miR-376a1, pri-miR-376a2, and miR-376a+b.237 ADAR2 inhibits the editing of 66 ADAR1 targets in the heart.238 There are two subtypes of ADAR1, p110 and p150, mainly distributed in the nucleus and the cytoplasm, respectively. In the nucleus, ADAR1p110 edits A-C mismatched base pairs of telomeric RNA: DNA hybrids, which facilitates RNase H2 processing of the telomere R-loop and promotes genomic stability.239 ADAR2 has a similar effect.240 Song et al.241 reported that ADAR2-mediated editing of COPA pre-mRNA results in an isoleucine-to-valine substitution at residue 164 in hepatocellular carcinoma, which produces a less stable protein and switches COPA from an oncogenic gene to a suppressor by deactivating PI3K/AKT/mTOR signaling. RNA editing alters the subcellular localization of the AZIN1 protein from the cytoplasm to the nucleus, with differentially interacting proteins,224,242 and the target of the edited miRNAs will also be changed.237,243
An overall decrease in edited dsRNA levels may result in a higher risk of diseases, such as inflammatory bowel disease (IBD), coronary artery disease (CAD), diabetes, and Parkinson’s disease (PD).244 ADAR1, for example, is essential in regulating the inflammatory response. ADAR1-mediated RNA editing is indispensable for inhibiting interferon (IFN) response to maintain homeostasis. To a certain degree, it may contribute to the restraint on innate RNA sensor MDA5 (encoded by IFIH1 gene) in recognition of unedited dsRNAs, as well as in the activation of PKR, ZBP1 and subsequent signaling.222,245,246,247
Pseudouridine (Ψ)
Pseudouridylation is considered the most abundant post-transcriptional modification in RNA and it is the isomerization product of a uridine by pseudouridine synthases (PUSs), which are different from the above-mentioned “writers”.248 Pseudouridylation promotes RNA stability. It is a hardly reversible procedure, because the C-C bond in Ψ is more stable than the C-N glycoside bond in uridine.249,250 Ψ can be formed in an RNA-independent or RNA-dependent manner. The RNA-independent mechanism means that PUS specifically recognizes and directly binds to the specific sequence or structure of RNA,251 whereas the RNA-dependent manner relies on the box H/ACA snoRNA complexed with proteins (known as snoRNPs), including PUSs (such as Dyskerin).252,253,254 U23 and U71 snoRNA target 18S rRNA via 5′- or 3′-hairpin elements and mediate the pseudouridylation of uridine residues at positions 97 and 410, respectively.255 Furthermore, Schwartz et al.256 found that in HEK293T cells and fibroblasts, the Ψ sites of mRNA are mainly enriched in GUUC and UGUAG motifs, which are consistent with the sites modified by Pus4 in yeast and Pus7 in other mammals, respectively. Moreover, some sites are modified by Dyskerin and are complementary to snoRNAs. A recent study, based on bisulfite-induced deletion sequencing (BID-seq), showed that the mRNA Ψ sites mainly distribute in CDS and 3′UTR.257 Environmental stimuli, such as serum starvation, significantly modulate mRNA pseudouridylation in HeLa cells, indicating Ψ may be involved in cellular growth state regulation.26
In mammalian, some PUSs are reported with biological function, including PUS1, PUS3, PUS7, PUS10, TRUB family (Pus4 homologs) and Dyskerin (a Cbf5p orthologue and coded by DKC1) (Fig. 7). The catalytic substrates of human PUS1 include tRNAs and mRNAs with structure specificity.251,258 Human PUS3 catalyzes tRNAs, usually mediating Ψ38 and Ψ39.259,260 PUS7 mainly targets the UGUAR motif in mRNA and ncRNA, especially snoRNA.5,261 Human PUS7 mediating Ψ-tRNA also relies on structural specificity, in which the T-arm and the anticodon arm are indispensable.262 PUS10 regulates Ψ54 and Ψ55 of tRNAs in the cytoplasm.263,264 Similarly, TRUB1, localized in mitochondria, is also responsible for forming Ψ55 of various tRNAs.265 TRUB1-KO causes conformation changes in these mt-tRNAs, making them more sensitive to nuclease and impair mitochondrial translation. Dyskerin mediates the pseudouridylation of rRNA and mRNA in an H/ACA snoRNA-dependent manner.266,267,268 Antonicka et al.269 identified three mitochondrial PUSs: TRUB2, RPUSD3, and RPUSD4. In human 143B cells, RPUSD4 mainly affects the pseudouridylation and stability of 16S rRNA, as well as the mitochondrial assembly, while TRUB2 and RPUSD3 mainly mediate the pseudouridylation of mRNA, thus affecting the synthesis of ATP6 and ATP8 subunits and the assembly of complex IV, respectively. Although PUS1, PUS7, RPUSD2, and TRUB2 can all mediate the Ψ-mRNA in humans, most modified sites are regulated by TRUB1 predominantly in nuclear,270,271 which Ψ-mRNAs.257 Also, it is important to note that the substrates of PUSs may be cell- and tissue- specific. Recently, methionine aminoacyl tRNAMet synthetase (MetRS) reportedly functions as a pseudouridylation “writer” in yeast, which recognizes and binds Ψ by PUS6 in tRNA and mRNA,272 suggesting that there may exist other Ψ “writers” in mammals waiting to be explored.

Pseudouridylation on RNAs and U34 modification on tRNA. The mammal pseudouridylation “writers” of RNA mainly include PUS1/3/7/10, PRUSD2-4, TRUB1/2, and Dyskerin. Dyskerin catalyzes pseudouridylation in a H/ACA snoRNA-dependent manner. Functionally, pseudouridylation is responsible for RNA processing, stability and functions. For tRNAs, U34 modifications, such as cm5U, τm5U, mcm5U, mcm5s2U and mcm5Um, play important roles in translation. ELP1-6 catalyzes cm5U methylated by ALKBH8 to produce mcm5U and its derivatives. GTPBP3 and MTO1 catalyze the τm5U of substrate mt-tRNAs, in which SHMT2 provides the raw material for methyl synthesis
A crucial biological function of Ψ is to regulate translation. It is reported that tRNA pseudouridylation regulates translation. First, PUS7 deletion significantly reduces specific tRNA-derived fragment (tRF) subgroups. For example, 5′-tRFs derive from tRNA containing a 5′-terminal oligoguanine (TOG), which are considered protein synthesis inhibitors by impairing eIF4E-mediated cap-dependent translation initiation.5,273 Cui et al.261 observed that PUS7-mediated tRNA Ψ inhibits codon-specific translation in glioblastoma stem cells (GSCs). In GSCs, PUS7-KO does not affect the overall translation efficacy, but significantly improves the translation of its target tRNA-Arg-CCG. Moreover, rRNA’s pseudouridylation also affects mRNA translation. The reduction of Ψ-rRNA in DKC1-deficient cells results in translation defects in specific mRNAs, such as p27, XIAP and Bcl-xL, in an IRES-dependent manner.274 The potential mechanism is that Ψ-rRNA directly affects the interaction between these mRNAs and the 40S subunit, thus regulating the assembly of 48S.275 In addition, the H/ACA snoRNA SNORA24-guided modification of 18S rRNA affects the ribosome’s efficiency of tRNA selection and the accuracy of translation in a codon-specific manner.276 Furthermore, the position of Ψ in mRNAs also determines different translation regulation mechanisms. In HEK293T cells, Ψ in mRNA codon increases the decoding rate of near-cognate tRNA and promotes protein synthesis.277 As an alternative translation regulation mechanism, Ψ in the termination codon significantly suppresses translation termination.257,278 Also, Ψ is beneficial to translation by inhibiting the activation of RNA-dependent protein kinase R (PKR), thus blocking the phosphorylation of eIF2A and promoting translation initiation.279 Furthermore, 2-thiouridine (s2U) and m5C have a similar effect, while 5-methyluridine (m5U) and m6A may play opposite role. Studies revealed that Ψ modification of snRNAs is important for their functions. For example, human Ψ-U1 helps a noncanonical uridine-pseudouridine interaction in the 5′-splice site and facilitates recognition.280 Ψ in U2 snRNA participates in spliceosome assembly280 and contributes to the binding and activity of the RNA-dependent ATPase Prp5,281 thus facilitating pre-mRNA splicing. Moreover, Martinez and colleagues282 observed that PUS1, PUS7 and RPUSD4-mediated Ψ enriches in the alternative splicing regions and near splice sites of pre-mRNA, which directly affects the splicing efficiency, which may be attributed to the Ψ sites overlapping with RBP sites (including splicing factors such as U2AF2) and the promoted RBP binding. Also, Ψ in 3′UTR probably regulates the cleavage and the polyadenylation of pre-mRNAs.
Modifications of U34 on tRNA
Anticodons are located at positions 34 to 36 in tRNAs, and identify codons in mRNA strictly following Watson-Crick pairing. There is one exception, however, where the anticodon at position 34 does not always obey the rule, which is known as wobble pairing. Modifications at position 34 are crucial in regulating wobble pairing, affecting protein translation and participation in some essential parts of life activities. Moreover, U34 enzymes-mediated tRNA modification is required to decode -AA codon and regulate codon-specific translation.6 The loss of U34 enzymes results in translation elongation defection, misfolded protein accumulation, and unfolded protein responses (UPRs).283,284 Tsutomu Suzuki31 mapped the post-transcriptional modifications of human tRNA and illustrated how aberrant tRNA modifications can lead to diseases in detail. Based on this and the modifications of tRNA mentioned above, this section will mainly focus on modifications, such as 5-methoxycarbonylmethyluridine (mcm5U) and its derivatives, 5-taurinomethyluridine (τm5U), and 5-carboxyaminomethyluridine (cmnm5U), at U34 on human tRNAs (Fig. 7).
Elongator complex (ELP1-6) catalyzes 5-carbamoylmethyluridine (cm5U) in the cytoplasm. Then, ALKBH8 is responsible for methylation, which corporately leads to the formation of mcm5U and its derivatives, including 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U) and 5-methoxycarbonylmethyl-2′-O-methyluridine (mcm5Um). The deficiency of ALKBH8 results in stop codon recoding and selenoprotein synthesis by impairing tRNASec(UGA).285 In breast cancer, ELP3-regulated mcm5s2U-tRNA modification of the Elongator subunit is required for IRES-dependent expression of genes associated with tumor cell invasion and metastasis.286 Moreover, ELP5, without enzymatic activity, is the critical subunit connecting ELP3 and ELP4. ELP5 depletion disrupts the integrity and stability of the Elongator complex, and ELP5-mediated the U34 tRNA modification cascade.287
In mitochondria, the point mutations cause mt-tRNALeu(UUR) lacking taurine modifications, including τm5U and 5-taurinomethyl-2-thiouridine (m5s2U), which significantly reduces UUG decoding with respiratory chain complex I deficiency by impairing the formation of codon-anticodon base pairs on the ribosomal A-site.288 GTPBP3 and MTO1 catalyze the τm5U of substrate mt-tRNAs. The human MTU1 (TRMU) is responsible for s2U34 on mt-tRNAs. Moreover, the τm5s2U is formed when both τm5U and s2U modifications exist. Mutations in GTPBP3 cause a decrease steady-state levels of respiratory complexes I and IV, as well as a defection in mitochondrial translation.289,290 The loss of mitochondrial folate enzyme serine hydroxymethyltransferase 2 (SHMT2) also affects the production of τm5U, and reduces the abundance of complex I, IV and V subunits because SHMT2-produced 5, 10-methylenetetrahydrofolate acid provides the raw material for methyl synthesis.30 However, MTU1 does not affect mitochondrial translation in human fibroblasts.291 Interestingly, mt-tRNAMet at U34 modified with 5-formylcytidine (f5C) can decode isoleucine codons AUU and AUC to methionine.292,293
Roles of RNA modification in cardiovascular diseases
Cardiovascular disease (CVD) refers to a series of diseases affecting the heart and blood vessels, which is the leading cause of global death and contributes to a large global disease burden. Prevalence and mortality of CVD increased significantly in patients over 40.294,295 Hypertension and atherosclerosis belong to the CVD and are important risk factors for other CVDs.296,297 The most common causes of CVD-related deaths include ischemic heart disease (IHD), atrial fibrillation (AF), cardiomyopathy, hypertensive heart disease, endocarditis, myocarditis, and others. The ischemic heart disease (IHD), also known as coronary heart disease (CHD), and stroke are the major culprits.297 Multiple studies revealed that RNA modifications and their regulators play essential roles in CVD (Table 1). This section mainly focuses on the heart (Fig. 8), while some vascular diseases, such as stroke and peripheral arterial disease (PAD), are not discussed.

RNA modifications in cardiovascular diseases. RNA modifications, especially m6A and A-to-I RNA editing, play crucial roles in cardiovascular diseases such as hypertension, atherosclerosis, CHD, HCM, and DCM. The total m6A levels in CHD significantly increase with the upregulation of m6A “writers” and downregulation of “erasers”. ADAR1 is increased in CVD, whereas ADAR2 is decreased. Functionally, RNA modifications participate in vascular inflammation, angiogenesis, cell death and proliferation, and cardiac functions. The red font represents upregulation/promotion, while the green font represents downregulation/inhibition. CVD cardiovascular disease, CHD coronary heart disease, HCM hypertrophic cardiomyopathy, DCM dilated cardiomyopathy, HF heart failure, PH pulmonary hypertension
Hypertension
Hypertension is the leading risk factor for CVD globally, but it is preventable.296 The occurrence of hypertension is strongly linked to genetic and environmental factors. The regulators of blood pressure (BP) are diverse and very complex, involving the renin-angiotensin-aldosterone system (RAAS), the sympathetic nervous system (SNS), the immune system, and oxidative stress,298 and regulate the function of vascular smooth muscle cells (VSCMs) together. ADAR2 is reported to be primarily responsible for FLNA editing in the cardiovascular system, where it regulates smooth muscle contraction. Mice with impaired FLNA pre-mRNA editing showed diastolic hypertension.299 Common genetic variants of FTO are reportedly associated with BP regulation in patients with hypertension.300 However, it remains unclear whether it acts in an m6A-dependent mechanism. Previous studies indirectly demonstrated that RNA modification may be involved in hypertension development. For example, HuR is a key executor for biological functions of m6A, uridylation and A-to-I RNA editing by interacting with modification sites or “readers”. HuR is reduced in the aorta of patients with hypertension, and specific HuR-cKO mice develop hypertension and cardiac hypertrophy.301 HuR reportedly binds to AREs in caveolin-1 mRNA and soluble guanylyl cyclase (sGC) mRNA and regulates BP.301,302
Atherosclerosis and coronary heart disease
Atherosclerosis is an inflammatory disease characterized by the accumulation of fat and fiber in the wall arteries, known as atherosclerotic plaque. Fibrosis and calcification occur in the coronary, leading to plaque bleeding, rupture, and thrombosis, which impedes blood flow and ultimately results in CHD. CHD mainly includes acute myocardial infarction (AMI), chronic stable angina pectoris, chronic CHD, and heart failure (HF) due to CHD.295 The m6A modification involves inflammatory cascades in extravascular sites and vascular endothelial cells (ECs), initiating and progressing atherosclerosis. The m6A “writers” METTL3 and METTL14 are significantly upregulated in atherosclerosis models and patients with CHD.303,304,305 Moreover, m6A promotes the adhesion of monocytes to ECs. Mechanistically, Chien et al.303 found that in an oscillatory shear (OS) stress-induced EC model, increased METTL3 expression promotes the stability of m6A-NLRP1 mRNA by YTHDF1, whereas it increases the degradation of m6A-KLF4 mRNA by YTHDF2. Furthermore, METTL14 mediates m6A modification of the transcription factor FOXO1 mRNA in TNF-α-induced ECs, and promotes FOXO1 mRNA translation by YTHDF1, thus activating VCAM-1 and ICAM-1transcription.304 The expression of VCAM-1 and ICAM-1 can also be regulated by lncRNA NEAT1 that is stabilized by the AUF1 (an RBP) in an ADAR1 mediated A-to-I RNA editing-dependent manner.306 However, the underlying mechanism is unclear. Macrophage inflammation is also regulated by m6A. METTL14-KD significantly reduces the stability of MyD88 mRNA in macrophages, which promotes macrophage M2 polarization and inhibits macrophage migration and adhesion by inhibiting NF-κB/IL-6 signaling.305 METTL3-m6A-YTHDF2 positively regulates oxidized low-density lipoprotein (oxLDL)-induced inflammation of macrophage and monocyte.307,308 ADAR1 and METTL3/14 also affect angiogenesis and vascular or valve calcification.231,309,310
In addition to inflammatory response and vascular function, RNA modification regulates cardiac function in CHD. For example, decreased expression of CUGBP1 in AMI models is attributed to cytoplasmic HuR re-localization and interaction with AREs in CUGBP1 3′UTR.214 CUGBP1 overexpression improves cardiac function in MI mice by modulating VEGF-A. PIWI-interacting RNA (piRNA) HAAPIR directly interacts with NAT10 and promotes CM apoptosis by enhancing ac4C acetylation of transcription factor EC (TFEC), which results in deteriorated cardiac function in MI.311 m6A levels increase with downregulated FTO expression in mammalian HF and CMs suffering from hypoxia, rather than cardiac fibroblasts (CFs) or ECs.312 FTO overexpression improves sarcomere contraction by regulating intracellular Ca2+ and cardiac function after MI, due to FTO-mediated demethylation of ATP2A2 mRNA (coding SERCA2a protein) in response to increased ATP2A2 mRNA levels. Similarly, ALKBH5 overexpression alleviates cardiac function post-MI and promotes CM proliferation via the m6A-YTHDF1-YAP axis.313 Furthermore, METTL3-m6A-HNRNPA2B1 promotes the biogenesis of extracellular vesicle (EV)-encapsulated miR-503 derived from ECs in AMI, which promotes CM apoptosis and cardiac dysfunction.314 Ischemia-reperfusion (I/R) injury is a severe cardiac ischemia complication resulting in CHD deterioration. The up-regulated METTL3 in the cardiac I/R injury model facilitates the binding of HNRNPD to m6A-TFEB mRNA with improved stability, impairs autophagy, and promotes apoptosis in CMs.315 Moreover, TFEB activates ALKBH5 transcription and inhibits METTL3 stability, thus forming a negative feedback loop. Additionally, the cardiac-specific METTL14-cKO alleviates I/R injury and cardiac dysfunction in mouse hearts with decreased expression of HF markers such as ANP, BNP, and β-MHC.316
Cardiomyopathy
Cardiomyopathy can be classified into two types: primary and secondary. Primary cardiomyopathy mainly includes hypertrophic cardiomyopathy (HCM), arrhythmogenic right ventricular cardiomyopathy (ARVC), dilated cardiomyopathy (DCM), restrictive cardiomyopathy (RCM) and myocarditis.317 Multiple genetic alterations that cause primary cardiomyopathy are identified and validated. Nearly 70% of patients with HCM have mutations in β-myosin heavy chain (MYH7) and myosin binding protein C (MYBPC3).318 Other related genes including troponin T (TNNT2), α-myosin heavy chain (MYH6), titin (TTN), muscle LIM protein (CSRP3), telethonin (TCAP), vinculin (VCL), and junctophilin 2 (JPH2), are also discovered. TTN, MYH7, and TNNT2 mutations also lead to DCM and RCM.319,320 Other DCM-related genes include LMNA, RBM20, and BAG3. GTPBP3 or MTO1 mutation-mediated impaired τm5U formation on mitochondrial tRNAs and mitochondrial dysfunction results in HCM.289,290,321 Cardiac-specific PTCD1-cKO causes DCM, because PTCD1 is essential for the Ψ on 16S rRNA at position 2509 and the stability of 16S rRNA, which affects the assembly of LSU, the mitochondrial function, and the following mTOR signaling.322 Gao et al.323 observed that cardiac-specific YTHDC1-cKO leads to DCM. Moreover, the YTHDC1 deletion results in an aberrant splicing of the m6A-modified TTN, with an increased ratio of N2BA:N2B isoforms. Gonzalez and colleagues246 reported that cardiac-specific ADAR1-cKO causes autoinflammatory myocarditis, eventually leading to DCM in mice. Furthermore, IFIH1 (coding MDA5) deficiency protects against DCM and HF by inhibiting the autoinflammatory processes caused by the absence of ADAR1-mediated RNA editing.
The etiology of secondary cardiomyopathy is usually acquired and includes diabetes, sepsis, and cardiotoxicity. In diabetic cardiomyopathy, METTL14 is down-regulated and mediates CM pyroptosis via the NLRP3-caspase-1 pathway.324 Functionally, METTL14-mediated hypermethylation of lncRNA TINCR promotes TINCR decay through YTHDF2, which weakens the inhibition of TINCR on the target NLRP3. LncRNA Airn alleviates cardiac fibrosis in diabetic cardiomyopathy by inhibiting ubiquitination degradation of IMP2 (encoded by IGF2BP2) and stabilizing p53 mRNA in an m6A-dependent manner.325 METTL14 seems to be a risk factor in doxorubicin (DOX)-induced cardiomyopathy by promoting ferroptosis.326
Cardiac hypertrophy and heat failure
Cardiac hypertrophy, both physiological and pathological, is the enlargement of the heart and CMs, due to the adaptation to cardiac stress.327 It is reported that in exercise-induced physiological cardiac hypertrophy, the total mRNA m6A levels in the heart are significantly reduced, possibly due to the METTL14 down-regulation.316 Pathological cardiac hypertrophy is a major contributor to HF with preserved ejection fraction (HFpEF) or reduced ejection fraction (HFrEF), occurring in various cardiac diseases. Cardiac-specific FTO-cKO deteriorates transverse aortic constriction (TAC) surgery-induced HFrEF in mice.328 In turn, overexpression of FTO attenuates TAC-induced cardiac hypertrophy and HF.329 Dorn et al.330 found that METTL3 overexpression induces a compensatory cardiac hypertrophy phenotype without pathological changes, even under stress stimuli such as pressure overload. Cardiac-specific METTL3-cKO results in HF or aggravates stress-induced HF in mice. The piRNA CHAPIR is identified as the upstream of METTL3.331 CHAPIR regulates cardiac hypertrophy by interacting with METTL3 and inhibiting its methylase activity, leading to the hypomethylation of the ADP-ribosyltransferase Parp10 mRNA and the inhibition of YTHDF2-mediated degradation. Moreover, MYH7, a marker of cardiac hypertrophy, is also a target of YTHDF2. YTHDF2 is up-regulated in HF and promotes the MYH7 stability in an m6A-dependent manner.332 Based on the sequencing technique, Kokot et al.227 explored the regulation of circRNAs by A-to-I RNA editing in human failing hearts. Their results showed that HF leads to decreased RNA editing with reduced ADAR2 protein and increased circRNA formation.
Others
Pulmonary hypertension (PH) can be divided into five groups. Group 2 PH is caused by left heart disease, which is typically complicated in more than 50% of patients with HFpEF or HFrEF.333 PH is characterized by the proliferation and apoptosis-resistance of pulmonary artery smooth muscle cells (PASMCs).334 This phenotypic switch leads to pulmonary vascular remodeling (PVR). In PH, increased total RNA m6A levels are accompanied by elevated expression of some “writers” and “readers”, such as METTL3/14 and YTHDF1/2.335,336 In these cases, METTL3/14 catalyzes the methylation of downstream genes associated with proliferation, while YTHDF1/2 promotes their translation and degradation. For example, the translation of MAGED1 is up-regulated by the METTL3-m6A-YTHDF1 axis.335 Functionally, MAGED1-KO inhibits hypoxia-induced PASMC proliferation and improves PVR and cardiac function in mice exposed to SU5416/hypoxia (Su/Hx). Chronic inflammation also contributes to PVR. YTHDF2 reportedly up-regulates in pulmonary macrophages of PH. YTHDF2 promotes PASMC proliferation by enhancing the m6A-HMOX1 degradation, which activates macrophage, inflammatory response, and oxidative stress. Myeloid-specific YTHDF2-cKO inhibits macrophage activation, PVR, and cardiac dysfunction in Su/Hx-induced PH mice.337
Severe arrhythmias, such as ventricular tachycardia (VT), ventricular fibrillation (VF) and AF, can lead to death. Among them, ventricular arrhythmias (VAs), including VT and VF, are the leading causes of sudden cardiac death (SCD), while AF is usually closely related to stroke and HF.338 Patients with MI or HF are often observed with increased sympathetic nerve activity which can lead to frequent arrhythmias.339 In MI animal models, the expression levels of METTL3 are upregulated in the heart and paraventricular nucleus (PVN), the cardiovascular regulatory region in the brain, and are responsible for receiving cardiac reflex information and regulating sympathetic nerve activity for feedback.340,341 It is associated with the activation of the NF-κB pathway, mediated by TRAF6 and TLR4, respectively. Conversely, METTL3 silencing inhibits sympathetic remodeling and hyperactivity, which is beneficial in attenuating MI and improving cardiac function. Similarly, elevated METTL3 expression also regulates cardiac fibrosis and AF in an m6A-dependent manner.342
Aortic dissection (AD) is a dangerous and fatal disease. Hypertension and atherosclerosis are major risk factors for AD.343 The loss of aortic smooth muscle cells (ASMCs) is one of the characteristics of AD. Li et al.344 reported that METTL3/14 expression is significantly increased with reduced FTO expression in the aortas of patients with Stanford type A AD (TAAD) compared to non-AD aortas. METTL3 may promote ferroptosis in human ASMCs by facilitating the degradation of m6A-modified SLC7A11 and FSP1 mRNA. However, more studies are required to confirm the functions and specific molecular mechanisms of RNA m6A modification in AD.
RNA modification-based therapeutics for cardiovascular disease
Regulators of RNA modification are involved in the initiation and development of CVD by affecting various phenotypes of different cells in the cardiovascular system and associated signaling pathways. The principles of RNA modification-based therapeutic strategies for CVD mainly include: inhibiting inflammatory response, reducing cell loss, attenuating damage, and promoting regeneration or alleviating remodeling. Moreover, RNA modification-based therapeutic strategies may act through multiple mechanisms and lead to better outcomes (Table 2).
Cardiac development and regeneration
The regenerative capacity of the adult mammalian heart is very limited. The loss of CMs can be permanent and difficult to repair. Thus, stimulating CM dedifferentiation and proliferation provides a promising perspective for cardiac regeneration strategies.345 NSUN5-mediated RNA m5C modification is required for the translation of TPM1, a cardiac development-related gene.346 NSUN5 mutation may cause decreased cell proliferation in the cardiac outflow tract (OFT) and OFT malformations in mice. Chen and colleagues223 explored the role of A-to-I RNA editing in the differentiation of human CMs. Their results showed that the proportion of editing sites in 3′UTRs decreases gradually and conversely, and the proportion of editing sites in introns increases during the process from undifferentiated cells to differentiated CMs. ADAR1 is essential for embryonic cardiac growth and development. Moore et al.347 observed that ADAR1-cKO results in apoptosis, reduced CM proliferation, and embryonic death. Han and colleagues348 revealed that total m6A levels increase with reduced ALKBH5 expression during the differentiation of mesoderm cells (MESs) into CMs. Consistently, Yang et al.349 found that the methylation peak in the heart of mice increases after birth and may plateau after 7 days. Furthermore, YTHDF1 and YTHDF3 are downregulated during the differentiation of embryonic stem cells (ESCs) into CMs.350 However, YTHDF1 and YTHDF3 regulate the expression of cardiac genes in CMs in opposite ways. Compared to embryonic hearts, m6A modification increases the expression of miR-133a and miR-499 in postnatal mouse hearts, thereby enhancing the inhibition of target genes involved in cardiac development.351 The cell cycle regulators, such as cyclins, cell cycle-independent kinases (CDKs) and proto-oncoproteins, promote CM proliferation.345 ALKBH5 promotes CM proliferation by positively regulating YAP expression, whereas METTL3 inhibits YAP expression via a negative regulation. Specifically, ALKBH5 is a demethylase of YAP and promotes its translation in a YTHDF1-dependent manner. Moreover, METTL3-mediated methylation of pri-miR-143 facilitates its maturation and inhibits the expression of YAP.313,352 In vivo injection of adeno-associated virus 9 (AAV9)-shMETTL3 promotes CM proliferation and heart regeneration in the injured heart but has no significant effect on the heart without injury.353 In addition, METTL14 is also thought to be beneficial to CM apoptosis and detrimental to CM proliferation by promoting the interaction of YTHDF1 with m6A-PHLPP2 mRNA with improved translation.316 However, ALKBH5 negatively regulates angiogenesis in cardiac microvascular endothelial cells (CMECs), detrimental to blood flow recovery in ischemic diseases.354
Preventing cell loss and remodeling
Cell loss is one of the critical pathologic bases in various CVDs. For example, the loss of CMs leads to fibrosis, decreased cardiac contraction, and pathological cardiac dilatation, known as pathological cardiac remodeling, which subsequently deteriorates these processes and ultimately results in HF.355 The leading causes of cell loss in CVDs include apoptosis, necrosis, ferroptosis, and pyroptosis.356 Cheng et al.357 designed a nanomedicine to ameliorate myocardial I/R injury based on the ALKBH5 inhibitor IOX1, called HSSS-I. One advantage of HSSS-I is that it can actively target the infarct area and inhibit apoptosis. In addition, exercise may significantly improve cardiac function by attenuating RNA editing and m6A-mediated CM apoptosis.229,358 Exercise also alleviates atherosclerosis by inhibiting m6A-regulated EC pyroptosis.359 Intermittent fasting (IF) may also inhibit CM apoptosis in high-fat diet-induced obesity cardiomyopathy through a similar mechanism.360 Although exercise and IF are unlikely to be treated clinically, they may benefit patients as health management strategies. The tail vein injection of lentivirus (LV)-shMETTL3 significantly inhibits pyroptosis in myocardial I/R injury mouse models.361 Liproxstatin-1, an inhibitor of ferroptosis, inhibits β-aminopropionitrile (BAPN)-induced medial degeneration and fragmentation of elastin in the aorta, significantly reducing morbidity and mortality.344 Similarly, MCC950, a pyroptosis inhibitor, partially attenuates cardiac injury in diabetic cardiomyopathy.324 It is unclear whether these inhibitors directly affect RNA modification or may act on the modified or downstream targets. We look forward to more studies to discover agents directly targeting RNA modification regulators and explore mechanisms and therapeutic effects. Additionally, AAV9-shMETTL3 or AAV9-FTO-overexpression via intravenous injection reportedly inhibits cardiac dysfunction and remodeling in mouse models of myocardial hypertrophy.329,362
Others
Cell proliferation is not always beneficial for CVD in all cases. For example, m6A-mediated aberrant proliferation of CFs results in cardiac fibrosis. Injection of LV-shMETTL3 or LV-shYTHDF2 significantly alleviates isoproterenol (ISO)-induced cardiac fibrosis in mice.325,342,363 Preventing ASMC proliferation and neointima formation after stenting or coronary artery bypass grafting improves patient outcomes. METTL3-mediated m6A modification promotes autophagosome formation to suppress ASMC proliferation, suggesting METTL3 as a potential therapeutic target.364 A clinical trial showed that arsenic trioxide (ATO) could be applied to drug-eluting stents with comparable efficacy and safety to traditional sirolimus-eluting ones.365 Yu et al.366 demonstrated that ATO selectively promotes apoptosis in ASMC by regulating m6A modification.
The application and mechanism of extracts from some natural plants or traditional Chinese medicines for CVD based on RNA modification have also received much attention. Maslinic acid (MA), a pentacyclic triterpenoid rich in olive pericarp, inhibits cardiac hypertrophy by decreasing total m6A levels and METTL3 expression.367 However, this mechanism has not been clarified. Tanshinone IIA (Tan IIA), an extract of traditional Chinese medicine Salvia miltiorrhiza Bunge, inhibits ALKBH5-mediated m6A modification of galectin-3, and may have a similar effect for CVD.368
Conclusions and perspectives
Post-transcriptional modifications widely exist in transcripts, regulating gene expression, affecting cell differentiation and organ development, and determining cell fate. These chemical processes are dynamic based on the roles of “writers” and “erasers”, which are reportedly reversible (such as m1A and m6A) or transformed (for example, from m5C to hm5C), and thus impact RNA metabolisms and biological functions. The dynamic feature is also reflected in changes in the RNA modification distribution and the regulator expression during cell growth, differentiation, and organ development. It suggests that changes in chemical modification may allow cells to adapt to internal or external stimuli. For example, serum starvation, glucose deprivation, hypoxia and pressure-overload stimulation alter the levels of m1A, m6A, and pseudouridylation and the expression of regulators.26,142,146,312,316 Cellular and tissue damage induced by these stimuli is a crucial therapeutic mechanism in CVDs, including MI and myocardial I/R injury. Chemical modification promotes or repeals the recruitment or accessibility of regulatory elements, especially RBPs, directly or with the synergy of “readers”, thus controlling downstream signaling, such as inflammatory response, an initial event or exacerbating factor of CVDs. These suggest that RNA modification can either respond to stimuli or lead to changes in the microenvironment, and there may be a complex cross-talk.
Despite tremendous progress, there are still many unknown or controversial issues. First, RNA modification regulators prefer substrates, sites, and functions. How regulators choose substrates and specific sites, how substrates are altered with condition changes, and how “readers” perform one of their functions need to be elucidated. Cell- or tissue-specific RNA modification patterns or regulators are observed. YTHDF1-3 are characterized as m6A “readers”. They are also candidates for m1A “readers” in Hela cell,24,369,370 while only YTHDF3 is reconfirmed in HEK293T and Raw264.7 cells,371 suggesting the potential cell specificity. However, false positive results cannot be excluded either. For instance, YTHDF2 reportedly mediates m1A-modified RNA degradation in an m6A-dependent manner rather than acts as an m1A “reader” in Hela cell.372 The direct binding of HRSP12 to the m1A motif promotes the binding of YTHDF2 to the m6A site, resulting in RNase P/MRP complex-mediated endoribonucleolytic cleavage. Cross-talk between chemical modification makes the regulatory mechanism even more intricate. We propose three types of cross-talk: 1) a transcript can be modified by different modifications with synergy or rejection effect by different regulators or a shared regulator (for example, m1A/m6A “eraser” FTO and m6A/m5C “reader” YTHDF2); 2) regulators of one modification are modified by another modification; 3) There are direct interactions between regulators of different modifications. Both m6A and A-to-I RNA editing are widely present in RNA, and developing techniques for detecting RNA modification may benefit CVD treatments. Although the mechanisms of m5C DNA methylation in myocardial infarction and dilated cardiomyopathy have been reported,373,374 there are few studies of m5C on RNA in CVD. Several studies have explored the role of RBPs in regulating the expression of genes enriched with AREs in CVD. However, the role of uridylyl transferases in the dynamic regulation of uridylation in CVD remains unclear. Effective prevention and intervention of CVD remains challenging; hence CVD continues to be a leading cause of death worldwide. The introduction of precision medicine and its application in CVD is expected to improve the prognosis of patients, based on the development of pan-omics, including genomics, transcriptomics, epigenomics, and proteomics.375 Fortunately, RNA modification-based therapeutic strategies have shown certain effects on CVD. Gene therapy using viral vectors to transfer genes to CVD is promising since several clinical trials have demonstrated its safety and efficacy.376 RNA modification-based gene therapy shows therapeutic effects in animal CVD models through various mechanisms. For example, interference with AAV9/LV-shMETTL3 promotes cardiomyocyte proliferation, inhibits cardiomyocyte death, and attenuates cardiac remodeling.330,342,363 It should be noted that the role of a regulator may be totally reversed in different cell types, and this could also be a potential problem for gene therapy, indicating the need for specific cell targeting to provide high therapeutic efficacy and avoid adverse effects. Nanotechnology could be a viable solution.377 Nanomedicine has the advantage of precise targeting with reduced side effects, facilitating combination therapy by a nanoplatform, and ultimately delivering better therapeutic effects at lower doses. Moreover, the potential of some FDA-approved drugs and compounds derived from natural plants or traditional Chinese herbal medicines for treating CVD should not be overlooked. They may be more accessible to clinical applications with potentially higher cost-effectiveness than nanomedicine, which remains an open clinical translation challenge, such as pharmaceutical metabolism in vivo. In addition, we expect to see the identification of more molecular inhibitors or activators targeting RNA modification regulators, particularly for CVD.
References
Deng, S. et al. RNA m(6)A regulates transcription via DNA demethylation and chromatin accessibility. Nat. Genet. 54, 1427–1437 (2022).
Takahashi, M. et al. The tumor suppressor kinase DAPK3 drives tumor-intrinsic immunity through the STING-IFN-β pathway. Nat. Immunol. 22, 485–496 (2021).
Karikó, K., Buckstein, M., Ni, H. & Weissman, D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23, 165–175 (2005).
Yoon, K. J. et al. Temporal control of mammalian cortical neurogenesis by m(6)A methylation. Cell 171, 877–889.e817 (2017).
Guzzi, N. et al. Pseudouridylation of tRNA-derived fragments steers translational control in stem cells. Cell 173, 1204–1216.e1226 (2018).
Rapino, F. et al. Codon-specific translation reprogramming promotes resistance to targeted therapy. Nature 558, 605–609 (2018).
Liu, Y. et al. tRNA-m(1)A modification promotes T cell expansion via efficient MYC protein synthesis. Nat. Immunol. 23, 1433–1444 (2022).
Yankova, E. et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature 593, 597–601 (2021).
Delaunay, S. et al. Mitochondrial RNA modifications shape metabolic plasticity in metastasis. Nature 607, 593–603 (2022).
Liu, H. et al. Targeting tumour-intrinsic N(7)-methylguanosine tRNA modification inhibits MDSC recruitment and improves anti-PD-1 efficacy. Gut 72, 1555–1567 (2023).
Jones, P. A., Issa, J.-P. J. & Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 17, 630–641 (2016).
Barbieri, I. & Kouzarides, T. Role of RNA modifications in cancer. Nat. Rev. Cancer 20, 303–322 (2020).
Bartee, D., Thalalla Gamage, S., Link, C. N. & Meier, J. L. Arrow pushing in RNA modification sequencing. Chem. Soc. Rev. 50, 9482–9502 (2021).
Wiener, D. & Schwartz, S. The epitranscriptome beyond m(6)A. Nat. Rev. Genet. 22, 119–131 (2021).
Boccaletto, P. et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 46, D303–d307 (2018).
Liu, N. et al. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518, 560–564 (2015).
Alarcón, C. R. et al. HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell 162, 1299–1308 (2015).
Wang, X. et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120 (2014).
Chen, X. et al. 5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs. Nat. Cell Biol. 21, 978–990 (2019).
Yang, X. et al. 5-methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Res. 27, 606–625 (2017).
Yang, Y. et al. RNA 5-methylcytosine facilitates the maternal-to-zygotic transition by preventing maternal mRNA decay. Mol. Cell 75, 1188–1202.e1111 (2019).
Zhang, Y. et al. Dnmt2 mediates intergenerational transmission of paternally acquired m etabolic disorders through sperm small non-coding RNAs. Nat. Cell Biol. 20, 535–540 (2018).
Dominissini, D. et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature 530, 441–446 (2016).
Wu, Y. et al. RNA m(1)A methylation regulates glycolysis of cancer cells through modulating ATP5D. Proc. Natl. Acad. Sci. USA 119, e2119038119 (2022).
Feng, Z. et al. The LINC00623/NAT10 signaling axis promotes pancreatic cancer progress ion by remodeling ac4C modification of mRNA. J. Hematol. Oncol. J. Hematol Oncol 15, 112 (2022).
Carlile, T. M. et al. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515, 143–146 (2014).
Ohira, T. et al. Reversible RNA phosphorylation stabilizes tRNA for cellular thermotolerance. Nature 605, 372–379 (2022).
Alexandrov, A. et al. Rapid tRNA decay can result from lack of nonessential modifications. Mol. Cell 21, 87–96 (2006).
Van Haute, L. et al. NSUN2 introduces 5-methylcytosines in mammalian mitochondrial tRNAs. Nucleic Acids Res. 47, 8720–8733 (2019).
Morscher, R. J. et al. Mitochondrial translation requires folate-dependent tRNA methylation. Nature 554, 128–132 (2018).
Suzuki, T. The expanding world of tRNA modifications and their disease relevance. Nat. Rev. Mol. Cell Biol. 22, 375–392 (2021).
Sharma, S. & Lafontaine, D. L. J. ‘View from a bridge’: a new perspective on eukaryotic rRNA base modification. Trends Biochem. Sci. 40, 560–575 (2015).
Liu, H. T. et al. lncRNA THAP7-AS1, transcriptionally activated by SP1 and post-transcriptionally stabilized by METTL3-mediated m6A modification, exerts oncogenic properties by improving CUL4B entry into the nucleus. Cell Death Differ. 29, 627–641 (2022).
Li, Z. X. et al. WTAP-mediated m(6)A modification of lncRNA DIAPH1-AS1 enhances its stability to facilitate nasopharyngeal carcinoma growth and metastasis. Cell Death Differ. 29, 1137–1151 (2022).
Chen, R. X. et al. N(6)-methyladenosine modification of circNSUN2 facilitates cytoplasmic export and stabilizes HMGA2 to promote colorectal liver metastasis. Nat. Commun. 10, 4695 (2019).
Wang, J. X. et al. Oxidative modification of miR-184 enables it to target Bcl-xL and Bcl-w. Mol. Cell 59, 50–61 (2015).
Jones, M. R. et al. Zcchc11-dependent uridylation of microRNA directs cytokine expression. Nat. Cell Biol. 11, 1157–1163 (2009).
Fan, J. et al. Long non-coding RNA ROR decoys gene-specific histone methylation to promote tumorigenesis. Genome Biol. 16, 139 (2015).
Sun, T. T. et al. LncRNA GClnc1 promotes gastric carcinogenesis and may act as a modular scaffold of WDR5 and KAT2A complexes to specify the histone modification pattern. Cancer Discov. 6, 784–801 (2016).
Tsai, M.-C. et al. Long noncoding RNA as modular scaffold of histone modification complexes. Science 329, 689–693 (2010).
Wei, C. M., Gershowitz, A. & Moss, B. Methylated nucleotides block 5’ terminus of HeLa cell messenger RNA. Cell 4, 379–386 (1975).
Furuichi, Y. et al. Methylated, blocked 5 termini in HeLa cell mRNA. Proc. Natl. Acad. Sci. USA 72, 1904–1908 (1975).
Dominissini, D. et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 (2012).
Meyer, K. D. et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 149, 1635–1646 (2012).
Zaccara, S., Ries, R. J. & Jaffrey, S. R. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol 20, 608–624 (2019).
Liu, J. et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10, 93–95 (2014).
Bokar, J. A., Shambaugh, M. E., Polayes, D., Matera, A. G. & Rottman, F. M. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 3, 1233–1247 (1997).
Ping, X. L. et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 24, 177–189 (2014).
Wang, P., Doxtader, K. A. & Nam, Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol. Cell 63, 306–317 (2016).
Wang, X. et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature 534, 575–578 (2016).
Warda, A. S. et al. Human METTL16 is a N(6)-methyladenosine (m(6)A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 18, 2004–2014 (2017).
Yue, Y. et al. VIRMA mediates preferential m(6)A mRNA methylation in 3’UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 4, 10 (2018).
Patil, D. P. et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537, 369–373 (2016).
van Tran, N. et al. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 47, 7719–7733 (2019).
Harper, J. E., Miceli, S. M., Roberts, R. J. & Manley, J. L. Sequence specificity of the human mRNA N6-adenosine methylase in vitro. Nucleic Acids Res. 18, 5735–5741 (1990).
Csepany, T., Lin, A., Baldick, C. J. Jr. & Beemon, K. Sequence specificity of mRNA N6-adenosine methyltransferase. J. Biol. Chem. 265, 20117–20122 (1990).
Bokar, J. A., Rath-Shambaugh, M. E., Ludwiczak, R., Narayan, P. & Rottman, F. Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J. Biol. Chem. 269, 17697–17704 (1994).
Jia, G. et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887 (2011).
Zheng, G. et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 49, 18–29 (2013).
Zhao, X. et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 24, 1403–1419 (2014).
Bartosovic, M. et al. N6-methyladenosine demethylase FTO targets pre-mRNAs and regulates alternative splicing and 3’-end processing. Nucleic Acids Res. 45, 11356–11370 (2017).
Xiao, W. et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol. Cell 61, 507–519 (2016).
Li, S. et al. Nuclear Aurora kinase A switches m(6)A reader YTHDC1 to enhance an oncogenic RNA splicing of tumor suppressor RBM4. Signal Transduct. Target. Ther. 7, 97 (2022).
Chen, L. et al. Nuclear m(6) A reader YTHDC1 suppresses proximal alternative polyadenylation sites by interfering with the 3’ processing machinery. EMBO Rep. 23, e54686 (2022).
Kasowitz, S. D. et al. Nuclear m6A reader YTHDC1 regulates alternative polyadenylation and splicing during mouse oocyte development. PLoS Genet. 14, e1007412 (2018).
Sun, S. et al. RNA binding protein NKAP protects glioblastoma cells from ferroptosis by promoting SLC7A11 mRNA splicing in an m(6)A-dependent manner. Cell Death Dis. 13, 73 (2022).
Alarcón, C. R., Lee, H., Goodarzi, H., Halberg, N. & Tavazoie, S. F. N6-methyladenosine marks primary microRNAs for processing. Nature 519, 482–485 (2015).
Zhang, J. et al. Excessive miR-25-3p maturation via N(6)-methyladenosine stimulated by cigarette smoke promotes pancreatic cancer progression. Nat. Commun. 10, 1858 (2019).
Ma, J. Z. et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary MicroRNA processing. Hepatology 65, 529–543 (2017).
Liu, Z. et al. A methyltransferase-like 14/miR-99a-5p/tribble 2 positive feedback circuit promotes cancer stem cell persistence and radioresistance via histone deacetylase 2-mediated epigenetic modulation in esophageal squamous cell carcinoma. Clin. Transl. Med. 11, e545 (2021).
Liu, N. et al. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 45, 6051–6063 (2017).
Zhou, K. I. et al. Regulation of co-transcriptional pre-mRNA splicing by m(6)A through the low-complexity protein hnRNPG. Mol. Cell 76, 70–81.e79 (2019).
Pendleton, K. E. et al. The U6 snRNA m(6)A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell 169, 824–835.e814 (2017).
Aoyama, T., Yamashita, S. & Tomita, K. Mechanistic insights into m6A modification of U6 snRNA by human METTL16. Nucleic Acids Res. 48, 5157–5168 (2020).
Tang, C. et al. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3’-UTR mRNAs in male germ cells. Proc. Natl. Acad. Sci. USA 115, E325–e333 (2018).
Wei, G. et al. Acute depletion of METTL3 implicates N (6)-methyladenosine in alternative intron/exon inclusion in the nascent transcriptome. Genome Res. 31, 1395–1408 (2021).
Roundtree, I. A. et al. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife 6, e31311 (2017).
Chen, X. et al. m(6)A modification of circSPECC1 suppresses RPE oxidative damage and maintains retinal homeostasis. Cell Rep. 41, 111671 (2022).
Edens, B. M. et al. FMRP modulates neural differentiation through m(6)A-dependent mRNA nuclear export. Cell Rep 28, 845–854.e845 (2019).
Batista, P. J. et al. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15, 707–719 (2014).
Ke, S. et al. A majority of m6A residues are in the last exons, allowing the potential for 3’ UTR regulation. Genes Dev 29, 2037–2053 (2015).
Slobodin, B. et al. Transcription dynamics regulate poly(A) tails and expression of the RNA degradation machinery to balance mRNA levels. Mol. Cell 78, 434–444.e435 (2020).
Du, H. et al. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat. Commun. 7, 12626 (2016).
Liu, L. et al. The N6-methyladenosine modification enhances ferroptosis resistance through inhibiting SLC7A11 mRNA deadenylation in hepatoblastoma. Clin. Transl. Med. 12, e778 (2022).
Zhang, G. et al. Dynamic FMR1 granule phase switch instructed by m6A modification contributes to maternal RNA decay. Nat. Commun. 13, 859 (2022).
Li, F. et al. N(6)-methyladenosine modulates nonsense-mediated mRNA Decay in human glioblastoma. Cancer Res. 79, 5785–5798 (2019).
Liu, J. et al. N (6)-methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science 367, 580–586 (2020).
Cheng, Y. et al. N(6)-Methyladenosine on mRNA facilitates a phase-separated nuclear body that suppresses myeloid leukemic differentiation. Cancer Cell 39, 958–972.e958 (2021).
Wojtas, M. N. et al. Regulation of m(6)A transcripts by the 3’→5’ RNA helicase YTHDC2 is essential for a successful meiotic program in the mammalian germline. Mol. Cell 68, 374–387.e312 (2017).
Jin, D. et al. m(6)A demethylase ALKBH5 inhibits tumor growth and metastasis by reducing YTHDFs-mediated YAP expression and inhibiting miR-107/LATS2-mediated YAP activity in NSCLC. Mol. Cancer 19, 40 (2020).
Zhou, C. et al. Genome-wide maps of m6A circRNAs identify widespread and cell-type-specific methylation patterns that are distinct from mRNAs. Cell Rep. 20, 2262–2276 (2017).
Shi, H. et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 27, 315–328 (2017).
Zaccara, S. & Jaffrey, S. R. A unified model for the function of YTHDF proteins in regulating m(6)A-modified mRNA. Cell 181, 1582–1595.e1518 (2020).
Lasman, L. et al. Context-dependent functional compensation between Ythdf m(6)A reader proteins. Genes Dev. 34, 1373–1391 (2020).
Xia, T. L. et al. N(6)-methyladenosine-binding protein YTHDF1 suppresses EBV replication and promotes EBV RNA decay. EMBO Rep. 22, e50128 (2021).
Li, J. et al. YTHDF1 promotes mRNA degradation via YTHDF1-AGO2 interaction and phase separation. Cell Prolif 55, e13157 (2022).
Ni, W. et al. Long noncoding RNA GAS5 inhibits progression of colorectal cancer by interacting with and triggering YAP phosphorylation and degradation and is negatively regulated by the m(6)A reader YTHDF3. Mol. Cancer 18, 143 (2019).
Hu, Y. et al. A reciprocal feedback between N6-methyladenosine reader YTHDF3 and lncRNA DICER1-AS1 promotes glycolysis of pancreatic cancer through inhibiting maturation of miR-5586-5p. J. Exp. Clin. Cancer Res. 41, 69 (2022).
Zhang, F. et al. Fragile X mental retardation protein modulates the stability of its m6A-marked messenger RNA targets. Hum. Mol. Genet. 27, 3936–3950 (2018).
Zhu, Y. et al. LncRNA LINC00942 promotes chemoresistance in gastric cancer by suppressing MSI2 degradation to enhance c-Myc mRNA stability. Clin. Transl. Med. 12, e703 (2022).
Huang, H. et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat. Cell Biol. 20, 285–295 (2018).
Zhu, S. et al. An oncopeptide regulates m(6)A recognition by the m(6)A reader IGF2BP1 and tumorigenesis. Nat. Commun. 11, 1685 (2020).
Bechara, R. et al. The m(6)A reader IMP2 directs autoimmune inflammation through an IL-17- and TNFα-dependent C/EBP transcription factor axis. Sci. Immunol. 6, eabd1287 (2021).
Müller, S. et al. IGF2BP1 promotes SRF-dependent transcription in cancer in a m6A- and miRNA-dependent manner. Nucleic Acids Res. 47, 375–390 (2019).
Müller, S. et al. The oncofetal RNA-binding protein IGF2BP1 is a druggable, post-transcriptional super-enhancer of E2F-driven gene expression in cancer. Nucleic Acids Res. 48, 8576–8590 (2020).
Liu, Y. et al. LncRNA-PACERR induces pro-tumour macrophages via interacting with miR-671-3p and m6A-reader IGF2BP2 in pancreatic ductal adenocarcinoma. J. Hematol. Oncol. 15, 52 (2022).
Zhou, L. et al. Hypoxia-induced lncRNA STEAP3-AS1 activates Wnt/β-catenin signaling to promote colorectal cancer progression by preventing m(6)A-mediated degradation of STEAP3 mRNA. Mol. Cancer 21, 168 (2022).
Chen, Z. et al. rtcisE2F promotes the self-renewal and metastasis of liver tumor-initiating cells via N(6)-methyladenosine-dependent E2F3/E2F6 mRNA stability. Sci. China Life Sci. 65, 1840–1854 (2022).
Yang, H. et al. Hypoxia inducible lncRNA-CBSLR modulates ferroptosis through m6A-YTHDF2-dependent modulation of CBS in gastric cancer. J. Adv. Res. 37, 91–106 (2022).
Zhu, P. et al. A novel hypoxic long noncoding RNA KB-1980E6.3 maintains breast cancer stem cell stemness via interacting with IGF2BP1 to facilitate c-Myc mRNA stability. Oncogene 40, 1609–1627 (2021).
Yang, L. et al. Hsa_circ_0004287 inhibits macrophage-mediated inflammation in an N(6)-methyladenosine-dependent manner in atopic dermatitis and psoriasis. J. Allergy Clin. Immunol. 149, 2021–2033 (2022).
Chen, Y. et al. WTAP facilitates progression of hepatocellular carcinoma via m6A-HuR-dependent epigenetic silencing of ETS1. Mol. Cancer 18, 127 (2019).
Yue, B. et al. METTL3-mediated N6-methyladenosine modification is critical for epithelial-mesenchymal transition and metastasis of gastric cancer. Mol. Cancer 18, 142 (2019).
Lan, T. et al. KIAA1429 contributes to liver cancer progression through N6-methyladenosine-dependent post-transcriptional modification of GATA3. Mol. Cancer 18, 186 (2019).
Zhang, Y. et al. The m(6)A demethylase ALKBH5-mediated upregulation of DDIT4-AS1 maintains pancreatic cancer stemness and suppresses chemosensitivity by activating the mTOR pathway. Mol. Cancer 21, 174 (2022).
Wang, Y. et al. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 16, 191–198 (2014).
Edupuganti, R. R. et al. N(6)-methyladenosine (m(6)A) recruits and repels proteins to regulate mRNA homeostasis. Nat. Struct. Mol. Biol. 24, 870–878 (2017).
Leppek, K., Das, R. & Barna, M. Functional 5’ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 19, 158–174 (2018).
Ishigaki, Y., Li, X., Serin, G. & Maquat, L. E. Evidence for a pioneer round of mRNA translation: mRNAs subject to nonsense-mediated decay in mammalian cells are bound by CBP80 and CBP20. Cell 106, 607–617 (2001).
Lin, S., Choe, J., Du, P., Triboulet, R. & Gregory, R. I. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell 62, 335–345 (2016).
Choe, J. et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature 561, 556–560 (2018).
Su, R. et al. METTL16 exerts an m(6)A-independent function to facilitate translation and tumorigenesis. Nat. Cell Biol. 24, 205–216 (2022).
Wang, F. et al. METTL16 promotes translation and lung tumorigenesis by sequestering cytoplasmic eIF4E2. Cell Rep. 42, 112150 (2023).
Wang, X. et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 161, 1388–1399 (2015).
Lin, X. et al. RNA m(6)A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of Snail. Nat. Commun. 10, 2065 (2019).
Mao, Y. et al. m(6)A in mRNA coding regions promotes translation via the RNA helicase-containing YTHDC2. Nat. Commun. 10, 5332 (2019).
Li, A. et al. Cytoplasmic m(6)A reader YTHDF3 promotes mRNA translation. Cell Res. 27, 444–447 (2017).
Coots, R. A. et al. m(6)A facilitates eIF4F-independent mRNA translation. Mol. Cell 68, 504–514.e507 (2017).
Yang, Y. et al. Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res. 27, 626–641 (2017).
Meyer, K. D. et al. 5’ UTR m(6)A promotes cap-independent translation. Cell 163, 999–1010 (2015).
Ma, H. et al. N(6-)Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat. Chem. Biol. 15, 88–94 (2019).
Pinto, R. et al. The human methyltransferase ZCCHC4 catalyses N6-methyladenosine modification of 28S ribosomal RNA. Nucleic Acids Res. 48, 830–846 (2020).
Ignatova, V. V. et al. The rRNA m(6)A methyltransferase METTL5 is involved in pluripotency and developmental programs. Genes Dev. 34, 715–729 (2020).
Rong, B. et al. Ribosome 18S m(6)A methyltransferase METTL5 promotes translation initiation and breast cancer cell growth. Cell Rep. 33, 108544 (2020).
Choi, J. et al. N(6)-methyladenosine in mRNA disrupts tRNA selection and translation-elongation dynamics. Nat. Struct. Mol. Biol. 23, 110–115 (2016).
Grozhik, A. V. et al. Antibody cross-reactivity accounts for widespread appearance of m(1)A in 5’UTRs. Nat. Commun. 10, 5126 (2019).
Bar-Yaacov, D. et al. Mitochondrial 16S rRNA is methylated by tRNA methyltransferase TRMT61B in all vertebrates. PLoS Biol. 14, e1002557 (2016).
Chujo, T. & Suzuki, T. Trmt61B is a methyltransferase responsible for 1-methyladenosine at position 58 of human mitochondrial tRNAs. RNA 18, 2269–2276 (2012).
Vilardo, E. et al. A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase–extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res. 40, 11583–11593 (2012).
Li, X. et al. Base-resolution mapping reveals distinct m(1)A methylome in nuclear- and mitochondrial-encoded transcripts. Mol. Cell 68, 993–1005.e1009 (2017).
Safra, M. et al. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature 551, 251–255 (2017).
Li, X. et al. Transcriptome-wide mapping reveals reversible and dynamic N(1)-methyladenosine methylome. Nat. Chem. Biol. 12, 311–316 (2016).
Woo, H. H. & Chambers, S. K. Human ALKBH3-induced m(1)A demethylation increases the CSF-1 mRNA stability in breast and ovarian cancer cells. Biochim. Biophys. Acta Gene Regul. Mech. 1862, 35–46 (2019).
Kuang, W. et al. ALKBH3-dependent m(1)A demethylation of Aurora A mRNA inhibits ciliogenesis. Cell Discov. 8, 25 (2022).
Chen, Z. et al. Transfer RNA demethylase ALKBH3 promotes cancer progression via induction of tRNA-derived small RNAs. Nucleic Acids Res. 47, 2533–2545 (2019).
Liu, F. et al. ALKBH1-mediated tRNA demethylation regulates translation. Cell 167, 816–828.e816 (2016).
Zhang, L. S. et al. ALKBH7-mediated demethylation regulates mitochondrial polycistronic RNA processing. Nat. Cell Biol. 23, 684–691 (2021).
Wei, J. et al. Differential m(6)A, m(6)A(m), and m(1)A demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol. Cell 71, 973–985.e975 (2018).
Guo, G. et al. Advances in mRNA 5-methylcytosine modifications: Detection, effectors, biological functions, and clinical relevance. Mol. Ther. Nucleic Acids 26, 575–593 (2021).
Squires, J. E. et al. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 40, 5023–5033 (2012).
Huang, T., Chen, W., Liu, J., Gu, N. & Zhang, R. Genome-wide identification of mRNA 5-methylcytosine in mammals. Nat. Struct. Mol. Biol. 26, 380–388 (2019).
Van Haute, L. et al. Deficient methylation and formylation of mt-tRNA(Met) wobble cytosine in a patient carrying mutations in NSUN3. Nat. Commun 7, 12039 (2016).
Liu, R. J., Long, T., Li, J., Li, H. & Wang, E. D. Structural basis for substrate binding and catalytic mechanism of a human RNA:m5C methyltransferase NSun6. Nucleic Acids Res. 45, 6684–6697 (2017).
Huang, Z. X. et al. Position 34 of tRNA is a discriminative element for m5C38 modification by human DNMT2. Nucleic Acids Res. 49, 13045–13061 (2021).
Janin, M. et al. Epigenetic loss of RNA-methyltransferase NSUN5 in glioma targets ribosomes to drive a stress adaptive translational program. Acta Neuropathol. 138, 1053–1074 (2019).
Liao, H. et al. Human NOP2/NSUN1 regulates ribosome biogenesis through non-catalytic complex formation with box C/D snoRNPs. Nucleic Acids Res. 50, 10695–10716 (2022).
Yang, L. et al. Nsun4 and Mettl3 mediated translational reprogramming of Sox9 promotes BMSC chondrogenic differentiation. Commun. Biol. 5, 495 (2022).
Spåhr, H., Habermann, B., Gustafsson, C. M., Larsson, N. G. & Hallberg, B. M. Structure of the human MTERF4-NSUN4 protein complex that regulates mitochondrial ribosome biogenesis. Proc. Natl. Acad. Sci. USA 109, 15253–15258 (2012).
Dai, X. et al. YTHDF2 Binds to 5-Methylcytosine in RNA and Modulates the Maturation of Ribosomal RNA. Anal. Chem. 92, 1346–1354 (2020).
Arguello, A. E. et al. Reactivity-dependent profiling of RNA 5-methylcytidine dioxygenases. Nat. Commun. 13, 4176 (2022).
Huang, W. et al. Formation and determination of the oxidation products of 5-methylcytosine in RNA. Chem. Sci. 7, 5495–5502 (2016).
Fu, L. et al. Tet-mediated formation of 5-hydroxymethylcytosine in RNA. J. Am. Chem. Soc. 136, 11582–11585 (2014).
Xue, C. et al. ALYREF mediates RNA m(5)C modification to promote hepatocellular carcinoma progression. Signal Transduct. Target. Ther. 8, 130 (2023).
Shen, Q. et al. Tet2 promotes pathogen infection-induced myelopoiesis through mRNA oxidation. Nature 554, 123–127 (2018).
Yang, W. L. et al. Nsun2 coupling with RoRγt shapes the fate of Th17 cells and promotes colitis. Nat. Commun. 14, 863 (2023).
Muthukrishnan, S., Both, G. W., Furuichi, Y. & Shatkin, A. J. 5′-Terminal 7-methylguanosine in eukaryotic mRNA is required for translation. Nature 255, 33–37 (1975).
Adams, J. M. & Cory, S. Modified nucleosides and bizarre 5′-termini in mouse myeloma mRNA. Nature 255, 28–33 (1975).
Aregger, M. et al. CDK1-cyclin B1 activates RNMT, coordinating mRNA cap methylation with G1 phase transcription. Mol. Cell 61, 734–746 (2016).
Létoquart, J. et al. Structural and functional studies of Bud23-Trm112 reveal 18S rRNA N7-G1575 methylation occurs on late 40S precursor ribosomes. Proc. Natl. Acad. Sci. USA 111, E5518–E5526 (2014).
Pandolfini, L. et al. METTL1 promotes let-7 microRNA processing via m7G methylation. Mol. Cell 74, 1278–1290.e1279 (2019).
Dai, Z. et al. N(7)-Methylguanosine tRNA modification enhances oncogenic mRNA translation and promotes intrahepatic cholangiocarcinoma progression. Mol. Cell 81, 3339–3355.e3338 (2021).
Zhang, L. S. et al. Transcriptome-wide mapping of internal N(7)-methylguanosine methylome in mammalian mRNA. Mol. Cell 74, 1304–1316.e1308 (2019).
Furuichi, Y., LaFiandra, A. & Shatkin, A. J. 5’-Terminal structure and mRNA stability. Nature 266, 235–239 (1977).
Konarska, M. M., Padgett, R. A. & Sharp, P. A. Recognition of cap structure in splicing in vitro of mRNA precursors. Cell 38, 731–736 (1984).
Hsu, C. L. & Stevens, A. Yeast cells lacking 5’–>3’ exoribonuclease 1 contain mRNA species that are poly(A) deficient and partially lack the 5’ cap structure. Mol. Cell. Biol. 13, 4826–4835 (1993).
Pabis, M. et al. The nuclear cap-binding complex interacts with the U4/U6·U5 tri-snRNP and promotes spliceosome assembly in mammalian cells. RNA 19, 1054–1063 (2013).
Niedzwiecka, A. et al. Biophysical studies of eIF4E cap-binding protein: recognition of mRNA 5’ cap structure and synthetic fragments of eIF4G and 4E-BP1 proteins. J. Mol. Biol. 319, 615–635 (2002).
Kiriakidou, M. et al. An mRNA m7G cap binding-like motif within human Ago2 represses translation. Cell 129, 1141–1151 (2007).
Pillai, R. S. et al. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science 309, 1573–1576 (2005).
Lin, S. et al. Mettl1/Wdr4-mediated m(7)G tRNA methylome is required for normal mRNA translation and embryonic stem cell self-renewal and differentiation. Mol. Cell 71, 244–255.e245 (2018).
Ma, J. et al. METTL1/WDR4-mediated m(7)G tRNA modifications and m(7)G codon usage promote mRNA translation and lung cancer progression. Mol. Ther. 29, 3422–3435 (2021).
Culjkovic, B., Topisirovic, I., Skrabanek, L., Ruiz-Gutierrez, M. & Borden, K. L. eIF4E promotes nuclear export of cyclin D1 mRNAs via an element in the 3’UTR. J. Cell Biol. 169, 245–256 (2005).
Xie, M. et al. Mammalian 5’-capped microRNA precursors that generate a single microRNA. Cell 155, 1568–1580 (2013).
Sheng, P. et al. Dicer cleaves 5’-extended microRNA precursors originating from RNA polymerase II transcription start sites. Nucleic Acids Res. 46, 5737–5752 (2018).
Dantsuji, S., Ohno, M. & Taniguchi, I. The hnRNP C tetramer binds to CBC on mRNA and impedes PHAX recruitment for the classification of RNA polymerase II transcripts. Nucleic Acids Res. 51, 1393–1408 (2023).
Ito, S. et al. Human NAT10 is an ATP-dependent RNA acetyltransferase responsible for N4-acetylcytidine formation in 18 S ribosomal RNA (rRNA). J. Biol. Chem. 289, 35724–35730 (2014).
Bortolin-Cavaillé, M. L. et al. Probing small ribosomal subunit RNA helix 45 acetylation across eukaryotic evolution. Nucleic Acids Res. 50, 6284–6299 (2022).
Sharma, S. et al. Yeast Kre33 and human NAT10 are conserved 18S rRNA cytosine acetyltransferases that modify tRNAs assisted by the adaptor Tan1/THUMPD1. Nucleic Acids Res. 43, 2242–2258 (2015).
Arango, D. et al. Acetylation of cytidine in mRNA promotes translation efficiency. Cell 175, 1872–1886.e1824 (2018).
Sas-Chen, A. et al. Dynamic RNA acetylation revealed by quantitative cross-evolutionary mapping. Nature 583, 638–643 (2020).
Arango, D. et al. Direct epitranscriptomic regulation of mammalian translation initiation through N4-acetylcytidine. Mol. Cell 82, 2797–2814.e2711 (2022).
Yu, X. M. et al. N4-acetylcytidine modification of lncRNA CTC-490G23.2 promotes cancer metastasis through interacting with PTBP1 to increase CD44 alternative splicing. Oncogene 42, 1101–1116 (2023).
Yang, W. et al. ac4C acetylation of RUNX2 catalyzed by NAT10 spurs osteogenesis of BMSCs and prevents ovariectomy-induced bone loss. Mol. Ther. Nucleic Acids 26, 135–147 (2021).
Wang, G. et al. NAT10-mediated mRNA N4-acetylcytidine modification promotes bladder cancer progression. Clin. Transl. Med. 12, e738 (2022).
Feng, Z. et al. The LINC00623/NAT10 signaling axis promotes pancreatic cancer progression by remodeling ac4C modification of mRNA. J. Hematol. Oncol. 15, 112 (2022).
Zheng, X. et al. N-acetyltransferase 10 promotes colon cancer progression by inhibiting ferroptosis through N4-acetylation and stabilization of ferroptosis suppressor protein 1 (FSP1) mRNA. Cancer Commun. 42, 1347–1366 (2022).
Jin, C. et al. Acetyltransferase NAT10 regulates the Wnt/β-catenin signaling pathway to promote colorectal cancer progression via ac(4)C acetylation of KIF23 mRNA. J. Exp. Clin. Cancer Res. 41, 345 (2022).
Liao, L. et al. Lysine 2-hydroxyisobutyrylation of NAT10 promotes cancer metastasis in an ac4C-dependent manner. Cell Res. 33, 355–371 (2023).
Nance, K. D. et al. Cytidine acetylation yields a hypoinflammatory synthetic messenger RNA. Cell Chem. Biol. 29, 312–320.e317 (2022).
Chang, H., Lim, J., Ha, M. & Kim, V. N. TAIL-seq: genome-wide determination of poly(A) tail length and 3’ end modifications. Mol. Cell 53, 1044–1052 (2014).
Lim, J. et al. Uridylation by TUT4 and TUT7 marks mRNA for degradation. Cell 159, 1365–1376 (2014).
Mullen, T. E. & Marzluff, W. F. Degradation of histone mRNA requires oligouridylation followed by decapping and simultaneous degradation of the mRNA both 5’ to 3’ and 3’ to 5’. Genes Dev 22, 50–65 (2008).
Schmidt, M. J., West, S. & Norbury, C. J. The human cytoplasmic RNA terminal U-transferase ZCCHC11 targets histone mRNAs for degradation. RNA 17, 39–44 (2011).
Yang, A. et al. TENT2, TUT4, and TUT7 selectively regulate miRNA sequence and abundance. Nat. Commun. 13, 5260 (2022).
Yi, H. et al. PABP cooperates with the CCR4-NOT complex to promote mRNA deadenylation and block precocious decay. Mol. Cell 70, 1081–1088.e1085 (2018).
Malecki, M. et al. The exoribonuclease Dis3L2 defines a novel eukaryotic RNA degradation pathway. EMBO J. 32, 1842–1854 (2013).
Ustianenko, D. et al. TUT-DIS3L2 is a mammalian surveillance pathway for aberrant structured non-coding RNAs. EMBO J. 35, 2179–2191 (2016).
Zhao, L. W. et al. Nuclear poly(A) binding protein 1 (PABPN1) mediates zygotic genome activation-dependent maternal mRNA clearance during mouse early embryonic development. Nucleic Acids Res. 50, 458–472 (2022).
Koppers-Lalic, D. et al. Nontemplated nucleotide additions distinguish the small RNA composition in cells from exosomes. Cell Rep. 8, 1649–1658 (2014).
Liu, X. et al. A MicroRNA precursor surveillance system in quality control of MicroRNA synthesis. Mol. Cell 55, 868–879 (2014).
Ripin, N. et al. Molecular basis for AU-rich element recognition and dimerization by the HuR C-terminal RRM. Proc. Natl. Acad. Sci. USA 116, 2935–2944 (2019).
Loh, X. Y. et al. RNA-binding protein ZFP36L1 suppresses hypoxia and cell-cycle signaling. Cancer Res. 80, 219–233 (2020).
Rataj, F. et al. Targeting AU-rich element-mediated mRNA decay with a truncated active form of the zinc-finger protein TIS11b/BRF1 impairs major hallmarks of mammary tumorigenesis. Oncogene 38, 5174–5190 (2019).
Gu, L. et al. Reconstitution of HuR-Inhibited CUGBP1 expression protects cardiomyocytes from acute myocardial infarction-induced injury. Antioxid. Redox Signal. 27, 1013–1026 (2017).
Lin, C. C. et al. Terminal uridyltransferase 7 regulates TLR4-triggered inflammation by controlling Regnase-1 mRNA uridylation and degradation. Nat. Commun. 12, 3878 (2021).
Heo, I. et al. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell 138, 696–708 (2009).
Rau, F. et al. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat. Struct. Mol. Biol. 18, 840–845 (2011).
Heo, I. et al. Mono-uridylation of pre-microRNA as a key step in the biogenesis of group II let-7 microRNAs. Cell 151, 521–532 (2012).
Kim, H. et al. A Mechanism for microRNA Arm Switching Regulated by Uridylation. Mol. Cell 78, 1224–1236.e1225 (2020).
Ansari, M. Y. et al. Genetic inactivation of ZCCHC6 suppresses interleukin-6 expression and reduces the severity of experimental osteoarthritis in mice. Arthritis Rheumatol. 71, 583–593 (2019).
Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 17, 83–96 (2016).
Chung, H. et al. Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell 172, 811–824.e814 (2018).
Chen, J. et al. Global RNA editing identification and characterization during human pluripotent-to-cardiomyocyte differentiation. Mol. Ther. Nucleic Acids 26, 879–891 (2021).
Wang, F. et al. A comprehensive RNA editome reveals that edited Azin1 partners with DDX1 to enable hematopoietic stem cell differentiation. Blood 138, 1939–1952 (2021).
Jiang, L. et al. ADAR1-mediated RNA editing links ganglioside catabolism to glioblastoma stem cell maintenance. J. Clin. Invest. 132, e143397 (2022).
Solomon, O. et al. RNA editing by ADAR1 leads to context-dependent transcriptome-wide changes in RNA secondary structure. Nat. Commun. 8, 1440 (2017).
Kokot, K. E. et al. Reduction of A-to-I RNA editing in the failing human heart regulates formation of circular RNAs. Basic Res. Cardiol. 117, 32 (2022).
Zipeto, M. A. et al. ADAR1 activation drives leukemia stem cell self-renewal by impairing Let-7 biogenesis. Cell Stem Cell 19, 177–191 (2016).
Wu, X. et al. ADAR2 increases in exercised heart and protects against myocardial infarction and doxorubicin-induced cardiotoxicity. Mol. Ther. 30, 400–414 (2022).
Hu, X., Zou, Q., Yao, L. & Yang, X. Survey of the binding preferences of RNA-binding proteins to RNA editing events. Genome Biol. 23, 169 (2022).
Stellos, K. et al. Adenosine-to-inosine RNA editing controls cathepsin S expression in atherosclerosis by enabling HuR-mediated post-transcriptional regulation. Nat. Med. 22, 1140–1150 (2016).
Shen, H. et al. ADARs act as potent regulators of circular transcriptome in cancer. Nat. Commun. 13, 1508 (2022).
Yang, C. C. et al. ADAR1-mediated 3’ UTR editing and expression control of antiapoptosis genes fine-tunes cellular apoptosis response. Cell Death Dis. 8, e2833 (2017).
Chalk, A. M., Taylor, S., Heraud-Farlow, J. E. & Walkley, C. R. The majority of A-to-I RNA editing is not required for mammalian homeostasis. Genome Biol. 20, 268 (2019).
Chan, T. H. et al. A disrupted RNA editing balance mediated by ADARs (Adenosine DeAminases that act on RNA) in human hepatocellular carcinoma. Gut 63, 832–843 (2014).
Chan, T. H. et al. ADAR-mediated RNA editing predicts progression and prognosis of gastric cancer. Gastroenterology 151, 637–650.e610 (2016).
van der Kwast, R. et al. Adenosine-to-inosine editing of vasoactive microRNAs alters their targetome and function in ischemia. Mol. Ther. Nucleic Acids 21, 932–953 (2020).
Tan, M. H. et al. Dynamic landscape and regulation of RNA editing in mammals. Nature 550, 249–254 (2017).
Shiromoto, Y., Sakurai, M., Minakuchi, M., Ariyoshi, K. & Nishikura, K. ADAR1 RNA editing enzyme regulates R-loop formation and genome stability at telomeres in cancer cells. Nat. Commun. 12, 1654 (2021).
Jimeno, S. et al. ADAR-mediated RNA editing of DNA:RNA hybrids is required for DNA double strand break repair. Nat. Commun. 12, 5512 (2021).
Song, Y. et al. RNA editing mediates the functional switch of COPA in a novel mechanism of hepatocarcinogenesis. J. Hepatol. 74, 135–147 (2021).
Chen, L. et al. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat. Med. 19, 209–216 (2013).
van der Kwast, R. et al. Adenosine-to-inosine editing of microRNA-487b alters target gene selection after ischemia and promotes neovascularization. Circ. Res. 122, 444–456 (2018).
Li, Q. et al. RNA editing underlies genetic risk of common inflammatory diseases. Nature 608, 569–577 (2022).
Liddicoat, B. J. et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349, 1115–1120 (2015).
Garcia-Gonzalez, C. et al. ADAR1 prevents autoinflammatory processes in the heart mediated by IRF7. Circ. Res. 131, 580–597 (2022).
de Reuver, R. et al. ADAR1 prevents autoinflammation by suppressing spontaneous ZBP1 activation. Nature 607, 784–789 (2022).
Karijolich, J., Yi, C. & Yu, Y. T. Transcriptome-wide dynamics of RNA pseudouridylation. Nat. Rev. Mol. Cell Biol. 16, 581–585 (2015).
Davis, D. R. Stabilization of RNA stacking by pseudouridine. Nucleic Acids Res. 23, 5020–5026 (1995).
Zhao, B. S. & He, C. Pseudouridine in a new era of RNA modifications. Cell Res. 25, 153–154 (2015).
Li, X. et al. Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat. Chem. Biol. 11, 592–597 (2015).
Ni, J., Tien, A. L. & Fournier, M. J. Small nucleolar RNAs direct site-specific synthesis of pseudouridine in ribosomal RNA. Cell 89, 565–573 (1997).
Ruggero, D. et al. Dyskeratosis congenita and cancer in mice deficient in ribosomal RNA modification. Science 299, 259–262 (2003).
Li, L. & Ye, K. Crystal structure of an H/ACA box ribonucleoprotein particle. Nature 443, 302–307 (2006).
Ganot, P., Bortolin, M. L. & Kiss, T. Site-specific pseudouridine formation in preribosomal RNA is guided by small nucleolar RNAs. Cell 89, 799–809 (1997).
Schwartz, S. et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 159, 148–162 (2014).
Dai, Q. et al. Quantitative sequencing using BID-seq uncovers abundant pseudouridines in mammalian mRNA at base resolution. Nat. Biotechnol. 41, 344–354 (2023).
Bykhovskaya, Y., Casas, K., Mengesha, E., Inbal, A. & Fischel-Ghodsian, N. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA). Am. J. Hum. Genet. 74, 1303–1308 (2004).
Shaheen, R. et al. A homozygous truncating mutation in PUS3 expands the role of tRNA modification in normal cognition. Hum. Genet. 135, 707–713 (2016).
Lin, T. Y. et al. Destabilization of mutated human PUS3 protein causes intellectual disability. Hum. Mutat. 43, 2063–2078 (2022).
Cui, Q. et al. Targeting PUS7 suppresses tRNA pseudouridylation and glioblastoma tumorigenesis. Nat Cancer 2, 932–949 (2021).
Guegueniat, J. et al. The human pseudouridine synthase PUS7 recognizes RNA with an extended multi-domain binding surface. Nucleic Acids Res. 49, 11810–11822 (2021).
McCleverty, C. J., Hornsby, M., Spraggon, G. & Kreusch, A. Crystal structure of human Pus10, a novel pseudouridine synthase. J. Mol. Biol. 373, 1243–1254 (2007).
Song, J. et al. Differential roles of human PUS10 in miRNA processing and tRNA pseudouridylation. Nat. Chem. Biol. 16, 160–169 (2020).
Jia, Z. et al. Human TRUB1 is a highly conserved pseudouridine synthase responsible for the formation of Ψ55 in mitochondrial tRNAAsn, tRNAGln, tRNAGlu and tRNAPro. Nucleic Acids Res. 50, 9368–9381 (2022).
Taoka, M. et al. Landscape of the complete RNA chemical modifications in the human 80S ribosome. Nucleic Acids Res. 46, 9289–9298 (2018).
Mochizuki, Y., He, J., Kulkarni, S., Bessler, M. & Mason, P. J. Mouse dyskerin mutations affect accumulation of telomerase RNA and small nucleolar RNA, telomerase activity, and ribosomal RNA processing. Proc. Natl. Acad. Sci. USA 101, 10756–10761 (2004).
Nir, R. et al. A systematic dissection of determinants and consequences of snoRNA-guided pseudouridylation of human mRNA. Nucleic Acids Res. 50, 4900–4916 (2022).
Antonicka, H. et al. A pseudouridine synthase module is essential for mitochondrial protein synthesis and cell viability. EMBO Rep. 18, 28–38 (2017).
Carlile, T. M. et al. mRNA structure determines modification by pseudouridine synthase 1. Nat. Chem. Biol. 15, 966–974 (2019).
Safra, M., Nir, R., Farouq, D., Vainberg Slutskin, I. & Schwartz, S. TRUB1 is the predominant pseudouridine synthase acting on mammalian mRNA via a predictable and conserved code. Genome Res. 27, 393–406 (2017).
Levi, O. & Arava, Y. S. Pseudouridine-mediated translation control of mRNA by methionine aminoacyl tRNA synthetase. Nucleic Acids Res. 49, 432–443 (2021).
Guzzi, N. et al. Pseudouridine-modified tRNA fragments repress aberrant protein synthesis and predict leukaemic progression in myelodysplastic syndrome. Nat. Cell Biol. 24, 299–306 (2022).
Yoon, A. et al. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science 312, 902–906 (2006).
Bellodi, C. et al. Loss of function of the tumor suppressor DKC1 perturbs p27 translation control and contributes to pituitary tumorigenesis. Cancer Res. 70, 6026–6035 (2010).
McMahon, M. et al. A single H/ACA small nucleolar RNA mediates tumor suppression downstream of oncogenic RAS. Elife 8, e48847 (2019).
Eyler, D. E. et al. Pseudouridinylation of mRNA coding sequences alters translation. Proc. Natl. Acad. Sci. USA 116, 23068–23074 (2019).
Karijolich, J. & Yu, Y. T. Converting nonsense codons into sense codons by targeted pseudouridylation. Nature 474, 395–398 (2011).
Anderson, B. R. et al. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res. 38, 5884–5892 (2010).
Tan, J. et al. Noncanonical registers and base pairs in human 5’ splice-site selection. Nucleic Acids Res. 44, 3908–3921 (2016).
Wu, G. et al. Pseudouridines in U2 snRNA stimulate the ATPase activity of Prp5 during spliceosome assembly. EMBO J. 35, 654–667 (2016).
Martinez, N. M. et al. Pseudouridine synthases modify human pre-mRNA co-transcriptionally and affect pre-mRNA processing. Mol. Cell 82, 645–659.e649 (2022).
Rapino, F. et al. Wobble tRNA modification and hydrophilic amino acid patterns dictate protein fate. Nat. Commun. 12, 2170 (2021).
Rosu, A. et al. Loss of tRNA-modifying enzyme Elp3 activates a p53-dependent antitumor checkpoint in hematopoiesis. J. Exp. Med. 218, e20200662 (2021).
Songe-Møller, L. et al. Mammalian ALKBH8 possesses tRNA methyltransferase activity required for the biogenesis of multiple wobble uridine modifications implicated in translational decoding. Mol. Cell. Biol. 30, 1814–1827 (2010).
Delaunay, S. et al. Elp3 links tRNA modification to IRES-dependent translation of LEF1 to sustain metastasis in breast cancer. J. Exp. Med. 213, 2503–2523 (2016).
Xu, S. et al. Genome-wide CRISPR screen identifies ELP5 as a determinant of gemcitabine sensitivity in gallbladder cancer. Nat. Commun. 10, 5492 (2019).
Kirino, Y. et al. Codon-specific translational defect caused by a wobble modification deficiency in mutant tRNA from a human mitochondrial disease. Proc. Natl. Acad. Sci. USA 101, 15070–15075 (2004).
Kopajtich, R. et al. Mutations in GTPBP3 cause a mitochondrial translation defect associated with hypertrophic cardiomyopathy, lactic acidosis, and encephalopathy. Am. J. Hum. Genet. 95, 708–720 (2014).
Chen, D. et al. Deletion of Gtpbp3 in zebrafish revealed the hypertrophic cardiomyopathy manifested by aberrant mitochondrial tRNA metabolism. Nucleic Acids Res. 47, 5341–5355 (2019).
Sasarman, F., Antonicka, H., Horvath, R. & Shoubridge, E. A. The 2-thiouridylase function of the human MTU1 (TRMU) enzyme is dispensable for mitochondrial translation. Hum. Mol. Genet. 20, 4634–4643 (2011).
Bilbille, Y. et al. The human mitochondrial tRNAMet: structure/function relationship of a unique modification in the decoding of unconventional codons. J. Mol. Biol. 406, 257–274 (2011).
Cantara, W. A., Murphy, F. V. T., Demirci, H. & Agris, P. F. Expanded use of sense codons is regulated by modified cytidines in tRNA. Proc. Natl. Acad. Sci. USA 110, 10964–10969 (2013).
Tsao, C. W. et al. Heart Disease and Stroke Statistics-2023 Update: A Report From the American Heart Association. Circulation 147, e93–e621 (2023).
Roth, G. A. et al. Global, Regional, and National Burden of Cardiovascular Diseases for 10 Causes, 1990 to 2015. J. Am. Coll. Cardiol. 70, 1–25 (2017).
Zhou, B., Perel, P., Mensah, G. A. & Ezzati, M. Global epidemiology, health burden and effective interventions for elevated blood pressure and hypertension. Nat Rev Cardiol. 18, 785–802 (2021).
Libby, P. et al. Atherosclerosis. Nat. Rev. Dis. Primers 5, 56 (2019).
Touyz, R. M. et al. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 114, 529–539 (2018).
Jain, M. et al. RNA editing of Filamin A pre-mRNA regulates vascular contraction and diastolic blood pressure. EMBO J. 37, e94813 (2018).
Marcadenti, A. et al. Effects of FTO RS9939906 and MC4R RS17782313 on obesity, type 2 diabetes mellitus and blood pressure in patients with hypertension. Cardiovasc. Diabetol. 12, 103 (2013).
Liu, S. et al. HuR (Human Antigen R) regulates the contraction of vascular smooth muscle and maintains blood pressure. Arterioscler. Thromb. Vasc. Biol. 40, 943–957 (2020).
Klöss, S., Rodenbach, D., Bordel, R. & Mülsch, A. Human-antigen R (HuR) expression in hypertension: downregulation of the mRNA stabilizing protein HuR in genetic hypertension. Hypertension 45, 1200–1206 (2005).
Chien, C. S. et al. METTL3-dependent N(6)-methyladenosine RNA modification mediates the atherogenic inflammatory cascades in vascular endothelium. Proc. Natl. Acad. Sci. USA 118, e2025070118 (2021).
Jian, D. et al. METTL14 aggravates endothelial inflammation and atherosclerosis by increasing FOXO1 N6-methyladeosine modifications. Theranostics 10, 8939–8956 (2020).
Zheng, Y. et al. Mettl14 mediates the inflammatory response of macrophages in atherosclerosis through the NF-κB/IL-6 signaling pathway. Cell. Mol. Life Sci. 79, 311 (2022).
Vlachogiannis, N. I. et al. Adenosine-to-inosine Alu RNA editing controls the stability of the pro-inflammatory long noncoding RNA NEAT1 in atherosclerotic cardiovascular disease. J. Mol. Cell. Cardiol. 160, 111–120 (2021).
Sun, Z. et al. Matr3 reshapes m6A modification complex to alleviate macrophage inflammation during atherosclerosis. Clin. Immunol. 245, 109176 (2022).
Zhang, X., Li, X., Jia, H., An, G. & Ni, J. The m(6)A methyltransferase METTL3 modifies PGC-1α mRNA promoting mitochondrial dysfunction and oxLDL-induced inflammation in monocytes. J. Biol. Chem. 297, 101058 (2021).
Chen, J. et al. METTL14-dependent m6A regulates vascular calcification induced by indoxyl sulfate. Life Sci 239, 117034 (2019).
Zhou, T. et al. Factors influencing osteogenic differentiation of human aortic valve interstitial cells. J. Thorac. Cardiovasc. Surg. 161, e163–e185 (2021).
Wang, K. et al. PIWI-interacting RNA HAAPIR regulates cardiomyocyte death after myocardial infarction by promoting NAT10-mediated ac(4) C acetylation of Tfec mRNA. Adv. Sci. 9, e2106058 (2022).
Mathiyalagan, P. et al. FTO-dependent N(6)-methyladenosine regulates cardiac function during remodeling and repair. Circulation 139, 518–532 (2019).
Han, Z. et al. ALKBH5 regulates cardiomyocyte proliferation and heart regeneration by demethylating the mRNA of YTHDF1. Theranostics 11, 3000–3016 (2021).
Sun, P. et al. Extracellular vesicle-packaged mitochondrial disturbing miRNA exacerbates cardiac injury during acute myocardial infarction. Clin. Transl. Med. 12, e779 (2022).
Song, H. et al. METTL3 and ALKBH5 oppositely regulate m(6)A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy 15, 1419–1437 (2019).
Wang, L. et al. METTL14 is required for exercise-induced cardiac hypertrophy and protects against myocardial ischemia-reperfusion injury. Nat. Commun. 13, 6762 (2022).
McKenna, W. J., Maron, B. J. & Thiene, G. Classification, epidemiology, and global burden of cardiomyopathies. Circ. Res. 121, 722–730 (2017).
Maron, B. J. & Maron, M. S. Hypertrophic cardiomyopathy. Lancet 381, 242–255 (2013).
Schultheiss, H. P. et al. Dilated cardiomyopathy. Nat. Rev. Dis. Primers 5, 32 (2019).
Muchtar, E., Blauwet, L. A. & Gertz, M. A. Restrictive cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ. Res. 121, 819–837 (2017).
Ghezzi, D. et al. Mutations of the mitochondrial-tRNA modifier MTO1 cause hypertrophic cardiomyopathy and lactic acidosis. Am. J. Hum. Genet. 90, 1079–1087 (2012).
Perks, K. L. et al. PTCD1 is required for 16S rRNA maturation complex stability and mitochondrial ribosome assembly. Cell Rep 23, 127–142 (2018).
Gao, S. et al. Depletion of m(6) A reader protein YTHDC1 induces dilated cardiomyopathy by abnormal splicing of Titin. J. Cell. Mol. Med. 25, 10879–10891 (2021).
Meng, L. et al. METTL14 suppresses pyroptosis and diabetic cardiomyopathy by downregulating TINCR lncRNA. Cell Death Dis. 13, 38 (2022).
Peng, T. et al. LncRNA Airn alleviates diabetic cardiac fibrosis by inhibiting activation of cardiac fibroblasts via a m6A-IMP2-p53 axis. Biol. Direct 17, 32 (2022).
Zhuang, S. et al. METTL14 promotes doxorubicin-induced cardiomyocyte ferroptosis by regulating the KCNQ1OT1-miR-7-5p-TFRC axis. Cell Biol Toxicol. (2021).
Nakamura, M. & Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 15, 387–407 (2018).
Berulava, T. et al. Changes in m6A RNA methylation contribute to heart failure progression by modulating translation. Eur. J. Heart Fail. 22, 54–66 (2020).
Zhang, B. et al. m6A demethylase FTO attenuates cardiac dysfunction by regulating glucose uptake and glycolysis in mice with pressure overload-induced heart failure. Signal Transduct. Target. Ther. 6, 377 (2021).
Dorn, L. E. et al. The N(6)-methyladenosine mRNA methylase METTL3 controls cardiac homeostasis and hypertrophy. Circulation 139, 533–545 (2019).
Gao, X. Q. et al. The piRNA CHAPIR regulates cardiac hypertrophy by controlling METTL3-dependent N(6)-methyladenosine methylation of Parp10 mRNA. Nat. Cell Biol. 22, 1319–1331 (2020).
Xu, H. et al. YTHDF2 alleviates cardiac hypertrophy via regulating Myh7 mRNA decoy. Cell Biosci. 11, 132 (2021).
Hoeper, M. M. et al. A global view of pulmonary hypertension. Lancet Respir. Med. 4, 306–322 (2016).
Schermuly, R. T., Ghofrani, H. A., Wilkins, M. R. & Grimminger, F. Mechanisms of disease: pulmonary arterial hypertension. Nat. Rev. Cardiol. 8, 443–455 (2011).
Hu, L. et al. YTHDF1 regulates pulmonary hypertension through translational control of MAGED1. Am. J. Respir. Crit. Care Med. 203, 1158–1172 (2021).
Liu, P. et al. m(6)A modification-mediated GRAP regulates vascular remodeling in hypoxic pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 67, 574–588 (2022).
Hu, L. et al. Ythdf2 promotes pulmonary hypertension by suppressing Hmox1-dependent anti-inflammatory and antioxidant function in alveolar macrophages. Redox Biol. 61, 102638 (2023).
Blackwell, D. J., Schmeckpeper, J. & Knollmann, B. C. Animal models to study cardiac arrhythmias. Circ. Res. 130, 1926–1964 (2022).
Herring, N., Kalla, M. & Paterson, D. J. The autonomic nervous system and cardiac arrhythmias: current concepts and emerging therapies. Nat. Rev. Cardiol. 16, 707–726 (2019).
Qi, L. et al. m(6)A methyltransferase METTL3 participated in sympathetic neural remodeling post-MI via the TRAF6/NF-κB pathway and ROS production. J. Mol. Cell. Cardiol. 170, 87–99 (2022).
Qi, L. et al. New insights into the central sympathetic hyperactivity post-myocardial infarction: Roles of METTL3-mediated m(6) A methylation. J. Cell. Mol. Med. 26, 1264–1280 (2022).
Zhou, Y. et al. METTL3 boosts glycolysis and cardiac fibroblast proliferation by increasing AR methylation. Int. J. Biol. Macromol. 223, 899–915 (2022).
Nienaber, C. A. et al. Aortic dissection. Nat. Rev. Dis. Primers 2, 16053 (2016).
Li, N. et al. Targeting ferroptosis as a novel approach to alleviate aortic dissection. Int. J. Biol. Sci. 18, 4118–4134 (2022).
Xin, M., Olson, E. N. & Bassel-Duby, R. Mending broken hearts: cardiac development as a basis for adult heart regeneration and repair. Nat. Rev. Mol. Cell. Biol. 14, 529–541 (2013).
Wang, Y. et al. Mutations in RNA methyltransferase gene NSUN5 confer high risk of outflow tract malformation. Front. Cell Dev. Biol. 9, 623394 (2021).
Moore, J. B. T. et al. The A-to-I RNA editing enzyme Adar1 is essential for normal embryonic cardiac growth and development. Circ. Res. 127, 550–552 (2020).
Han, Z. et al. ALKBH5-mediated m(6)A mRNA methylation governs human embryonic stem cell cardiac commitment. Mol. Ther. Nucleic Acids 26, 22–33 (2021).
Yang, Y. et al. Dynamic patterns of N6-methyladenosine profiles of messenger RNA correlated with the cardiomyocyte regenerability during the early heart development in mice. Oxid. Med. Cell. Longev. 2021, 5537804 (2021).
Wang, S. et al. Differential roles of YTHDF1 and YTHDF3 in embryonic stem cell-derived cardiomyocyte differentiation. RNA Biol. 18, 1354–1363 (2021).
Qian, B., Wang, P., Zhang, D. & Wu, L. m6A modification promotes miR-133a repression during cardiac development and hypertrophy via IGF2BP2. Cell Death Discov. 7, 157 (2021).
Gong, R. et al. Loss of m(6)A methyltransferase METTL3 promotes heart regeneration and repair after myocardial injury. Pharmacol. Res. 174, 105845 (2021).
Jiang, F. Q. et al. Mettl3-mediated m(6)A modification of Fgf16 restricts cardiomyocyte proliferation during heart regeneration. Elife 11, e77014 (2022).
Zhao, Y. et al. Loss of m6A demethylase ALKBH5 promotes post-ischemic angiogenesis via post-transcriptional stabilization of WNT5A. Clin. Transl. Med. 11, e402 (2021).
Hashimoto, H., Olson, E. N. & Bassel-Duby, R. Therapeutic approaches for cardiac regeneration and repair. Nat. Rev. Cardiol. 15, 585–600 (2018).
Del Re, D. P., Amgalan, D., Linkermann, A., Liu, Q. & Kitsis, R. N. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol. Rev. 99, 1765–1817 (2019).
Cheng, P. et al. Amelioration of acute myocardial infarction injury through targeted ferritin nanocages loaded with an ALKBH5 inhibitor. Acta Biomater. 140, 481–491 (2022).
Liu, K. et al. Exercise training ameliorates myocardial phenotypes in heart failure with preserved ejection fraction by changing N6-methyladenosine modification in mice model. Front. Cell Dev. Biol. 10, 954769 (2022).
Yang, Q. et al. Exercise mitigates endothelial pyroptosis and atherosclerosis by downregulating NEAT1 through N6-methyladenosine modifications. Arterioscler. Thromb Vasc. Biol. 43, 910–926 (2023).
Xu, Z., Qin, Y., Lv, B., Tian, Z. & Zhang, B. Intermittent fasting improves high-fat diet-induced obesity cardiomyopathy via alleviating lipid deposition and apoptosis and decreasing m6A methylation in the heart. Nutrients 14, 251 (2022).
Wang, X., Li, Y., Li, J., Li, S. & Wang, F. Mechanism of METTL3-mediated m(6)A modification in cardiomyocyte pyroptosis and myocardial ischemia-reperfusion injury. Cardiovasc. Drugs Ther. 37, 435–448 (2023).
Zhang, R. et al. METTL3 mediates Ang-II-induced cardiac hypertrophy through accelerating pri-miR-221/222 maturation in an m6A-dependent manner. Cell. Mol. Biol. Lett. 27, 55 (2022).
Li, T. et al. Silencing of METTL3 attenuates cardiac fibrosis induced by myocardial infarction via inhibiting the activation of cardiac fibroblasts. FASEB J. 35, e21162 (2021).
Fang, Z. M. et al. Methyltransferase-like 3 suppresses phenotypic switching of vascular smooth muscle cells by activating autophagosome formation. Cell Prolif. 56, e13386 (2023).
Shen, L. et al. Nine-month angiographic and two-year clinical follow-up of novel biodegradable-polymer arsenic trioxide-eluting stent versus durable-polymer sirolimus-eluting stent for coronary artery disease. Chin. Med. J. (Engl) 128, 768–773 (2015).
Yu, H. et al. Arsenic trioxide activates yes-associated protein by lysophosphatidic acid metabolism to selectively induce apoptosis of vascular smooth muscle cells. Biochim. Biophys. Acta Mol. Cell Res. 1869, 119211 (2022).
Fang, M., Deng, J., Zhou, Q., Hu, Z. & Yang, L. Maslinic acid protects against pressure-overload-induced cardiac hypertrophy by blocking METTL3-mediated m(6)A methylation. Aging (Albany N. Y.) 14, 2548–2557 (2022).
Zhang, M. et al. Tanshinone IIA alleviates cardiac hypertrophy through m6A modification of galectin-3. Bioengineered 13, 4260–4270 (2022).
Seo, K. W. & Kleiner, R. E. YTHDF2 recognition of N(1)-methyladenosine (m(1)A)-modified RNA is associated with transcript destabilization. ACS Chem. Biol. 15, 132–139 (2020).
Dai, X., Wang, T., Gonzalez, G. & Wang, Y. Identification of YTH domain-containing proteins as the readers for N1-methyladenosine in RNA. Anal. Chem. 90, 6380–6384 (2018).
Zheng, Q. et al. Cytoplasmic m(1)A reader YTHDF3 inhibits trophoblast invasion by downregulation of m(1)A-methylated IGF1R. Cell Discov. 6, 12 (2020).
Boo, S. H., Ha, H. & Kim, Y. K. m(1)A and m(6)A modifications function cooperatively to facilitate rapid mRNA degradation. Cell Rep. 40, 111317 (2022).
Olsen, M. B. et al. NEIL3-dependent regulation of cardiac fibroblast proliferation prevents myocardial rupture. Cell Rep. 18, 82–92 (2017).
Tabish, A. M. et al. Association of intronic DNA methylation and hydroxymethylation alterations in the epigenetic etiology of dilated cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 317, H168–h180 (2019).
Leopold, J. A. & Loscalzo, J. Emerging role of precision medicine in cardiovascular disease. Circ. Res. 122, 1302–1315 (2018).
Ylä-Herttuala, S. & Baker, A. H. Cardiovascular gene therapy: past, present, and future. Mol. Ther. 25, 1095–1106 (2017).
Saeed, S. et al. Nanoparticle: a promising player in nanomedicine and its theranostic applications for the treatment of cardiovascular diseases. Curr. Probl. Cardiol. 48, 101599 (2023).
Acknowledgements
The authors thank John Angles (University at Albany, State University of New York) for editorial assistance. This work was supported by the National Natural Science Foundation of China (Grant No. 82073260, 82003203, 81972837, 81572280) and the Natural Science Foundation of Hunan Province (Grant No. 2023JJ30766, 2023JJ10091, 2021JJ41058, 2019JJ40420).
Author information
Authors and Affiliations
Contributions
H.L., W.L., and C.W. conceived, supervised, and revised the paper. C.W., X.H., H.L., and W.L. organized figures and formatted the paper. Q.G., H.Z., L.Z., X.H., C.W., L.L., J.L., F.L., H.L., and W. L. participated in different parts of writing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, C., Hou, X., Guan, Q. et al. RNA modification in cardiovascular disease: implications for therapeutic interventions. Sig Transduct Target Ther 8, 412 (2023). https://doi.org/10.1038/s41392-023-01638-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-023-01638-7
This article is cited by
-
Exercise training decreases lactylation and prevents myocardial ischemia–reperfusion injury by inhibiting YTHDF2
Basic Research in Cardiology (2024)
