
Abstract
Background
Paternally inherited loss-of-function mutations in MKRN3 underlie central precocious puberty (CPP). We describe clinical and genetic features of CPP patients with paternally inherited MKRN3 mutations in two independent families.
Methods
The single coding exon of MKRN3 was analyzed in three patients with CPP and their family members, followed by segregation analyses. Additionally, we report the patients’ responses to GnRH analog treatment.
Results
A paternally inherited novel heterozygous c.939C>G, p.(Ile313Met) missense mutation affecting the RING finger domain of MKRN3 was found in a Finnish girl with CPP (age at presentation 6 years). Two Polish siblings (a girl presenting with B2 at the age of 4 years and a boy with adult size testes at the age of 9 years) had inherited a novel heterozygous MKRN3 mutation c.1237_1252delGGAGACACATGCTTTT p.(Gly413Thrfs*63) from their father. The girls were treated with GnRH analogs, which exhibited suppression of the hypothalamic–pituitary–gonadal axis. In contrast, the male patient was not treated, yet he reached his target height.
Conclusions
We describe two novel MKRN3 mutations in three CPP patients. The first long-term data on a boy with CPP due to an MKRN3 mutation questions the role of GnRH analog treatment in augmenting adult height in males with this condition.
Impact
-
We describe the genetic cause for central precocious puberty (CPP) in two families.
-
This report adds two novel MKRN3 mutations to the existing literature. One of the mutations, p.(Ile313Met) affects the RING finger domain of MKRN3, which has been shown to be important for repressing the promoter activity of KISS1 and TAC3.
-
We describe the first long-term observation of a male patient with CPP due to a paternally inherited MKRN3 loss-of-function mutation. Without GnRH analog treatment, he achieved an adult height that was in accordance with his mid-parental target height.
Similar content being viewed by others

Introduction
Environmental and genetic factors affect the timing of puberty.1,2 Precocious activation of the hypothalamic–pituitary–gonadal axis, i.e., central precocious puberty (CPP), results in testis growth in boys before the age of 9 years and in breast development in girls before the age of 8 years. The incidence of CPP has been estimated to be 37 per 100,000.3 Early puberty is associated with increased risk of cardiovascular disease and delinquent behavior in adulthood.4,5 In the UK biobank study, earlier timing of normal puberty was associated with 48 adverse health outcomes such as breast cancer, short stature, and depression.6 Our understanding of the CPP genetics was expanded in 2013, when Abreu et al. showed that paternally inherited mutations in the makorin ring finger protein 3 (MKRN3) underlie CPP.7 MKRN3 encodes a putative ubiquitin ligase, and MKRN3 protein interacts with proteins implicated in the timing of puberty and congenital hypogonadotropic hypogonadism.8 The mechanisms by which MKRN3 regulates puberty are rapidly emerging.9,10,11 The latest major advance in the field shows that, in mice, Mkrn3 is coexpressed in kisspeptin neurons, human MKRN3 binds to KISS1 and TAC3 promoters, and mutations affecting the RING finger domain of the protein reduce MKRN3 ubiquitination activity and compromise the ability of MKRN3 to repress KISS1 and TAC3 promoter activity.11 Describing the phenotype, penetrance, and long-term follow-up of CPP patients with loss-of-function MKRN3 mutations are thus expected to increase our understanding on the elusive functions of the MKRN3 and to reveal possible genotype–phenotype correlations.
This study describes genotypic and phenotypic features of two independent CPP families with paternally inherited MKRN3 mutations as well as their growth parameters, responses to gonadotropin-releasing hormone (GnRH) analog treatment, and summarizes the reported mutations in MKRN3. While the majority of the MKRN3 mutations described to date lead to truncated proteins, missense mutations affecting the RING finger domain are of particular interest.11 Also, such mutations pose a diagnostic challenge since they are easily classified as variants of unknown significance, whereas the demonstration of paternal inheritance of such a mutation to a patient with CPP is the key to set the correct diagnosis.
Materials and methods
Patients
We analyzed phenotypes and MKRN3 carrier status of two independent families who originated from Finland (one female CPP proband, investigated in the New Children’s Hospital at the Helsinki University Hospital, Finland) and from Poland (brother and sister who were evaluated at the Karol Jonscher’s Clinical Hospital, Poznan, Poland). The detailed clinical phenotypes of the patients and their hormonal data are described in the “Results” section.
Hormone assays, radiological studies, and clinical measurements
In the Finnish patient, serum luteinizing hormone (LH) and follicle-stimulating hormone (FSH) levels were measured with immunoelectrochemiluminometric assay on an automated immunoanalyser (Modular Analytics E170 Roche Diagnostics, Mannheim, Germany). The detection limit of the LH assay was 0.1 IU/l, intra-assay coefficient of variation (CV) was <2%, and inter-assay CV was <6%, whereas the detection limit of the FSH assay was 0.1 IU/l with intra-assay CV <3% and inter-assay CV <5%. GnRH stimulation test was performed by intravenous injection of a GnRH analog (3.5 μg/kg [max. 100 μg], Relefact® LH-RH 0.1 mg; Aventis Pharma, Frankfurt, Germany).
In Polish patients, LH, FSH, and estradiol (E2) were assessed with chemiluminescent microparticle immunoassays by ARCHITECT (Abbott Laboratories, Lisnamuck, Ireland). The detection limit of the LH assay was 0.03 IU/l, intra-assay CV was <3.6%, and inter-assay CV was <8.7%. The detection limit of the FSH assay was 0.02 IU/l with intra-assay CV <3.3% and inter-assay CV <4.6%, whereas the detection limit of the E2 assay was 36.7 pmol/l, intra-assay CV varied from 1.4 to 6.4%, and inter-assay CV varied from 1.8 to 7.4%. Testosterone was determined with TESTO-RIA-CT (DIAsource ImmunoAssays S.A., Louvain-la-Neuve, Belgium) with the lowest detection limit of 0.1 nmol/l, intra-assay CV was <8.9%, and inter-assay CV varied from 5.2% to 11.6%. GnRH stimulation test was conducted with an intravenous injection of 2.5 μg/kg of GnRH (Relefact LH-RH®, Sanofi-Aventis, Frankfurt, Germany) (max. 100 μg) with blood drawn prior to the test and at 30 min and at 60 min after injection.
Bone age was determined from X-ray of the left hand and wrist with an automated bone age assessment (BoneXpert) or by a pediatric endocrinologist.12 Brain magnetic resonance imaging (MRI) was performed with 1.5 or 3 T platforms.
To calculate height standard deviation scores (SDS), we used the most recent national reference data.13,14 In all patients, the age-adjusted body mass index (BMI) values (ISO-BMI) were calculated from documented growth measurements by using the Finnish reference data.13
Genetic testing
The genetic evaluation of the Finnish index patient was performed with Blueprint Genetics MKRN3 single-gene test Plus. Peripheral blood or saliva samples were collected from her parents and from the Polish family for DNA extraction. The coding exon and exon–intron boundaries of MKRN3 (ENSG00000179455, ENST00000314520, Ensembl release 97) were then PCR amplified. The PCR conditions and primers are available upon request. The PCR products were purified with ExoProStar treatment (GE Healthcare Life Sciences, Chicago, IL, USA) and sequenced from both directions using the ABI BigDyeTerminator Cycle Sequencing Kit (v3.1) and ABI Prism 3730xl DNA Analyzer automated sequencer (Applied Biosystems, Foster City, CA, USA). The DNA sequences were aligned and read with the Sequencher® 4.9 software (Gene Codes Corporation, AnnArbor, MI, USA).
Allele frequencies of the identified variants were validated from the Genome Aggregation Database (gnomAD) (http://gnomad.broadinstitute.org/) and SISu Database (http://sisuproject.fi).15,16 The gnomAD contains whole-genome sequencing and exome data from 141,456 individuals including 12,562 Finnish samples. Effects of the identified variants on transcripts were predicted with SIFT (http://sift.jcvi.org/),17 Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/), and MutationTaster (http://www.mutationtaster.org/) in silico tools.18,19
Ethics
The study was approved by the Ethics Committee of the Hospital District of Helsinki and Uusimaa. Written informed consents were obtained from the patients and their parents.
Results
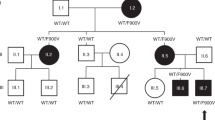
The Finnish index patient (Patient 1) was born at term and was appropriate for gestational age. Her early developmental milestones were unremarkable. At the age of 6 years, she presented with breast development and was referred at the age of 6.4 years to Pediatric Endocrine Outpatient clinic. Her mother’s age for menarche was 13 years and she was 171 cm tall (+0.5 SDS), whereas the father had a history of precocious puberty. His pubertal growth had ended at the age of 12 years, and his adult height was 160 cm (−3.4 SDS). He had another daughter (half-sister to the index patient) who also had been diagnosed and treated for CPP at the age of 5.5 years. The pedigree is presented in Fig. 1a.

Filled symbols denote central precocious puberty. a The Finnish pedigree; b the Polish pedigree. Arrow, index patient. N/A phenotype not available.
Due to her familial CPP, MKRN3 was analyzed and a heterozygous missense c.939C>G, p.(Ile313Met) variant that was predicted to be deleterious according to the used in silico tools (SIFT score 0.01, “deleterious”; Polyphen-2 score 1.000, “probably damaging”; and MutationTaster score 29, “disease-causing”) was found. The mutation was absent from the gnomAD database and from SISu (the national Finnish variant database). Her father with CPP carried the same mutation, whereas the mother did not (Fig. 1a). Index patient’s half-sister and cousin presented early puberty; however, their genotypes were not available. On physical examination, the index patient had Tanner breast stage 2 and pubic hair stage 1; otherwise, her physical examination was unremarkable. She displayed accelerated growth (growth velocity 7.8 cm/year, Fig. 2) and her bone age at the calendar age of 6.4 was 6.8 years. Her baseline serum LH was 0.3 IU/l and FSH concentration 5.2 IU/l. GnRH stimulation test revealed a clearly pubertal LH response (max. 10.4 IU/l). Brain MRI scan showed normal pituitary region. She was started on GnRH analog (Leuprorelin 3.75 mg every 4 weeks) at the age of 6.5 years, and at the age of 7.9 years, she had breast stage 1 and pubic hair stage 1. Her linear growth and ISO-BMI curves before and during the treatment are shown in Figs. 2 and 3. Her weight was normal before and after the initiation of the treatment.

Arrows, initiation of the GnRH analog treatment. TH mid-parental target height.

ISO-BMI values were calculated by using the Finnish reference data for all patients. Arrows, initiation of GnRH analog treatment.
The Polish family included two siblings who both had CPP (Fig. 1b). Their mother had menarche at the age of 13 years and her adult height was 170 cm (+0.7 SDS). The father was 186 cm (+1.2 SDS) tall and had normal pubertal timing. However, because both the children presented with familial CPP, MKRN3 gene was sequenced in all the family members. The father and both children had a novel heterozygous deletion in MKRN3 that was predicted to lead to frameshift and premature stop codon (c.1237_1252delGGAGACACATGCTTTT p.(Gly413Thrfs*63)). The variant was absent from the gnomAD database. The boy (Patient 2) was diagnosed with CPP at the age of 9 years and 3 months when he presented with adult-sized testes (20 ml) and tall stature with not observed acceleration of his growth velocity (Fig. 2). He had pubarche at the age of 7 years and axillarche at the age of 9 years. His testicular ultrasound was normal, testosterone level was 3.6 nmol/l, and GnRH-stimulated maximal LH and FSH responses were 8.9 and 1.7 IU/l, respectively. His brain MRI showed an incidental small pineal cyst. His bone age was 11.2 years, and his mid-parental target height was 184.5 cm (+1.0 SDS). First signs of pubertal spurt were noticed at the age of 9 years and 9 months. He did not receive treatment for the CPP. He was followed up until the age of 15.4 years, when he had reached adult height of 180.5 cm, which was 4 cm below his mid-parental target (Fig. 2). During follow-up, he was overweight, based on the ISO-BMI values (Fig. 3).
His younger sister (Patient 3, Fig. 1b) was diagnosed with CPP at the age of 4.5 years. She presented with thelarche and tall stature with no acceleration of her growth velocity at the age of 4 and 4.5 years, respectively (Fig. 2). She was overweight prior to the initiation of the treatment, and thereafter ISO-BMI values were within normal limits (Fig. 3). Her bone age was 5 years, and her mid-parental target height was 171.5 cm (+1.0 SDS). Her maximal LH response (12.6 IU/l) to GnRH stimulation test was consistent with CPP. Her serum estradiol level was 91.8 pmol/l. Brain MRI scan was unremarkable, and no tumor was found in transabdominal ultrasound. She was started on GnRH analog (Triptorelin 3.75 mg every 4 weeks) at the age of 4.5 years, and at the age of 5.3 years, the breast examination was Tanner I showing regression of breast tissue. GnRH analog treatment is continued until now.
Discussion
This report describes two families with precocious puberty and novel MKRN3 variants: a Finnish family with a c.939C>G, p.(Ile313Met) missense variant and a Polish family with a c.1237_1252delGGAGACACATGCTTTT p.(Gly413Thrfs*63) frameshift variant. These families underwent genetic testing since both pedigrees contained multiple affected persons. Since only paternally inherited loss-of-function mutations in MKRN3 cause CPP, segregation analysis was of crucial importance. Indeed, both novel MKRN3 variants reported herein were inherited from the fathers to patients 1–3 and were absent from gnomAD. The Finnish family variant p.(Ile313Met) was also absent from the national Finnish variant database SISu. Both variants were predicted to be deleterious according to the used in silico tools. The p.(Ile313Met) variant is located in the C3HC4 RING zinc finger motif where at least seven previous variants, two of which affecting nearby codons, have been linked to CPP (Fig. 4).7,20,21,22,23,24 It was recently shown that MKRN3 mutations that affect the RING finger domain exhibit reduced E3 ubiquitin ligase activity and disrupt the ability of MKRN3 to transrepress KISS1 and TAC3 promoters.11 We speculate that these molecular mechanisms also cause CPP in Patient 1. In turn, the Polish family variant c.1237_1252delGGAGACACATGCTTTT, p.(Gly413Thrfs*63) is located in the last C3H1 zinc finger motif of the MKRN3 protein, likewise known to contain at least four previous variants associated with precocious puberty (Fig. 4).25,26,27,28 The frameshift mutation is predicted to lead to a premature stop codon and to produce a truncated protein.

C3H1 C3H1-type zinc finger motif, MKRN3 CYS-HIS Makorin-type Cys-His motif, C3HC4 RING C3HC4 RING-type zinc finger motif. Adapted from Valadares et al.20 and Chen et al.32 The promoter region mutations were reported in Ramos et al.,9 Fanis et al.,33 and Lu et al.34 and are indicated in relation to the translation initiation codon.
In our report, the boy with the MKRN3 mutation presented advanced signs of puberty at the age of 9 years, whereas the girls had thelarche at the ages of 6 and 4.5 years. This sex difference in the timing of the puberty is in agreement with previous patient series20,23,29 and that the initiation of puberty in girls is approximately 2 years earlier than in the male mutation carriers.23,29 To the best of our knowledge, this is the first report on the long-term effects of an MKRN3 mutation in a male patient. Interestingly, Patient 2 did not receive any treatment for the condition and yet reached adult height that was in accordance with his mid-parental target height. We speculate that his favorable height outcome reflects the relatively mild initial advancement of pubertal timing. To this end, his bone age was only approximately 2 years ahead of his chronological age, and his growth had not accelerated at the time of diagnosis. This was rather unexpected, since he displayed adult-size testes at the age of 9 years, suggesting rapid tempo of sexual maturation after the loss of central gonadotropin secretion restraint. On the other hand, the father of the Finnish family, who carried the missense MKRN3 mutation, had also CPP but was relatively short. Of note, two of the patients were overweight in early childhood, and previous studies have reported high BMI values at the time of puberty onset in CPP patients with or without MKRN3 mutations10,30 and that in mice Mkrn3 functions independently of leptin action.31 At the same time, we acknowledge that our study included four patients with CPP and an MKRN3 mutation and that detailed phenotypes were available only in three of them.
In conclusion, we describe two CPP families with multiple members carrying paternally inherited MKRN3 mutations. The long-term health consequences of MKRN3 mutations, if any, are currently unknown, and further investigations are needed to evaluate whether they bear predictive value in terms of clinical management of these patients.
References
Parent, A. S. et al. The timing of normal puberty and the age limits of sexual precocity: variations around the world, secular trends, and changes after migration. Endocr. Rev. 24, 668–693 (2003).
Elks, C. E. et al. Thirty new loci for age at menarche identified by a meta-analysis of genome-wide association studies. Nat. Genet. 42, 1077–1085 (2010).
Soriano-Guillen, L. et al. Central precocious puberty in children living in Spain: incidence, prevalence, and influence of adoption and immigration. J. Clin. Endocrinol. Metab. 95, 4305–4313 (2010).
Lakshman, R. et al. Early age at menarche associated with cardiovascular disease and mortality. J. Clin. Endocrinol. Metab. 94, 4953–4960 (2009).
Xhrouet-Heinrichs, D. et al. Longitudinal study of behavioral and affective patterns in girls with central precocious puberty during long-acting triptorelin therapy. Acta Paediatr. 86, 808–815 (1997).
Day, F. R., Elks, C. E., Murray, A., Ong, K. K. & Perry, J. R. Puberty timing associated with diabetes, cardiovascular disease and also diverse health outcomes in men and women: the UK Biobank study. Sci. Rep. 5, 11208 (2015).
Abreu, A. P. et al. Central precocious puberty caused by mutations in the imprinted gene MKRN3. N. Engl. J. Med. 368, 2467–2475 (2013).
Yellapragada, V. et al. MKRN3 interacts with several proteins implicated in puberty timing but does not influence GNRH1 expression. Front. Endocrinol. 10, 48 (2019).
Ramos, C. O. et al. Outcomes of patients with central precocious puberty due to loss-of-function mutations in MKRN3 gene after treatment with gonadotropin-releasing hormone analog. Neuroendocrinology https://doi.org/10.1159/000504446 (2019).
Abreu, A. P. & Kaiser, U. B. Pubertal development and regulation. Lancet Diabetes Endocrinol. 4, 254–264 (2016).
Abreu, A. P. et al. MKRN3 inhibits the reproductive axis through actions in kisspeptin-expressing neurons. J. Clin. Invest. 130, 4486–4500 (2020).
Thodberg, H. H., Kreiborg, S., Juul, A. & Pedersen, K. D. The BoneXpert method for automated determination of skeletal maturity. IEEE Trans. Med. Imaging 28, 52–66 (2009).
Saari, A. et al. New Finnish growth references for children and adolescents aged 0 to 20 years: Length/height-for-age, weight-for-length/height, and body mass index-for-age. Ann. Med. 43, 235–248 (2011).
Kułaga, Z. et al. Polish 2010 growth references for school-aged children and adolescents. Eur. J. Pediatr. 170, 599–609 (2011).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Lim, E. T. et al. Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genet. 10, e1004494 (2014).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 (2009).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362 (2014).
Valadares, L. P. et al. MKRN3 mutations in central precocious puberty: a systematic review and meta-analysis. J. Endocr. Soc. 3, 979–995 (2019).
Kansakoski, J., Raivio, T., Juul, A. & Tommiska, J. A missense mutation in MKRN3 in a Danish girl with central precocious puberty and her brother with early puberty. Pediatr. Res. 78, 709–711 (2015).
Settas, N. et al. Central precocious puberty in a girl and early puberty in her brother caused by a novel mutation in the MKRN3 gene. J. Clin. Endocrinol. Metab. 99, E647–E651 (2014).
Simon, D. et al. Mutations in the maternally imprinted gene MKRN3 are common in familial central precocious puberty. Eur. J. Endocrinol. 174, 1–8 (2016).
Neocleous, V. et al. In silico analysis of a novel MKRN3 missense mutation in familial central precocious puberty. Clin. Endocrinol. 84, 80–84 (2016).
Macedo, D. B. et al. Central precocious puberty that appears to be sporadic caused by paternally inherited mutations in the imprinted gene makorin ring finger 3. J. Clin. Endocrinol. Metab. 99, E1097–E1103 (2014).
de Vries, L., Gat-Yablonski, G., Dror, N., Singer, A. & Phillip, M. A novel MKRN3 missense mutation causing familial precocious puberty. Hum. Reprod. 29, 2838–2843 (2014).
Grandone, A. et al. Molecular screening of MKRN3, DLK1, and KCNK9 genes in girls with idiopathic central precocious puberty. Horm. Res. Paediatr. 88, 194–200 (2017).
Lee, H. S. et al. Low frequency of MKRN3 mutations in central precocious puberty among Korean girls. Horm. Metab. Res. 48, 118–122 (2016).
Bessa, D. S. et al. High frequency of MKRN3 mutations in male central precocious puberty previously classified as idiopathic. Neuroendocrinology 105, 17–25 (2017).
Pasquino, A. M. et al. Long-term observation of 87 girls with idiopathic central precocious puberty treated with gonadotropin-releasing hormone analogs: impact on adult height, body mass index, bone mineral content, and reproductive function. J. Clin. Endocrinol. Metab. 93, 190–195 (2008).
Roberts, S. A. et al. The peripubertal decline in Makorin ring finger protein 3 expression is independent of leptin action. J. Endocr. Soc. 4, bvaa059 (2020).
Chen, T. et al. Low frequency of MKRN3 and DLK1 variants in Chinese children with central precocious puberty. Int. J. Endocrinol. 2019, 9879367 (2019).
Fanis, P. et al. Central precocious puberty caused by novel mutations in the promoter and 5’-UTR region of the imprinted MKRN3 gene. Front. Endocrinol. 10, 677 (2019).
Lu, W. et al. A novel mutation in 5’-UTR of Makorin ring finger 3 gene associated with the familial precocious puberty. Acta Biochim. Biophys. Sin. 50, 1291–1293 (2018).
Acknowledgements
This work was supported by the Foundation for Pediatric Research, Sigrid Juselius Foundation, the Academy of Finland, and Helsinki University Hospital Research Funds.
Author information
Authors and Affiliations
Contributions
T.V., A.-P.I., J.K., K.W., and K.V. were involved in acquisition of data, analyzed the data, and participated in manuscript drafting. M.H. and P.J.M. did the critical analysis of the manuscript. M.N. and T.R. made the concept and design of the study and drafted and critically analyzed the manuscript. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Varimo, T., Iivonen, AP., Känsäkoski, J. et al. Familial central precocious puberty: two novel MKRN3 mutations. Pediatr Res 90, 431–435 (2021). https://doi.org/10.1038/s41390-020-01270-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-020-01270-z
This article is cited by
-
Genetische und epigenetische Einflüsse auf den Pubertätsverlauf in Bezug auf Pubertas praecox vera und Pubertas tarda
Gynäkologische Endokrinologie (2023)
-
A new DLK1 defect in a family with idiopathic central precocious puberty: elucidation of the male phenotype
Journal of Endocrinological Investigation (2022)
