
Abstract
Bronchopulmonary dysplasia (BPD) is the most common complication of preterm birth. Up to 1/3 of children with BPD develop pulmonary hypertension (PH). PH increases mortality, the risk of adverse neurodevelopmental outcome and lacks effective treatment. Current vasodilator therapies address symptoms, but not the underlying arrested vascular development. Recent insights into placental biology and novel technological advances enabling the study of normal and impaired lung development at the single cell level support the concept of a vascular phenotype of BPD. Dysregulation of growth factor pathways results in depletion and dysfunction of putative distal pulmonary endothelial progenitor cells including Cap1, Cap2, and endothelial colony-forming cells (ECFCs), a subset of vascular progenitor cells with self-renewal and de novo angiogenic capacity. Preclinical data demonstrate effectiveness of ECFCs and ECFC-derived particles including extracellular vesicles (EVs) in promoting lung vascular growth and reversing PH, but the mechanism is unknown. The lack of engraftment suggests a paracrine mode of action mediated by EVs that contain miRNA. Aberrant miRNA signaling contributes to arrested pulmonary vascular development, hence using EV- and miRNA-based therapies is a promising strategy to prevent the development of BPD-PH. More needs to be learned about disrupted pathways, timing of intervention, and mode of delivery.
Impact
-
Single-cell RNA sequencing studies provide new in-depth view of developmental endothelial depletion underlying BPD-PH.
-
Aberrant miRNA expression is a major cause of arrested pulmonary development.
-
EV- and miRNA-based therapies are very promising therapeutic strategies to improve prognosis in BPD-PH.
Similar content being viewed by others


Introduction
Bronchopulmonary dysplasia (BPD) is the most common long-term complication of preterm birth.1 The incidence of BPD is inversely correlated with gestational age and ranges from 23% in children born at 28 weeks of gestation to 73% in extremely preterm babies born at 23 weeks.2 Additional risk factors for the development of BPD include male sex, chorioamnionitis, intrauterine growth restriction (IUGR), prolonged use of mechanical ventilation, exposure to high concentrations of oxygen, perinatal asphyxia, patent ductus arteriosus, and sepsis.1,3 Pulmonary hypertension (PH) complicates the course of BPD in 17–37% of preterm infants. Furthermore, 2% of children who do not meet the current criteria for BPD are diagnosed with PH.4
The pathogenesis of PH associated with BPD (BPD-PH) is multifactorial and involves maternal, placental, fetal, and postnatal factors that disrupt the fetal maturation of the pulmonary vascular bed and interfere with the postnatal decline in pulmonary vascular resistance, hinting to the possibility of a specific vascular phenotype of BPD. These observations are further supported by a recent meta-analysis that identified prenatal predictors of BPD-PH including oligohydramnios, small for gestational age status and multiple gestations, as well as postnatal indicators—birth weight and gestational age, severity of BPD, history of sepsis, use of high frequency ventilation, duration of mechanical ventilation, duration of neonatal intensive care unit stay.5
BPD-PH is associated with additional morbidity, and adverse neurodevelopmental outcome in prematurity survivors. The reported mortality in children with BPD-PH is up to 50% in the first 2 years of life.6 Currently, there is no treatment with proven efficacy in improving the prognosis of BPD-PH.
In this narrative review, we outline the limitations of currently available treatment methods for BPD-PH, summarize recent advances in understanding of pulmonary vascular maldevelopment, identify new promising treatment modalities with a particular focus on interventions that promote vascular growth, with an outlook on clinical translation.
Traditional vasodilator therapies
Regulation of the vascular tone
Pioneering work in the early 1980s identified three metabolic pathways involved in the regulation of pulmonary vascular tone. These have led to targeted medications for PH. These pathways include the endothelium-derived factors: nitric oxide (NO), prostacyclin, and endothelin. NO activates soluble guanyl cyclase (GC) which increases intracellular cGMP to promote relaxation of vascular smooth muscle cells. The concentration of cGMP is tightly regulated, and it is broken down by phosphodiesterase 5 (PDE5). Prostacyclin, in turn, activates adenyl cyclase (AC) to increase the concentration of cAMP in smooth muscle cells, leading to vasorelaxation. Endothelin is one of the most potent endogenous vasoconstrictors and acts through ETA and ETB receptors in smooth muscle cells. These metabolic pathways are the target of all medications promoting pulmonary vasodilation used in PH.
Therapies targeting vascular tone and their shortcomings
Inhaled NO (iNO) is a potent and selective pulmonary vasodilator with well-documented efficacy in term and near-term newborns with persistent pulmonary hypertension of the newborn (PPHN).7 The use of iNO in preterm infants <34 weeks of gestation is still under investigation and may be most advantageous in those with confirmed PH.8
Sildenafil, a PDE5 inhibitor, is the most widely used medication in the treatment of BPD-PH. However, data on its efficacy in newborns and infants based on randomized controlled trials are lacking. Small retrospective studies demonstrate that sildenafil decreases estimated pulmonary artery pressure,9,10,11,12 but improvement in clinically relevant endpoints has not been universally reported. Recently, results of a phase I study have been published, which demonstrate that sildenafil is well tolerated, and its use is only occasionally associated with relevant side effects, including hypotension.13 Two larger, placebo-controlled studies evaluating the safety of sildenafil in preterm babies are currently recruiting patients (NCT04447989, NCT03142568). Sildenafil has been approved by the European Medicines Agency (EMA) for clinical use in children >1 year of age with pulmonary arterial hypertension (PAH), but not in babies with BPD-PH. Its use is frequently associated with other side effects, such as gastroesophageal reflux.
Riociguat, a recently developed GC stimulator demonstrates promising efficacy in preclinical studies.14,15 Its off-label use has been occasionally reported in children with PH,16 including case reports that demonstrate efficacy in severe, treatment-resistant PH.17 It has not yet been tested in infants with BPD-PH.
The use of prostacyclin derivatives—epoprostenol,18 iloprost,19,20,21 treprostinil22 has only occasionally been reported in infants with BPD-PH, with no randomized controlled trials published thus far and no ongoing studies registered in ClinicalTrials.gov database.
Similarly, experience with endothelin receptor antagonists—bosentan,23 macicentan, ambrisentan in BPD-PH is limited. Bosentan is mostly used in combination with sildenafil in infants with severe PH, in whom sildenafil does not provide control of symptoms.24 It is frequently associated with side effects, including liver toxicity.
Traditional vasodilator therapies remain the mainstay of treatment in infants with established BPD-PH, even though they lack data on safety and efficacy. These medications are only effective in mitigating symptoms. They do not address the underlying developmental pathology in the pulmonary vasculature. These limitations support the search for new therapeutic avenues. Recent advances grounded in preclinical research provide a rationale for endothelial progenitor cell-based therapies to specifically target interrupted lung vascular development with the promise to improve the prognosis in BPD-PH.
Pulmonary vascular maldevelopment as a target for therapeutic interventions
Evidence over the past 20 years indicates the critical role of pulmonary vascular development in normal and impaired lung growth. Fetal pulmonary vascular development is classically driven by two processes: vasculogenesis – the de novo formation of blood vessels from angioblasts, and angiogenesis—formation of blood vessels from existing ones. Developing airways not only constitute a structural framework for vascular bed, but epithelial cells send paracrine signals directly stimulating endothelial development, and normal lung development is most likely determined by interactions between both.
Angiogenic growth factors
Pulmonary vascular development is tightly regulated by several growth factors. Vascular endothelial growth factor (VEGF), its receptors—FLK1 and FLT1, hypoxia inducible factor (HIF) family, NF-κB, nitric oxide and nitric oxide synthase (NOS), amongst others, play a crucial role in pulmonary vascular development and parenchymal maturation. Genetic and environmental factors that cause disruption of these pathways result in early demise or a severe phenotype associated with arrested pulmonary vascular development and parenchymal simplification.25,26,27,28,29
The role of alterations in bone morphogenetic protein receptor type 2 (BMPR2) related signaling has been well described in adult and pediatric PAH.30,31 Data from preclinical neonatal models confirm these observations in BPD-PH.32 Higher concentrations of BMP10, a member of the proangiogenic BMP family, were reported in preterm newborns who subsequently developed BPD-PH.33 Furthermore, impaired BMPR2 signaling is a hallmark of early pulmonary vascular injury.34
Many growth factors govern the fetal lung development: TGF-β1 overexpression leads to phenotype similar to BPD,35 and its downstream target—caveolin 1 is a key regulator of endothelial nitric oxide synthase (eNOS) and extracellular matrix remodeling. Cav1−/− mice demonstrate reduced lung compliance and airway resistance.36 Other examples include connective tissue growth factor (CTGF)—another downstream target of TGF-β1 and a potent stimulator of vascular smooth muscle cell proliferation,37 and fibroblast growth factor 10 (FGF10)—a key stimulator of early branching morphogenesis. Their role was first established in normal lung development. Data from animal models of BPD and genetic knock-out studies confirmed the causative role of abnormal signaling in arrested pulmonary development. The imbalance in other pro- or antiangiogenic molecules, such as angiopoietin 1 and endostatin also impair vascular maturation.38,39,40 All of these have been proposed as targets for therapeutic interventions; however, to date, no clinical trials based on modification of these pathways have been initiated. Moreover, several antenatal interventions have been considered as candidate strategies to prevent arrested pulmonary development. Gestational vitamin D deficiency has been linked to pulmonary diseases in children41 and abnormal lung development.42 Treatment with vitamin D during the critical period for lung development has recently been demonstrated to attenuate hyperoxia-induced alveolar simplification through increased HIF- 1α and VEGF signaling.43 Other chemical HIF stabilizers have been investigated as potential protective factors in early lung development.44,45
Endothelial progenitor cells
Evidence for endothelial progenitor cell depletion and dysfunction
The “new” BPD is characterized by alveolar simplification and vascular paucity typical for arrested lung development. Angiogenic growth factors released during the acute injury stimulate the function and migration of endothelial progenitor cells (EPC) to the injury site and promote repair mechanisms.46 That makes these cells appealing therapeutic candidates for treatment of BPD-PH. The discovery of adult circulating EPCs by Asahara in 1997 with subsequent characterization of umbilical cord blood-derived endothelial colony-forming cells (ECFCs) and resident pulmonary microvascular endothelial progenitor cells paved the way for characterization of their role in pulmonary vascular development.47,48,49 These cells are highly proliferative, have self-renewal capacity and robust angiogenic properties. Animal model data confirm that hyperoxia reduces the number of bone marrow, circulating and lung residual endothelial progenitors.50 Furthermore, ECFCs have been identified in greater numbers in the cord blood of preterm babies compared to term babies, notably though the exposure to hyperoxia impaired the function of preterm ECFCs.51 Even more intriguing, the number of ECFCs in the cord blood of preterm infants who developed BPD was reduced compared to preterm infants without BPD.52 This finding supports the role of the antenatal origins of vascular progenitor cell dysfunction in the pathogenesis of BPD. Resident ECFCs were also identified in developing human and rat lungs.53 Exposure of these cells to hyperoxia alters their vasculogenic properties, which in turn contributes to arrested pulmonary vascular development resulting in the BPD-PH phenotype in these animals. Recently Ren et al. identified a specific population of endothelial progenitors expressing cKIT (cKIT+) that stimulate alveolar maturation and are dependent on FOXF1—a transcription factor that regulates a network of genes involved in pulmonary angiogenesis. These cells lose their proangiogenic properties when exposed to hyperoxia.54
Antenatal triggers of ECFC dysfunction
Previous work has highlighted the critical role of placental vascular abnormalities and the link with BPD48,49 supporting the vascular hypothesis.50 The role of ECFCs in vascular development has been explored in the context of adverse prenatal events, such as preeclampsia and IUGR—both known risk factors for neonatal PH. Munoz-Hernandez et al. demonstrated that newborns whose mothers suffer from preeclampsia carry a lower number of ECFCs in their cord blood compared to the control group. They were unable to detect ECFC dysfunction.55 Differences in the number of ECFCs in cord blood were further confirmed by Gumina et al., moreover, impaired growth and migration of ECFCs in the preeclampsia group were reported.56 Unlike these two studies, which do not offer any insight into possible mechanisms of this association, recently Schroeder-Heurich et al. identified down-regulation of miR-1270 as the mechanism that explains ECFC dysfunction in babies of women with preeclampsia.57 Similarly, term ECFCs in cord blood of babies born with IUGR, irrespective of the etiology are less abundant and their functional capacity is diminished.58 These findings demonstrate that dysfunction of endothelial progenitors originates in antenatal period and may be the missing link between both early complications, such as PH, but also long-term cardiovascular sequelae.
Novel endothelial cell revelations when looking at lung development at the single-cell level
The dissemination of single-cell-based analytical methods has revolutionized our understanding of the complex processes involved in pulmonary vascular development. This resulted in identification of endothelial cell subtypes and their developmental trajectories in the immature lung. Vila Ellis et al.29 were the first to identify the transcriptionally distinct population of endothelial cells that produce carbonic anhydrase 4. These cells were described by Gillich et al.59 as aerocytes (aCap) together with a second population of pulmonary capillary cells—general capillary cells (gCap). The first are large, complex cells, spanning across multiple alveoli, equipped with pores. gCap cells, on the other hand, are much smaller, have fewer pores, but are more abundant. Not only do these cells have different morphologies, but they also perform distinct functions. Aerocytes build the surface area for gas exchange and are involved in leukocyte trafficking, whereas gCap cells have the capacity of antigen-presenting cells and serve as progenitor cells to generate aCap cells after injury. Lineage tracing demonstrated that these cells indeed derived from a common progenitor cell. Negretti et al. provided even more in-depth insight into the developmental trajectories of these newly recognized populations of endothelial cells.60 In their single-cell atlas of lung development they demonstrated that aCap cells first appear in the initial phase of the saccular stage of lung development (E18 in mouse lung) – at the same time as alveolar type 1 (AT1) cells emerge and persist through the alveolar stage in stable numbers. Furthermore, they confirmed that aerocytes arise from gCap cells and that the expression of the vascular endothelial growth factor receptor (VEGFR) increases over time in these cells, much like the expression of VEGF in the neighboring AT1 cells. Dramatic changes in endothelial cell composition of the maturing lungs were demonstrated in a mouse model of hyperoxia-induced lung injury.61 gCap cells were severely depleted, whereas the number of aCap cells increased, albeit both cell types demonstrated aberrant gene expression. gCap dell depletion may be a hint into the irreversibility of damage to the developing pulmonary microvasculature and further support a rationale for endothelial-cell-derived therapy.
Recently, knowledge gained through single-cell RNA sequencing studies has been synthesized to create a standardized set of lung cell cards that provide functional annotations of all cell types in the healthy lung.62 The authors suggest changing the terminology of pulmonary endothelial cells and replace the term gCap cells with Cap 1 cells (capillary type 1) and aCap cells with Cap 2 cells (capillary type 2). These exciting discoveries provide unprecedented close-up information on the development of the pulmonary endothelium and open new research directions that aim to characterize the role of abnormal endothelial maturation in lung vascular disease and identify new repair strategies.
ECFCs as a therapeutic modality
Functional impairment of ECFCs offers a strong rationale for studying the effectiveness of ECFCs in the prevention / treatment of perinatal pulmonary vascular complications. A growing number of preclinical studies demonstrate the beneficial effects of ECFCs in disease models, mainly related to ischemia.63,64,65,66,67 The vasculogenic properties of ECFCs were harnessed in these studies to rescue the limb ischemia,63,64 ischemic myocardium,65 acute ischemic kidney injury66 or endothelial injury of the carotid artery.67 ECFCs reduced limb tissue necrosis,63,64 increased capillary density, improved left ventricular function,65 attenuated renal tubular necrosis, macrophage infiltration and apoptosis66 and augmented regeneration of the carotid endothelium.67
In the case of BPD models, the effectiveness of these cells has only been tested in two controlled preclinical studies so far (Table 1). ECFCs demonstrated capacity to reverse oxygen induced alveolar simplification and PH in newborn mice and rats.53 Furthermore, quantification of the exogenous human ECFCs in the rodent lungs with qPCR revealed a very low level of engraftment. Subsequent experiments confirmed a similar beneficial effect with the administration of ECFC-conditioned medium (ECFC-CM), which strongly suggests that ECFCs exert their regenerative activity through a paracrine mechanism. Similarly, ECFC-CM was effective in enhancing the growth and angiogenesis of pulmonary artery endothelial cells (PAECs) and improving RVH in a rat model.68 So far, these findings have not been replicated in other studies using similar or large animal models of BPD-PH. Furthermore, the results of a recent systematic review69 indicate that, although the body of evidence supporting the effectiveness of ECFCs in preclinical models of ischemic diseases is growing rapidly, these studies are often fraught with reporting biases and heterogeneity of cell product characterization. The authors suggest that future studies ought to be conducted with greater rigour to facilitate translation to clinical practice.
ECFCs are immunogenic. This hinders their use as an allogeneic source of cell-based therapies. Many potential solutions are being investigated, including ex vivo genetic modifications, alternative cell sources. Induced pluripotent stem cells (iPSC) have been used to differentiate patient-specific ECFCs (iPSC-ECFCs) with robust angiogenic capabilities.70 These cells are particularly attractive, as they are phenotype-specific, offer a stable endothelial phenotype and may enable the derivation of clinically relevant numbers of vessel-forming cells for endothelial repair. Recently a FOXF1 + cKIT+ gCap cells for the treatment of a mouse model of the alveolar capillary dysplasia with misalignment of pulmonary veins was generated using blastocyst complementation and interspecies mouse-rat chimeras as bioreactors.71 Finally, ECFCs are present in umbilical and peripheral blood in small number, preventing their routine clinical use without implementing standardized protocols for effective ex vivo expansion.
The potential risk of tumor formation is another limitation of using ECFCs as a therapeutic product. The concern regarding oncologic potential of ECFCs stems from the proliferative capabilities of these cells and associations of higher proliferative potential of these cells in patients with vascular malformations, e.g., Kaposi sarcoma72 or infantile haemangiona.73 To our knowledge this has never been corroborated in preclinical studies. Long-term safety in rodents was confirmed with follow-up up to 10 months.53
Enhancing rigor in defining endothelial progenitors
Preclinical studies of ECFCs are frequently burdened with poor characterization and lack of unified reporting. To address this issue, a consensus statement on the nomenclature of endothelial progenitors has been published.74 To improve the quality of data reported in future controlled trials, the authors suggest a set of minimal criteria for the cells demonstrating pro-angiogenic properties (summarized in Table 2.). For ECFCs, these criteria include: endothelial cell phenotype, significant proliferative potential, capacity to self-assemble into functional blood vessels in vivo, and a precise immunophenotype defined as positive for: CD31, VE-Cadherin, von Willebrand factor, CD146, VEGFR2 and negative for CD45 and CD14. These cells are the only true endothelial progenitors and should be distinguished from myeloid angiogenic cells (MACs). While pro-angiogenic, these cells have a haematopoietic origin and do not give rise to endothelial cells. This document offers a set of standard, minimal criteria for defining ECFC and is by no means exhaustive. Recent advances in cell characterization and single-cell techniques will most likely result in further evolution of this definition, as recently reviewed.75
Cell-free therapies
Extracellular vesicles: from trash-can to cell-cell communicator to therapy
The discovery of paracrine mechanisms involved in the beneficial effects of stem cells generated a lot of interest. Utilizing a cell-free therapeutic product that offers the same clinical benefit allows to overcome immunogenicity of allogeneic cells and the risk of embolus or tumor formation.67 Extracellular vesicles (EVs), first described in 1981, are small, membrane-coated spherical particles. They carry a wide array of bioactive agents, including proteins, enzymes, lipids, DNA, mRNA, and miRNA. Originally believed to be the cell’s trash-can, EVs are now recognized as critical in cell-to-cell communication. The molecular mechanisms involved in EV-mediated intercellular crosstalk are not yet fully understood. Exosomes, a subpopulation of small EVs (30–100 nm in diameter) are secreted through endosomal pathway and have been implicated as mediators of paracrine activity of stem cells.
The data on ECFC-derived EVs, their role in conveying their angiogenic capacity, and the precise characterization of their cargo, are limited. ECFCs-CM has been demonstrated to ameliorate hyperoxia-induced and bleomycin-induced pulmonary hypertension in animal models in two controlled studies.53,68 These studies were the first to demonstrate that the therapeutic effect of ECFCs in PH associated with perinatal insults is mediated by paracrine mechanisms. However, neither of these studies identified EVs as the carrier of the therapeutic cargo. Importantly, the long-term safety and efficacy of ECFC therapy was demonstrated based on preserved alveolar architecture, improved exercise capacity and echocardiographic parameters in hyperoxic animals treated with ECFCs at 10 months of age, as well as lack of structural organ abnormalities in room air-housed animals treated with ECFCs.53
EVs originating from endothelial progenitors promote angiogenesis by improving cell proliferation, migration, and tube formation based on in vitro evaluation of pulmonary microvascular endothelial cells exposed to hyperoxia.76 In a more recent study cord blood derived EVs of preterm babies enhanced angiogenic activity of human umbilical vein endothelial cells.77 EVs from cord blood of babies who later developed BPD were isolated and their cargo compared with babies who did not develop BPD. Based on differentially expressed miRNAs the authors created a miRNA-mRNA network and transfected three miRNA mimics: miR-103a-3p, miR-185–5p and miR-200a-3p to the endothelial cells, demonstrating that overexpression of miR103a-3p and miR-185-5p promotes endothelial cell proliferation, migration, and tube formation, whereas overexpression of miR200-a-3p has an opposite effect. These results confirm that the EVs present in the cord blood of preterm babies who develop BPD inhibit endothelial angiogenic properties. That in turn hints to the antenatal origins of arrested pulmonary vascular development. Second, this effect is mediated by differentially expressed miRNA. Finally, modifying miRNA expression in endothelial cells alone could be a promising therapeutic target in the prevention of BPD-PH. Importantly, the isolation of EVs in the cord blood does not allow to confirm their cellular origin and these results need to be replicated with a more optimally characterized therapeutic product.
MiRNA—the long-awaited intermediary?
MicroRNAs, first described in 199378 are short (21–25 bp), single-stranded, noncoding RNA molecules. They are involved in posttranscriptional control of gene expression by binding with target sequences within 3’UTR region of mRNA and blocking its expression. An individual miRNA can bind to multiple mRNAs and repress the biosynthesis of several proteins. This makes miRNAs unique regulators of multiple pathways.
The differential expression of miRNA in the blood of infants with BPD79 and the lung tissue of rodent pups exposed to hyperoxia80,81 point to potential biomarkers and therapeutic targets. Downstream targets for differentially expressed miRNAs in the lung tissue of hyperoxia-exposed rodents included brain acid soluble protein 1 (BASP-1), connective tissue growth factor (CTGF), glycoprotein nonmetastatic melanoma protein B (GPNMB), and insulin-like growth factor 1 (IGF-1). The authors confirmed a causative relationship between increased miR-150 and the downregulation of GPNMB, a particle involved in angiogenesis. The role of disrupted miRNA expression and their target downstream growth factor pathways (in brackets) in BPD was confirmed in several subsequent publications, including: miR-489 (IGF-1),82 miR-17∼92 cluster (TGF-ß),83 miR-34a (PDGFRα),84 miR199a-5p (caveolin-1),85 miR-154 (TGF-ß),86 miR-421 (FGF10),87 miR-547-3p (adrenomedullin)88 and others.89 Gain/loss-of-function evaluation in selected studies demonstrates the causative relationship between disrupted miRNA expression and the BPD phenotype. This in turn supports the robust regulatory potential of miRNA. Although none of these studies included PH as an endpoint, the role of miRNAs in the development of PH should not be ruled out. PH in this case is not a separate entity, but rather an extreme end of the spectrum associated with severe BPD.
Summary of data on the link between arrested pulmonary vascular development and altered miRNA expression is presented in Table 3. Syed et al.90 elegantly demonstrated that miR34-a was up-regulated in the lung tissue of mice exposed to hyperoxia. MiR34-a inhibition attenuated parenchymal injury and pulmonary vascular disease, by increasing the expression of angiopoietin 1. Several miRNAs inhibit VEGF-A expression. Cheng et al. reported that miR-203a-3p was up-regulated in hyperoxia-induced lung injury in rats.91 The lung tissue damage could be rescued by suppressing miR-203a-3p, which in turn resulted in VEGF-A up-regulation. Gilfillan et al. described a similar relationship between miR-451, macrophage migration inhibitory factor (MIF), and VEGF-A.92
The dysregulation of miRNA in infants with BPD was concurrently demonstrated in selected studies. Cheng et al. confirmed the up-regulation of miR-203a-3p in the serum of infants who developed BPD corresponding with lower VEGF-A and HIF1α mRNA.91 Similarly, Freeman et al. demonstrated the up-regulation of miR-219-5p in tracheal aspirates of infants with BPD.93 These human studies describe association between disrupted miRNA signaling and abnormal lung development. The results must be interpreted with caution to avoid attributing causation prematurely. On the other hand, inclusion of animal and in vitro verification experiments supports possible causal nature of this relationship.
The data summarized above demonstrate central regulatory role of miRNA in pulmonary vascular development and confirm that hyperoxia disrupts miRNA physiology. Correcting miRNA signals in developing lungs holds promise to prevent or reverse BPD-PH. Thus, synthetic nanoparticles and EVs derived from ECFCs that contain a therapeutic combination of miRNA particles represent a promising therapeutic avenue. The causal relationship between aberrant miRNA expression and perinatal ECFC dysfunction needs to be demonstrated in future studies. Novel techniques, including single-cell miRNA-mRNA co-sequencing will help test this hypothesis.94,95
Where do we go from here?
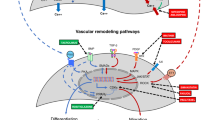
Looking at the developing pulmonary vascular system in a single-cell resolution reveals early origins of BPD-PH. Dysfunction of ECFCs and depletion of capillary endothelial cells in preterm infants with BPD may explain the limitation of current traditional vasodilator therapies and form the rationale for proposing endothelial progenitor cell-based therapies. Capitalizing on the scRNAseq data will also help enhance the rigour in defining EPCs. Cell-free ECFC-EV- and miRNA-based therapies are two most promising treatment modalities currently being explored. (summarized in Fig. 1.) Both strategies require further preclinical exploration. Isolation and characterization methods of ECFC-EVs need to be standardized to ensure reproducibility. Strategies to scale-up the production of EVs must be developed. Better understanding of the functional cargo is necessary, including gain of function and loss of function experiments. Other biologically active particles in EV cargo and the impact of ex-vivo priming of ECFCs on angiogenic properties of ECFC-EVs should also be investigated.

Created with Biorender.com. Arrested lung development in BPD is a result of disrupted growth factor signaling coupled with a disbalance in regulatory miRNA. This leads to alveolar simplification, vascular paucity, endothelial depletion and dysfunction. ECFCs as vascular progenitors are a promising therapeutic strategy to reverse these changes. Their therapeutic effect is mediated by paracrine mechanisms, with EVs containing miRNA. Thus, correcting aberrant miRNA signaling may reverse/prevent BPD-PH development. VEGF vascular endothelial growth factor, TGFβ transforming growth factor beta, FGF10 fibroblast growth factor 10, BMPR2 bone morphogenetic protein receptor type 2, CTGF connective tissue growth factor, PDGF platelet derived growth factor, ECFC endothelial colony forming cell, ECFC-EVs endothelial colony forming cell-derived extracellular vesicles, EVs extracellular vesicles, Cap1 capillary type 1, Cap2 capillary type 2 cell, AT1 alveolar type 1 cell, AT2 alveolar type 2 cell.
MiRNA-based therapies represents a novel direction in BPD-PH research. Comprehensive understanding of miRNA – mRNA interactions in the developing lung is critical and may enable the design of personalized therapies tailored to aberrant miRNA signaling phenotype. Optimal route of delivery, dose and timing are yet to be determined, as is the source of miRNA and the vehicle facilitating its delivery. Our understanding of miRNA biology and its role in BPD-PH is still in its infancy. However, recent discoveries provide a compelling rationale for further well-designed preclinical trials that can eventually bring new therapies to patients. What is at the end of this journey, might just be the first intervention able to improve the prognosis of a disease with no effective treatment.
Data availability
Data sharing is not applicable as no datasets were generated or analyzed for this review.
References
Thébaud, B. et al. Bronchopulmonary dysplasia. Nat. Rev. Dis. Primers. 5, 1–23 (2019).
Stoll, B. J. et al. Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics 126, 443–456 (2010).
Jobe, A. H., Bancalari, E. Bronchopulmonary dysplasia. In: Am. J. Respir. Crit. Care Med. Vol 163. American Lung Association; 2001:1723-1729.
Arjaans, S. et al. Identification of gaps in the current knowledge on pulmonary hypertension in extremely preterm infants: A systematic review and meta-analysis. Paediatr. Perinat. Epidemiol. 32, 258–267 (2018).
Nagiub, M., Kanaan, U., Simon, D. & Guglani, L. Risk factors for development of pulmonary hypertension in infants with bronchopulmonary dysplasia: systematic review and meta-analysis. Paediatr. Respir. Rev. 23, 27–32 (2017).
Khemani, E. et al. Pulmonary artery hypertension in formerly premature infants with bronchopulmonary dysplasia: Clinical features and outcomes in the surfactant era. Pediatrics 120, 1260–1269 (2007).
Barrington, K. J., Finer, N., Pennaforte, T., Altit, G. Nitric oxide for respiratory failure in infants born at or near term. Cochrane Database Syst. Rev. 2017, CD000399 (2017).
Stritzke, A., Bhandari, V. & Lodha, A. Use of inhaled nitric oxide in preterm infants: is there sufficient evidence? Indian J. Pediatr. 89, 262–266 (2022).
Mourani, P. M., Sontag, M. K., Ivy, D. D. & Abman, S. H. Effects of long-term sildenafil treatment for pulmonary hypertension in infants with chronic lung disease. J. Pediatr. 154, 379–384 (2009).
Nyp, M., Sandritter, T., Poppinga, N., Simon, C. & Truog, W. E. Sildenafil citrate, bronchopulmonary dysplasia and disordered pulmonary gas exchange: Any benefits? J. Perinatol. 32, 64–69 (2012).
Tan, K., Krishnamurthy, M. B., O’Heney, J. L., Paul, E. & Sehgal, A. Sildenafil therapy in bronchopulmonary dysplasia-associated pulmonary hypertension: a retrospective study of efficacy and safety. Eur. J. Pediatr. 174, 1109–1115 (2015).
Trottier-Boucher, M. N. et al. Sildenafil for the treatment of pulmonary arterial hypertension in infants with bronchopulmonary dysplasia. Pediatr. Cardiol. 36, 1255–1260 (2015).
Jackson, W. et al. Safety of sildenafil in extremely premature infants: a phase I trial. J. Perinatol. 42, 31–36 (2022).
Katsuragi, S. et al. Riociguat can ameliorate bronchopulmonary dysplasia in the SU5416 induced rat experimental model. Exp. Lung Res. 47, 382–389 (2021).
Donda, K. et al. Riociguat prevents hyperoxia-induced lung injury and pulmonary hypertension in neonatal rats without effects on long bone growth. PLoS One 13, e0199927 (2018).
Jeremiasen, I., Naumburg, E., Westöö, C. G., Weismann, C. & Tran-Lundmark, K. Vasodilator therapy for pulmonary hypertension in children: a national study of patient characteristics and current treatment strategies. Pulm Circ. 11, 20458940211057891 (2021).
Spreemann, T., Bertram, H., Happel, C. M., Kozlik-Feldmann, R. & Hansmann, G. First-in-child use of the oral soluble guanylate cyclase stimulator riociguat in pulmonary arterial hypertension. Pulm Circ. 8, 2045893217743123 (2018).
Rugolotto, S. et al. Weaning of epoprostenol in a small infant receiving concomitant bosentan for severe pulmonary arterial hypertension secondary to bronchopulmonary dysplasia. Minerva Pediatr. 58, 491–494 (2006).
Gürakan, B., Kayiran, P., Öztürk, N., Kayiran, S. M. & Dindar, A. Therapeutic combination of sildenafil and iloprost in a preterm neonate with pulmonary hypertension. Pediatr. Pulmonol. 46, 617–620 (2011).
Hwang, S. K., O, Y. C., Kim, N. S., Park, H. K. & Yum, M. K. Use of inhaled iloprost in an infant with bronchopulmonary dysplasia and pulmonary artery hypertension. Korean Circ. J. 39, 342–345. (2009).
Piastra, M. et al. Nebulized iloprost and noninvasive respiratory support for impending hypoxaemic respiratory failure in formerly preterm infants: a case series. Pediatr. Pulmonol. 47, 757–762 (2012).
Ferdman, D. J., Rosenzweig, E. B., Zuckerman, W. A. & Krishnan, U. Subcutaneous treprostinil for pulmonary hypertension in chronic lung disease of infancy. Pediatrics 134, e274–e278 (2014).
Kadmon, G. et al. Pulmonary hypertension specific treatment in infants with bronchopulmonary dysplasia. Pediatr. Pulmonol. 52, 77–83 (2017).
Migdał, A. et al. Children with bronchopulmonary dysplasia-associated pulmonary hypertension treated with pulmonary vasodilators-the pediatric cardiologist point of view. Children (Basel) 8, 326–338 (2021).
Le Cras, T. D., Markham, N. E., Tuder, R. M., Voelkel, N. F. & Abman, S. H. Treatment of newborn rats with a VEGF receptor inhibitor causes pulmonary hypertension and abnormal lung structure. Am. J. Physiol. Lung Cell Mol. Physiol. 283, L555–L562 (2002). 3.
Nicolls, M. R. et al. New models of pulmonary hypertension based on VEGF receptor blockade-induced endothelial cell apoptosis. Pulm. Circ. 2, 434–442 (2012).
Kunig, A. M. et al. Recombinant human VEGF treatment enhances alveolarization after hyperoxic lung injury in neonatal rats. Am. J. Physiol. Lung Cell Mol. Physiol. 289, L529–L535 (2005). 4.
Thébaud, B. et al. Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury: Evidence that angiogenesis participates in alveolarization. Circulation 112, 2477–2486 (2005).
Vila Ellis, L. et al. Epithelial vegfa specifies a distinct endothelial population in the mouse lung. Dev. Cell 52, 617–630.e6 (2020).
Orriols, M., Gomez-Puerto, M. C. & ten Dijke, P. BMP type II receptor as a therapeutic target in pulmonary arterial hypertension. Cell. Mol. Life Sci. 74, 2979–2995 (2017).
Garcia-Rivas, G., Jerjes-Sánchez, C., Rodriguez, D., Garcia-Pelaez, J. & Trevino, V. A systematic review of genetic mutations in pulmonary arterial hypertension. BMC Med. Genet 18, 82 (2017).
Yee, M. et al. Neonatal hyperoxia causes pulmonary vascular disease and shortens life span in aging mice. Am. J. Pathol. 178, 2601–2610 (2011).
Arjaans, X. S. et al. Early angiogenic proteins associated with high risk for bronchopulmonary dysplasia and pulmonary hypertension in preterm infants. Am. J. Physiol. Lung Cell Mol. Physiol. 318, L644–L654 (2020).
Heydarian, M. et al. Relationship between impaired BMP signalling and clinical risk factors at early-stage vascular injury in the preterm infant. Thorax. Published online May 17, 2022:thoraxjnl-2021-218083.
Gauldie, J. et al. Transfer of the active form of transforming growth factor-β1 gene to newborn rat lung induces changes consistent with bronchopulmonary dysplasia. Am. J. Pathol. 163, 2575–2584 (2003).
Le Saux, O. et al. The role of caveolin-1 in pulmonary matrix remodeling and mechanical properties. Am. J. Physiol. Lung Cell Mol. Physiol. 295, L1007–L1017 (2008).
Fan, W. H., Pech, M. & Karnovsky, M. J. Connective tissue growth factor (CTGF) stimulates vascular smooth muscle cell growth and migration in vitro. Eur. J. Cell Biol. 79, 915–923 (2000).
Thomas, W. et al. Airway concentrations of angiopoietin-1 and endostatin in ventilated extremely premature infants are decreased after funisitis and unbalanced with bronchopulmonary dysplasia/death. Pediatr. Res. 65, 468–473 (2009).
Mohamed, W. A. W., Niyazy, W. H. & Mahfouz, A. A. Angiopoietin-1 and endostatin levels in cord plasma predict the development of bronchopulmonary dysplasia in preterm infants. J. Trop. Pediatr. 57, 385–388 (2011).
Kim, D. H. & Kim, H. S. Serial changes of serum endostatin and angiopoietin-1 levels in preterm infants with severe bronchopulmonary dysplasia and subsequent pulmonary artery hypertension. Neonatology 106, 55–61 (2014).
Lai, S. H. et al. Low cord-serum 25-hydroxyvitamin D levels are associated with poor lung function performance and increased respiratory infection in infancy. PLoS One 12, e0173268 (2017).
Foong, R. E. et al. The effects of in utero Vitamin D deficiency on airway smooth muscle mass and lung function. Am. J. Respir. Cell Mol. Biol. 53, 664–675 (2015).
Wang, Y. & Jiang, L. Role of vitamin D-vitamin D receptor signaling on hyperoxia-induced bronchopulmonary dysplasia in neonatal rats. Pediatr. Pulmonol. 56, 2335–2344 (2021).
Groenman, F. A. et al. Effect of chemical stabilizers of hypoxia-inducible factors on early lung development. Am. J. Physiol. Lung Cell Mol. Physiol. 293, L557–L567 (2007).
Shimoda, L. A. & Semenza, G. L. HIF and the lung: role of hypoxia-inducible factors in pulmonary development and disease. Am. J. Respir. Crit. Care Med. 183, 152–156 (2011).
Peplow, P. V. Influence of growth factors and cytokines on angiogenic function of endothelial progenitor cells: a review of in vitro human studies. Growth Factors 32, 83–116 (2014).
Asahara, T. et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science 275, 964–967 (1997).
Ingram, D. A. et al. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood 104, 2752–2760 (2004).
Alvarez, D. F. et al. Lung microvascular endothelium is enriched with progenitor cells that exhibit vasculogenic capacity. Am. J. Physiol. Lung Cell Mol. Physiol. 294, L419–L430 (2008).
Balasubramaniam, V., Mervis, C. F., Maxey, A. M., Markham, N. E. & Abman, S. H. Hyperoxia reduces bone marrow, circulating, and lung endothelial progenitor cells in the developing lung: Implications for the pathogenesis of bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell Mol. Physiol. 292, L1073–L1084 (2007).
Baker, C. D. et al. Endothelial colony-forming cells from preterm infants are increased and more susceptible to hyperoxia. Am. J. Respir. Crit. Care Med. 180, 454–461 (2009).
Baker, C. D. et al. Cord blood angiogenic progenitor cells are decreased in bronchopulmonary dysplasia. Eur. Respir. J. 40, 1516–1522 (2012).
Alphonse, R. S. et al. Existence, functional impairment, and lung repair potential of endothelial colony-forming cells in oxygen-induced arrested alveolar growth. Circulation 129, 2144–2157 (2014).
Ren, X. et al. Postnatal alveologenesis depends on FOXF1 signaling in c-KIT1 endothelial progenitor cells. Am. J. Respir. Crit. Care Med. 200, 1164–1176 (2019).
Muñoz-Hernandez, R. et al. Decreased level of cord blood circulating endothelial colony-forming cells in preeclampsia. Hypertension 64, 165–171 (2014).
Gumina, D. L., Black, C. P., Balasubramaniam, V., Winn, V. D. & Baker, C. D. Umbilical cord blood circulating progenitor cells and endothelial colony-forming cells are decreased in preeclampsia. Reprod. Sci. 24, 1088–1096 (2017).
Schröder‐Heurich, B. et al. Downregulation of miR‐1270 mediates endothelial progenitor cell function in preeclampsia: Role for ATM in the Src/VE‐cadherin axis. FASEB J. 36 (2022).
Sipos, P. I. et al. Endothelial colony-forming cells derived from pregnancies complicated by intrauterine growth restriction are fewer and have reduced vasculogenic capacity. J. Clin. Endocrinol. Metab. 98, 4953–4960 (2013).
Gillich, A. et al. Capillary cell-type specialization in the alveolus. Nature 586, 785–789 (2020).
Negretti, N. M. et al. A single-cell atlas of mouse lung development. Development (Cambridge) 148, dev199512 (2021).
Hurskainen, M. et al. Single cell transcriptomic analysis of murine lung development on hyperoxia-induced damage. Nat. Commun. 12, 1565 (2021).
Sun, X. et al. A census of the lung: CellCards from LungMAP. Dev. Cell. 57, 112–145.e2 (2022).
Sarlon, G. et al. Therapeutic effect of fucoidan-stimulated endothelial colony-forming cells in peripheral ischemia. J. Thromb. Haemost. 10, 38–48 (2012).
Goto, K. et al. Intravenous administration of endothelial colony-forming cells overexpressing integrin β 1 augments angiogenesis in ischemic legs. Stem Cells Transl. Med 5, 218–226 (2016).
Tan, Q. et al. Transplantation of healthy but not diabetic outgrowth endothelial cells could rescue ischemic myocardium in diabetic rabbits. Scand. J. Clin. Lab. Invest. 70, 313–321 (2010).
Burger, D. et al. Human endothelial colony-forming cells protect against acute kidney injury role of exosomes. Am. J. Pathol. 185, 2309–2323 (2015).
Xia, W. H. et al. BMP4/Id2 signaling pathway is a novel therapeutic target for late outgrowth endothelial progenitor cell-mediated endothelial injury repair. Int. J. Cardiol. 228, 796–804 (2017).
Baker, C. D. et al. Endothelial colony-forming cell conditioned media promote angiogenesis in vitro and prevent pulmonary hypertension in experimental bronchopulmonary dysplasia. Am. J. Physiol. Lung Cell Mol. Physiol. 305, L73–L81 (2013).
Liao, G., Zheng, K., Shorr, R. & Allan, D. S. Human endothelial colony-forming cells in regenerative therapy: A systematic review of controlled preclinical animal studies. Stem Cells Transl. Med 9, 1344–1352 (2020).
Prasain, N. et al. Differentiation of human pluripotent stem cells to cells similar to cord-blood endothelial colony-forming cells. Nat. Biotechnol. 32, 1151–1157 (2014).
Wang, G. et al. Generation of pulmonary endothelial progenitor cells for cell-based therapy using interspecies mouse-rat chimeras. Am. J. Respir. Crit. Care Med. 204, 326–338 (2021).
Calcaterra, F. et al. Increased frequency and vasculogenic potential of endothelial colony-forming cells in patients with Kaposi’s sarcoma. J. Invest. Dermatol. 137, 1533–1540 (2017).
Campanelli, R. et al. Kinetic and angiogenic activity of circulating endothelial colony forming cells in patients with infantile haemangioma receiving propranolol. Thromb. Haemost. 119, 274–284 (2019).
Medina, R. J. et al. Endothelial progenitors: A consensus statement on nomenclature. Stem Cells Transl. Med 6, 1316–1320 (2017).
Salybekov, A. A., Kobayashi, S. & Asahara, T. Characterization of endothelial progenitor cell: past, present, and future. Int. J. Mol. Sci. 23, 7697 (2022).
Zhang, X. et al. Exosomes secreted by endothelial progenitor cells improve the bioactivity of pulmonary microvascular endothelial cells exposed to hyperoxia in vitro. Ann. Transl. Med. 7, 254–254 (2019).
Zhong, X. Q. et al. Umbilical cord blood-derived exosomes from very preterm infants with bronchopulmonary dysplasia impaired endothelial angiogenesis: roles of exosomal MicroRNAs. Front. Cell Dev. Biol. 9, 637248 (2021).
Lee, R. C., Feinbaum, R. L. & Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75, 843–854 (1993).
Wu, Y. T. et al. MicroRNA expression aberration associated with bronchopulmonary dysplasia in preterm infants: A preliminary study. Respir. Care 58, 1527–1535 (2013).
Zhang, X. et al. MicroRNA expression profile in hyperoxia-exposed newborn mice during the development of bronchopulmonary dysplasia. Respir. Care 56, 1009–1015 (2011).
Bhaskaran, M. et al. Identification of microRNAs changed in the neonatal lungs in response to hyperoxia exposure. Physiol. Genomics. 44, 970–980 (2012).
Olave, N. et al. Regulation of alveolar septation by microRNA-489. Am. J. Physiol. Lung Cell Mol. Physiol. 310, L476–L487 (2016).
Rogers, L. K. et al. Attenuation of MIR-17∼92 cluster in bronchopulmonary dysplasia. Ann. Am. Thorac. Soc. 12, 1506–1513 (2015).
Ruiz‐Camp, J. et al. Targeting miR‐34a/ Pdgfra interactions partially corrects alveologenesis in experimental bronchopulmonary dysplasia. EMBO Mol Med. 11, e9448 (2019).
Alam, M. A., Betal, S. G. N., Aghai, Z. H. & Bhandari, V. Hyperoxia causes miR199a-5p-mediated injury in the developing lung. Pediatr. Res 86, 579–588 (2019).
Chao, C. M. et al. Failure to down-regulate miR-154 expression in early postnatal mouse lung epithelium suppresses alveologenesis, with changes in Tgf-β signaling similar to those induced by exposure to hyperoxia. Cells 9, 849 (2020).
Yuan, H. S., Xiong, D. Q., Huang, F., Cui, J. & Luo, H. MicroRNA-421 inhibition alleviates bronchopulmonary dysplasia in a mouse model via targeting Fgf10. J. Cell. Biochem. 120, 16876–16887 (2019).
Gong, X., Qiu, J., Qiu, G. & Cai, C. Adrenomedullin regulated by miRNA-574-3p protects premature infants with bronchopulmonary dysplasia. Biosci. Rep. 40, BSR20191879 (2020).
Hu, Y. Inhibition of microRNA-29a alleviates hyperoxia-induced bronchopulmonary dysplasia in neonatal mice via upregulation of GAB1. Mol. Med. 26, 1–12 (2019).
Syed, M. et al. Hyperoxia causes MIR-34a-mediated injury via angiopoietin-1 in neonatal lungs. Nat. Commun. 8, 1–17 (2017).
Cheng, H. et al. Knockdown of miR-203a-3p alleviates the development of bronchopulmonary dysplasia partly via the up-regulation of vascular endothelial growth factor A. J. Bioenerg. Biomembr. 53, 13–23 (2021).
Gilfillan, M., Das, P., Shah, D., Alam, M. A., Bhandari, V. Inhibition of microRNA-451 is associated with increased expression of Macrophage Migration Inhibitory Factor and mitigation of the cardio-pulmonary phenotype in a murine model of Bronchopulmonary Dysplasia. Respir. Res. 21 (2020).
Freeman, A. et al. MicroRNA 219-5p inhibits alveolarization by reducing platelet derived growth factor receptor-alpha. Respir. Res. 22, 1–9 (2021).
Zhang, J. et al. Exploring cell-specific miRNA regulation with single-cell miRNA-mRNA co-sequencing data. BMC Bioinform 22, 578 (2021).
Chakraborty, C., Bhattacharya, M. & Agoramoorthy, G. Single-cell sequencing of miRNAs: A modified technology. Cell Biol. Int. 44, 1773–1780 (2020).
Mukherjee, D. et al. Fetal pulmonary hypertension: dysregulated microRNA-34c-Notch1 axis contributes to impaired angiogenesis in an ovine model. Pediatr. Res. Published online 2022.
Wang, C. et al. Integrated MicroRNA-mRNA analyses of distinct expression profiles in hyperoxia-induced bronchopulmonary dysplasia in neonatal mice. Am J Perinatol. Published online 2021.
Funding
W.D. is financially supported by the Bekker Programme from the Polish National Agency for Academic Exchange – (Number BPN/BEK/2021/1/00329/ U/00001). B.T. is supported by the Canadian Institutes of Health Research (CIHR) and the Stem Cell Network.
Author information
Authors and Affiliations
Contributions
Concept and design: W.D. and B.T.; interpretation of relevant literature: W.D.; drafting the manuscript: W.D.; review and final approval of the manuscript: B.T. All authors have read and agreed to the published version of the manuscript
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Durlak, W., Thébaud, B. The vascular phenotype of BPD: new basic science insights—new precision medicine approaches. Pediatr Res (2022). https://doi.org/10.1038/s41390-022-02428-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41390-022-02428-7
This article is cited by
-
Adequate nutrition for bronchopulmonary dysplasia, but do not forget oxygen
Pediatric Research (2024)
