
Abstract
This was a double-blind, randomized, phase 2 study of adults (18–64 years) with DSM−5 diagnosis of major depressive disorder (MDD), with moderate-to-severe episode severity (Montgomery–Åsberg Depression Rating Scale [MADRS] ≥25) despite an adequate course with ongoing antidepressant for ≥6 weeks to ≤12 months. Following a double-blind placebo lead-in period (up to 3 weeks), participants were randomized to receive once daily aticaprant 10 mg or continue placebo, added to their ongoing treatment, for 6 weeks. Of 184 participants enrolled, 169 were included in safety analyses (aticaprant n = 85, placebo n = 84) and 166 in full intent-to-treat (fITT) efficacy analyses; 121 placebo lead-in non-responders (<30% reduction in MADRS total score) in fITT were included in enriched ITT (eITT) analyses. Improvement (least squares mean difference [upper limit 1-sided 80% CI] versus placebo) in MADRS total score at week 6 for aticaprant was significant versus placebo (eITT: −2.1 [−1.09], 1-sided p = 0.044; effect size (ES) 0.23; fITT −3.1 [2.21], 1-sided p = 0.002; ES 0.36). The between-group difference was larger among participants with Snaith–Hamilton Pleasure Scale (SHAPS) score greater/equal to versus less than baseline median SHAPS. The most common treatment-emergent adverse events reported for aticaprant (versus placebo) were headache (11.8% versus 7.1%), diarrhea (8.2% versus 2.4%), nasopharyngitis (5.9% versus 2.4%), and pruritus (5.9% versus 0%). One participant (1.2%) in each arm discontinued treatment due to an adverse event. In this study of participants with MDD and inadequate response to SSRI/SNRI, adjunctive treatment with aticaprant significantly reduced depressive symptoms versus placebo, without resulting in significant safety signals, supporting further investigation in larger trials.
Similar content being viewed by others


Introduction
Major depressive disorder (MDD) is a leading cause of disability, affecting approximately 280 million individuals worldwide [1]. MDD is associated with increased mortality and risk for suicide [1, 2]. As previously described [3], anhedonia, the lack of reactivity to pleasurable stimuli, is a cardinal feature of MDD and has received renewed interest as a potential endophenotype of this debilitating disease [4].
Inadequate response to first-line standard-of-care antidepressant treatment for MDD remains a significant problem [5,6,7], leaving many patients with substantial, persistent impairment [8]. While switching antidepressants and using adjunctive treatments may improve response, almost 40% of patients remain symptomatic and fail to achieve remission despite multiple treatment trials [9, 10]. During treatment the persistence of core depressive symptoms, including anhedonia, leads to longer time to remission, reduces the chance of achieving full recovery, and increases the vulnerability of MDD patients to future depressive episodes [11,12,13,14]. Furthermore, reward processing deficits in patients with MDD contribute to functional impairment, which putatively contribute to persistent and prolonged anhedonia [15]. Additional treatments, targeting this core depressive symptom, are needed for patients with partial or no response to current antidepressants [14].
MDD has been associated with dysregulation of the endogenous mu- and kappa-opioid systems [16, 17]. Kappa opioid receptors (KOR) and the endogenous peptide ligand dynorphin are expressed in brain regions/circuits involved with stress and reward and may play a key role in the pathophysiology of mood, stress, reward mechanisms, and addictive disorders [16, 18, 19]. Emerging evidence implicates KOR system overactivation in mediating depressive symptoms [20], especially anhedonia [21]. KOR antagonism has been tested in preclinical models of depression and anhedonia, and found to have meaningful effects that may translate to therapeutic benefit in humans with mood disorders, especially by modulating the negative affective state associated with responses to stress [16, 22].
We conducted a phase 2 study of aticaprant, a high-affinity, selective kappa receptor antagonist (KRA), with the aim of determining its efficacy and safety as an adjunctive therapy in patients with MDD. This clinical development study follows on single- and multi-dose phase 1 studies of aticaprant [23] and a positron emission tomography scan occupancy study which identified a clinical dose of 10 mg as achieving saturation of brain KOR at maximal concentrations, while providing a suitable safety margin [19]. Furthermore, in the FAST-fail trial in Mood and Anxiety Spectrum disorders (FAST-MAS), a proof-of-mechanism study of participants with anhedonia and a history of MDD or anxiety disorder, treatment with aticaprant 10 mg daily for 8 weeks increased activity in the ventral striatum during a reward anticipation task, improved reward learning, and reduced anhedonic symptoms assessed by the Snaith–Hamilton Pleasure Scale (SHAPS) [24, 25]. In the current study, we tested the hypothesis that this aticaprant dose, administered adjunctively to a monoamine reuptake inhibiting antidepressant (to which participants with MDD had partially responded despite remaining moderate-to-severely depressed), would produce antidepressant effects assessed by change from baseline in the Montgomery–Åsberg Depression Rating Scale [26] (MADRS) score. We further tested a secondary hypothesis that aticaprant’s antidepressant effect would be greater in the subgroup with more severe anhedonia symptoms at baseline, assessed using the SHAPS as a surrogate marker for the impaired reward processing thought to reflect elevated dynorphin-kappa receptor signaling. Thus, the participants were stratified at randomization by baseline SHAPS score.
Patients and methods
Ethical practices
An independent review board (United States [US]) or ethics committee (ex-US sites) at each study site approved the study protocol and its amendments. The study was conducted in accordance with ethical principles that originated in the Declaration of Helsinki, current guidelines on Good Clinical Practices, and applicable regulatory and country-specific requirements. All individuals voluntarily provided written informed consent before participating in the study.
Study design
This phase 2, multicenter (28 sites in the US, 11 in Russia, 5 in the United Kingdom, 6 in Ukraine, 2 in Germany, 1 in Moldova), randomized, double-blind, placebo-controlled study (clinicaltrials.gov: NCT03559192) was conducted between July 2018 and May 2020. The study consisted of a 5-week screening phase and an 11-week double-blind treatment phase, the latter consisting of 3 periods: (1) a double-blind placebo lead-in period of variable duration (up to 3 weeks); (2) a 6-week double-blind treatment period; and (3) a withdrawal period during which (only) participants who completed double-blind treatment received placebo for the remaining time of the treatment phase. Investigators and participants were informed that the variable duration of treatment with placebo during the lead-in period could last from 0 to 3 weeks, and that participants could be randomly assigned to treatment with either aticaprant or continued placebo during the 6-week double-blind period.
Study population
Study participants were between 18 and 64 years of age with a diagnosis of MDD per DSM−5 criteria [27], without psychosis; the current episode was to be 18 months or shorter in duration. Eligible participants had been treated with a selective serotonin reuptake inhibitor (SSRI) or serotonin-norepinephrine reuptake inhibitor (SNRI), at an adequate dosage for at least 6 continuous weeks, but no more than 12 months for the current depressive episode of moderate-to-severe severity, and had inadequate response documented at screening (i.e., based on MADRS [26] total score ≥25).
Individuals who had failed (≤25% improvement based on the Massachusetts General Hospital Antidepressant Treatment Response Questionnaire [MGH-ATRQ] [28]) three or more antidepressants, including the current SSRI/SNRI, during the current episode of depression, despite adequate dose and duration (≥6 weeks), were not eligible for study participation. The study excluded individuals with potentially confounding psychiatric and general medical comorbidities. All inclusion and exclusion criteria are presented in the Supplement (Table S1).
Randomization and double-blind study drug
Eligible participants were randomly assigned (1:1), based on a computer-generated randomization schedule, to receive double-blind treatment with either 10 mg aticaprant (JNJ−67953964) or to continue matching placebo, each once daily (in fasting condition, before breakfast) for 6 weeks during the double-blind treatment period. Randomization was balanced by using randomly permuted blocks of 4, stratified by placebo lead-in response status and SHAPS score (≥20 versus <20). Treatment codes were assigned by a centralized interactive web response system. Participants continued taking the same SSRI/SNRI antidepressant/dose that they had received prior to study entry, with no changes permitted during the study. The use of quetiapine (≤100 mg) was allowed, primarily as a sedative, when it had been used in a stable dose for ≥8 weeks prior to screening and was continued unchanged during the study. Participants, investigators/site personnel, those assessing outcomes, and those analyzing the data were blinded to treatment group assignment.
Adherence was tracked by the site, with study drug dispensing and return recorded in the electronic case report form. Adherence to study drug was also monitored using a smart phone app that recorded study drug ingestion.
Efficacy assessments
Severity of depression was assessed by site-based, trained, certified, and blinded raters using the MADRS [26]. Severity of depressive illness was also assessed by investigators using the Clinical Global Impression–Severity (CGI-S; rated on a 7-point scale from 1 [normal – not at all ill] to 7 [among the most extremely ill patients]) [29].
Participants rated their hedonic capacity using the SHAPS, a reliable and validated 14-item instrument developed for use in MDD (score range 14–56; rating according Franken et al. [30]), with higher score indicating greater severity of anhedonia) [30, 31].
Investigators assessed anxiety using the Structured Interview Guide for the Hamilton Anxiety scale (SIGH-A; comprised of 14 items, each scored from 0 [not present] to 4 [maximum degree]) [32, 33] and the 6-item Hamilton Anxiety Subscale (HAM-A6; comprised of 5 psychic anxiety items [anxious mood, psychic tension, fears, intellectual disturbances, and anxious behavior] and 1 somatic item [muscular tension], each scored from 0 to 4) [34].
To assess the effect of study drug on aspects of cognitive and executive function, participants were asked to complete the Massachusetts General Hospital Cognitive and Physical Function Questionnaire (CPFQ) [35]. The CPFQ includes 7 questions about attention, energy, memory, mental acuity, and motivation, scored on a 6-point Likert scale, with higher values indicating worse function.
Samples were collected for exploratory plasma biomarkers and salivary cortisol; results will be reported elsewhere.
Safety assessments
Adverse events and other standard safety assessments (i.e., hematology, serum chemistry, urinalysis, physical and neurological examination, vital signs, electrocardiogram [ECG]) were monitored throughout the study. Investigators classified adverse events as mild (i.e., easily tolerated, caused minimal discomfort and did not interfere with everyday activities), moderate (sufficient discomfort to cause interference with normal activity), or severe (i.e., caused extreme distress, significant impairment of functioning or incapacitation, and prevented normal everyday activities).
At each study visit, investigators administered the Columbia Suicide Severity Rating Scale (C-SSRS) questionnaire [36] to solicit the occurrence, severity, and frequency of suicide-related ideation and behaviors.
Statistical methods
All randomized participants who received ≥1 dose of study drug during the double-blind treatment period were included in the safety analysis dataset, of whom those having ≥1 post-baseline MADRS assessment during the double-blind treatment period were included in a ‘full’ intent-to-treat (fITT) analysis dataset. Efficacy data were also analyzed for an “enriched” intent-to-treat (eITT) analysis dataset, which included all participants in the fITT analysis dataset who were non-responders at the end of the placebo lead-in period (i.e., <30% improvement in MADRS total score from screening or entry baseline). Investigators were blind to the definition of the improvement threshold. The 30% improvement threshold for detection of possible placebo response was selected based partly on a previous trial of ALKS−5461 (a combination of buprenorphine, a partial mu-opioid receptor agonist and KOR antagonist, and samidorphan, a potent mu-opioid receptor antagonist) [37], which used 50% improvement after 4 weeks of placebo (the first stage of their Sequential Parallel Comparison Design), and partly on our own adjustment for the shorter duration of the placebo lead-in, which was informed by a comparison of the prevalence of 30% and 50% improvement in participants assigned to placebo in previous Johnson & Johnson-sponsored placebo-controlled studies of other candidate antidepressants in MDD. The goal was to balance the response definition, while adjusting for the duration of the lead-in phase, and allowing for a typical attrition rate. The eITT analysis dataset was prespecified as the primary efficacy analysis dataset.
Statistical analyses were conducted using SAS, version 9.4. Analyses of efficacy endpoints were performed at a significance level of 0.20 (primary endpoint and other endpoints related to MADRS, 1-sided; secondary endpoints and other endpoints not pertaining to MADRS, 2-sided). Adjustment for multiple comparisons was not performed.
Efficacy endpoints and statistical analyses
The primary efficacy endpoint – change from treatment baseline to treatment week 6 in MADRS total score – was analyzed using a mixed-effects model using repeated measures (MMRM). The model included baseline MADRS score as a covariate; treatment (aticaprant, placebo), country, time, and time-by-treatment interaction as fixed effects; and a random patient effect.
In a pre-specified subgroup analysis, the impact of baseline anhedonia level (above versus below the baseline median SHAPS score) on the primary endpoint was summarized descriptively.
The overall differences between treatment groups based on the proportion of responders (defined by ≥30% and by ≥50% improvement from treatment baseline MADRS total score) and the proportion of participants in remission (MADRS ≤ 10) at the end of the 6-week double-blind treatment period were analyzed using Chi-square tests.
MADRS items most closely reflecting anhedonia symptoms (i.e., apparent sadness, reported sadness, concentration difficulties, inability to feel, and lassitude, referred to as the MADRS 5-item anhedonia factor subscale [38,39,40]; total score range 0–30) were examined post hoc according to the MMRM model described above for the primary efficacy endpoint analysis, but using the baseline MADRS 5-item anhedonia factor subscale score as covariate. Change in the MADRS 5-item anhedonia factor subscale score was also analyzed by baseline anhedonia level.
The same MMRM model, with respective baseline score as covariate, was also used to compare the treatment groups based on SHAPS score, SIGH-A total score, and HAM-A6 subscale score.
Analysis of safety endpoints
Treatment-emergent adverse events and other measures of safety were summarized descriptively for each treatment group.
Sample size determination
The sample size planned for this study was calculated assuming a treatment effect size of 0.45 at treatment week 6 for mean change from baseline in MADRS total score between aticaprant and placebo. The assumed effect size and an estimated standard deviation (SD) of 11 were derived from clinical trials of ALKS−5461 as adjunctive treatment in patients with MDD who had inadequate response to one or two courses of antidepressants [37, 41]. Based on an overall 1-sided significance level of 0.2 and SD of 11, randomization of about 90 individuals – 96 when adjusted for an anticipated 5% drop-out rate during the treatment period – was required to achieve 90% power. After adjusting for an estimated placebo response rate of 25% and drop-out rate of 10% during the placebo lead-in period, 142 participants were to enter the placebo lead-in period.
In accordance with the protocol, a blinded sample size re-estimation was conducted due to a higher-than-anticipated lead-in placebo response of 26.6% (Table S2), resulting in the inclusion of 181 participants.
The choice of alpha and beta (1-power) for this phase 2 study was intended to increase sensitivity for detecting a therapeutic signal while also maintaining a modest sample size. Thus, for the purpose of sample size estimation the power was set to a high value (power = 90%; beta = 0.10) but the type 1 error rate was specified at 1-sided alpha=0.20, as proposed by Lindborg and co-authors [42].
Consistent with the study design, the results for the analyses based on MADRS are characterized by 1-sided upper 80% CI, and those based on SHAPS by 2-sided 80% CI.
Study results
Of 324 individuals screened, 181 were enrolled and entered the double-blind placebo lead-in period, of whom 169 were randomized and received ≥1 dose of study drug, and thus were included in the safety analysis dataset (Fig. S1). Three of these participants had no MADRS assessment during the treatment period, consequently 166 participants were included in the fITT analysis dataset, 124 (74.7%) of whom were placebo lead-in non-responders, while 42 (25.3%) were deemed placebo lead-in responders; 121 were included in the eITT analysis dataset. Of the 169 randomized participants, 161 (95.3%) completed double-blind treatment and entered the withdrawal period; 3 participants randomized to placebo (1 each due to adverse event, participant decision, and relocation) and 5 participants randomized to aticaprant (1 each due to adverse events [described in Safety section], lack of efficacy, noncompliance, protocol deviation, and self-imposed isolation during coronavirus pandemic) did not complete 6 weeks of double-blind treatment.
The mean (SD) exposure to placebo during the lead-in was 21.0 (1.31) days. Duration of the withdrawal period was 13.9 (1.88) days.
The treatment groups were similar with respect to demographic and baseline clinical characteristics (Table S2). As required for inclusion, all participants had been treated with an SSRI (131/169, 77.5%) or SNRI for the current episode prior to enrollment and continued them throughout the treatment period.
Adherence to study drug exceeded 90% for the trial, based on tracking in clinic or video recording via smart phone, and did not differ between the treatment groups.
Efficacy
Change in depression severity
In the eITT, prespecified primary efficacy analysis dataset, mean MADRS total score decreased from baseline to week 6, with greater improvement among those treated with aticaprant as compared to placebo, each adjunctive to ongoing antidepressant treatment (Table 1; least squares (LS) mean difference [upper limit 1-sided 80% CI]: −2.1 [−1.09], 1-sided p = 0.044; effect size (ES) 0.23). Improvement from baseline in MADRS score favored aticaprant over placebo at all time points during the 6-week double-blind treatment period, beginning at week 1 (eITT −1.2 [−0.19]; fITT −1.6 [−0.70]) and increasing with continuous dosing (Fig. 1a). The results of sensitivity analyses of the primary endpoint, conducted to evaluate the potential impact of COVID−19, are reported in the Supplement.

eITT enriched intent-to treat, fITT full intent-to-treat, LS least squares, MADRS Montgomery–Åsberg Depression Rating Scale, SE standard error, SNRI serotonin-norepinephrine reuptake inhibitor, SSRI selective serotonin reuptake inhibitor. Note: MADRS total score ranges from 0 to 60; a higher score indicates a more severe condition. Negative change in score indicates improvement. Negative difference favors aticaprant.
In the fITT analysis dataset, mean MADRS total score decreased from baseline to week 6, with greater improvement observed among those in the aticaprant group as compared with the placebo group (Table 1; LS mean difference [upper limit 1-sided 80% CI]: −3.1 [−2.21], 1-sided p = 0.002; ES 0.36) and over the 6-week treatment period (Fig. 1b).
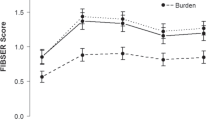
In a subgroup analysis of the MADRS data, the between-group difference was larger among participants with high versus low anhedonia level (Fig. 2a [eITT] and Fig. 2b [fITT]), with the magnitude at week 6 (LS mean difference [upper limit 1-sided 80% CI]) smaller in the eITT versus fITT analysis dataset (high anhedonia level: eITT, −3.2 [−1.38], 1-sided p = 0.068, ES 0.36; fITT, −5.1 [−3.65], 1-sided p = 0.0015, ES 0.56; low anhedonia level: eITT, −0.6 [0.72], 1-sided p = 0.349, ES 0.07; fITT, −1.9 [−0.73], 1-sided p = 0.083, ES 0.24).

eITT enriched intent-to treat, fITT full intent-to-treat, LS least squares, MADRS Montgomery–Åsberg Depression Rating Scale, SE standard error, SHAPS Snaith–Hamilton Pleasure Scale, SNRI serotonin-norepinephrine reuptake inhibitor, SSRI selective serotonin reuptake inhibitor. Note: MADRS total score ranges from 0 to 60; a higher score indicates a more severe condition. Negative change in score indicates improvement. Negative difference favors aticaprant.
The proportion of participants who achieved remission and the proportion who were responders increased over time during the double-blind treatment period, to a greater extent in the aticaprant group than the placebo group (Figs. S2, S3). At week 6, the difference between groups was statistically significant, favoring aticaprant, for response rate in the fITT analysis dataset, but not for remission rate in either efficacy analysis dataset (Figs. S2, S3).
Consistent with the MADRS assessments, mean CGI-S score numerically improved from baseline to week 6 (mean [SD]) in both the eITT analysis dataset (aticaprant: baseline 4.3 [0.48], change from baseline −0.9 [1.04]; placebo: baseline 4.3 [0.62], change from baseline −0.8 [0.86]) and in the fITT analysis dataset (aticaprant: baseline 3.9 [0.84], change from baseline −0.9 [1.01]; placebo: baseline 4.0 [0.80], change from baseline −0.7 [0.88]). The proportion of aticaprant-treated participants in the eITT analysis dataset with moderate or severe depressive illness (i.e., CGI-S score of ≥4) decreased progressively over the 6-week treatment period, to a numerically greater extent than among participants in the placebo group (Fig. 3). The same trend was observed in the fITT analysis dataset (Fig. 4).

CGI-S Clinical Global Impression—Severity, eITT enriched intent-to treat.

CGI-S Clinical Global Impression—Severity, fITT full intent-to-treat.
Change in anhedonia severity
Mean (SD) change in SHAPS score from treatment baseline to week 6 was −4.6 (6.23) for aticaprant and −4.2 (5.04) for placebo in the eITT analysis dataset (Fig. S4a; LS mean between-group difference [2-sided 80% CI]: −0.7 [−1.81, 0.41], 2-sided p = 0.419; ES 0.07). Treatment effect was similar in the fITT analysis dataset (Fig. S4b; LS mean between-group difference [2-sided 80% CI]: −0.8 [−1.79, 0.10], 2-sided p = 0.250; ES 0.08).
In a post hoc analysis of anhedonia severity based on the SHAPS score, improvement in participant self-reported anhedonia was numerically greater with aticaprant compared to placebo (LS mean change [2-sided 80% CI]) in the subgroup of participants with higher baseline anhedonia severity (defined by SHAPS score greater/equal to baseline median) (eITT: −1.70 [−3.74, 0.34], 2-sided p = 0.284; fITT: −2.09 [−3.95, −0.22], 2-sided p = 0.152); improvement in severity of anhedonia was minimal in the subgroup of participants with lower baseline anhedonia severity and comparable between the treatment groups (eITT: 0.04 [−1.19, 1.27], 2-sided p = 0.966; fITT: −0.12 [−1.13, 0.89], 2-sided p = 0.878) (Table S3).
The prevalence of more severe anhedonia, characterized by SHAPS score greater than/equal to baseline median, was lower at week 6 than at baseline in both groups (eITT: aticaprant, 43.3% [baseline] versus 13.6% [week 6]; placebo, 44.3% versus 22.0%; fITT: aticaprant, 36.1% versus 10.4%; placebo, 41.0% versus 18.5%).
Results of post hoc analysis of anhedonia severity based on clinician-assessed 5-item MADRS anhedonia factor subscale score were consistent with those from participant-reported SHAPS score. Improvement in the 5-item MADRS anhedonia factor was greater with aticaprant compared to placebo (LS mean difference [upper limit 1-sided 80% CI]) among the participants with higher baseline anhedonia severity (eITT: −2.7 [−1.64], 1-sided p = 0.015; fITT: −3.6 [−2.59], 1-sided p = 0.001) and smaller for both treatment groups among the participants with lower baseline anhedonia severity (eITT: −0.4 [0.46], 1-sided p = 0.349; fITT: −1.2 [−0.57], 1-sided p = 0.059) (Table S4).
Change in anxiety severity
Mean SIGH-A total score decreased from baseline to week 6, with greater improvement of anxiety seen in the fITT analysis dataset (LS mean difference [80% CI] eITT dataset: −0.7 [−1.90, 0.41], 2-sided p = 0.410; fITT dataset: −1.4 [−2.31, −0.44], 2-sided p = 0.060) among participants treated with aticaprant compared to placebo, each adjunctive to SSRI/SNRI antidepressant. Consistent with these SIGH-A findings, treatment effect on anxiety at week 6 was also demonstrated based on HAM-A6 (eITT dataset: −0.6 [−1.21, 0.07], 2-sided p = 0.148; fITT dataset: −1.1 [−1.56, −0.62], 2-sided p = 0.003) (Table S5).
Change in cognition
Results of the CPFQ assessments are presented in the Supplement. The results indicate a numerically greater reduction in CPFQ total scores for aticaprant compared to placebo, although baseline to endpoint changes were not significantly different.
Safety
Adverse events
During the placebo lead-in period, the only treatment-emergent adverse event reported in ≥5% of participants was headache (11/169, 6.5%) (Table S6).
During the double-blind treatment period, the most common treatment-emergent adverse events (incidence ≥5.0%) reported for aticaprant adjunctive to SSRI/SNRI were headache, diarrhea, nasopharyngitis, and pruritus (Table 2). Most events were of mild or moderate severity (248 of 251) and transient. No difference between treatment groups was observed in the incidence (2.4% each) of adverse events suggestive of abuse potential (defined in Table S7).
During the withdrawal period, new-onset adverse events were reported for 5 (of 85, 5.9%) and 4 (of 84, 4.8%) participants who had been treated with aticaprant and placebo, respectively, in the double-blind treatment period (Table S8); none of these adverse events were considered by investigators to be related to withdrawal of study drug.
Serious treatment-emergent adverse events were reported for 2 participants: one during the placebo lead-in period (adverse event of suicidal ideation) and one in the placebo group during the double-blind treatment period (adverse event of acute cholecystitis). The latter participant and one other in the aticaprant group (adverse events of diarrhea, nausea, vomiting, and headache) discontinued the study drug prematurely due to adverse events. No deaths were reported in this study.
Suicidal ideation/behavior
A comparable proportion of participants in each treatment group reported suicidal ideation (i.e., C-SSRS score between 1 and 5) during the double-blind treatment period: 4.8% (4/83) and 3.6% (3/83) of participants in the aticaprant and placebo groups, respectively, at double-blind treatment endpoint. No participant in either treatment group experienced suicidal behavior in any study period.
Other safety assessments
There were no clinically meaningful changes in laboratory tests, vital signs, body weight (Table S9), or ECG. Results of laboratory testing, vital sign measurements, and ECG are summarized in the Supplement.
Discussion
In this phase 2 study of aticaprant for patients with MDD who were inadequately responding to ongoing SSRI/SNRI antidepressants, adjunctive treatment with aticaprant led to statistically significantly greater reduction in depressive symptoms severity on the MADRS compared to placebo added to the ongoing antidepressant. The proportion of responders (≥30% and ≥50%) was also significantly higher with aticaprant. Remission rates did not differ to an extent that was statistically significant, although the difference in remission rates between aticaprant and placebo arms (31.2% and 22.2%, respectively) in the fITT analysis dataset was within the range commonly observed for clinical trials of approved antidepressants and adjunctive therapies for MDD. Moreover, a treatment period longer than 6 weeks may have been necessary to sensitively evaluate impact on remission [43]. The reliability of the data is supported by a low discontinuation rate during randomized treatment and high levels of adherence to treatment. Multiple approaches were used to encourage adherence, which may have enhanced participants’ engagement in the trial.
The greater treatment effect on the MADRS in the subgroup of participants with elevated anhedonia at baseline is noteworthy, suggesting that patients with MDD and more severe anhedonia may have greater benefit from adjunctive aticaprant. The greater effect of aticaprant in depressed participants with elevated anhedonia supports the hypothesized dysregulation of reward circuitry in depression and anhedonia [44]. Modulating dynorphin activity by a KRA putatively offers a means for restoring motivation and ability to experience pleasure in depression, as reflected previously by the mechanistic results of the FAST-MAS trial [24, 25] and herein, especially by improvement in MADRS anhedonia factor subscale scores.
The treatment effect in the fITT analysis dataset exceeded that in the eITT analysis dataset. A larger treatment effect and effect size had been predicted for the eITT analysis dataset that included only participants who were non-responders during the placebo lead-in period. However, among non-responders during the placebo lead-in period, the placebo response was 45.8%, showing that the lead-in period did not reduce the placebo response, which was slightly lower (44.4%) in the sample that included both responders and non-responders to the placebo lead-in period. Our findings are consistent with those of the placebo-controlled trials of aripiprazole in MDD, in which the MDD patients with MADRS below the median at the end of the lead-in phase had a greater effect size than the MDD patients with MADRS total score above the median [45]. The use of percent improvement from baseline has been used in previous trials [e.g., 37, 46] and was utilized in our trial at the end of the double-blind placebo lead-in phase. Notably, it did not include a minimum threshold score for severity at the end of the lead-in period (MADRS score ≥25 was required for inclusion). This could have resulted in excluding individuals from the eITT who achieved ≥30% improvement from baseline but still had at least moderate depression severity, and potential for further improvement. For example, a participant with MADRS total score of 35 and improvement of 30% would have a MADRS score of 24, reflecting moderate depression severity. Such an individual, who still had significant symptoms after the placebo lead-in and potential to respond, was excluded from the eITT yet included in the fITT analysis dataset.
In this study, improvement in participant-reported anhedonia, based on the SHAPS score, was observed in both treatment groups, although the between-group difference was not significant. In contrast, a significant treatment effect of aticaprant on the SHAPS was seen in the FAST-MAS trial [24, 25]. The divergent findings may be explained by the longer treatment period in the FAST-MAS trial, differences in severity of illness, or ongoing SSRI/SNRI use. The difference in anhedonia severity, rated by the SHAPS, was greater in the participants with higher baseline anhedonia severity, although the treatment difference was not significant. Moreover, analysis of anhedonia severity based on clinician-assessed 5-item MADRS anhedonia factor subscale score showed greater improvement with aticaprant compared to placebo among participants with higher baseline anhedonia severity in both the eITT and fITT datasets (Table S4).
Finally, the treatment effect on anxiety symptoms was greater in the fITT analysis dataset than in the eITT dataset as observed in the change in the HAM-A6 subscale and the full SIGH-A scale scores (Table S5).
In this study we tested the antidepressant efficacy of aticaprant administered adjunctively to SSRI/SNRI treatment to which participants had proven partially – but inadequately – responsive, evidenced by persistence of moderate-to-severe depressive symptoms. We hypothesized that the combination of the KRA and monoamine reuptake inhibitor mechanisms may exert synergistic effects on monoamine transmission [22]. The increased synthesis and release of dynorphin, induced under conditions of chronic stress or hypothalamic-pituitary-adrenal axis activation and some other physiological stressors, results in kappa receptor activation, which inhibits dopamine release from ventral tegmental area neurons during the processing of reward-related stimuli, putatively contributing to negative affective states and impaired reward learning [16, 17, 21]. Dynorphin-kappa receptor activation also reduces serotonin release from dorsal raphe nucleus projections to the nucleus accumbens, hippocampus, and other limbic regions in preclinical stress models, putatively contributing to anxiety and depression-like behaviors [17]. By blocking kappa receptor signaling, aticaprant may allow dopamine and serotonin release to return to adaptive levels during stress and reward processing, thereby producing antidepressant and anti-anhedonia effects. Crucially, by restoring normal release of monoamines, aticaprant may augment the efficacy of monoamine-reuptake inhibiting antidepressants, which can only increase synaptic levels of monoamine neurotransmitters after their release. Such a complementary effect of aticaprant to the effects of monoamine reuptake inhibiting antidepressants has been demonstrated in preclinical studies, which showed that antidepressant-like effects produced by concurrent administration of sub-active doses of aticaprant (3 mg/kg PO) and imipramine (5 mg/kg IP) were comparable with those produced by 15 mg/kg IP of imipramine [47, 48]. In these studies, synergy also was observed for the combination of aticaprant (3 mg/kg PO) and citalopram (3 mg/kg IP). While these data appear compatible with the clinical results reported herein, our study design cannot establish whether the antidepressant efficacy of aticaprant used adjunctively to monoamine reuptake inhibiting antidepressants in patients who previously had experienced inadequate antidepressant responses to such agents reflects a synergistic mechanism or only additive effects.
Safety results in this study were similar to the safety profile of aticaprant reported over an 8-week exposure period [24]. During the double-blind treatment period, the most common adverse events were headache, diarrhea, nasopharyngitis, and pruritus, each with incidence higher in the aticaprant group compared to the placebo group. Pruritus was previously reported in participants treated with aticaprant [24, 49]. Diarrhea was not reported in the FAST-MAS study with aticaprant monotherapy administered at 10 mg daily for 8 weeks [24] although it was observed in phase 1 studies of healthy volunteers at higher doses (described as ‘loose stools’ of mild severity which did not require treatment; unpublished data [NCT04185051]) and in persons diagnosed with cocaine dependence in early abstinence who received 10 mg daily for 4 days [49]. One (1.2%) participant in each group had adverse events leading to discontinuation of study drug, and one serious adverse event was reported (acute cholecystitis for a participant in the placebo group). Aticaprant may offer a more favorable safety profile compared to other adjunctive treatments to SSRI/SNRI antidepressants, and especially compared to approved adjunctive treatments for MDD, which currently are limited to atypical antipsychotic agents. Some of the latter agents commonly produce side effects such as weight gain, metabolic changes, extrapyramidal symptoms, or akathisia, none of which have been observed with aticaprant.
The generalizability of the study findings may be limited by the exclusion of participants with treatment-resistant depression, significant psychiatric co-morbidities, or substance dependence and by the preponderance of white study participants. This early-phase clinical trial aimed to assess the short-term efficacy and safety of aticaprant. Maintenance of antidepressant effect and long-term safety will be evaluated in phase 3 clinical development studies.
In conclusion, the favorable antidepressant effect and safety profile observed in this phase 2 study of aticaprant for patients with MDD and anhedonia, inadequately treated with SSRI/SNRI antidepressants, support further investigation of aticaprant in larger trials in MDD. Confirmatory trials of aticaprant as adjunct treatment for MDD and anhedonia are ongoing.
Data availability
The data sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
References
World Health Organization. Depressive disorder (depression). 31 March 2023. https://www.who.int/news-room/fact-sheets/detail/depression. Accessed 18 March 2024.
Walker ER, McGee RE, Druss BG. Mortality in mental disorders and global disease burden implications: a systematic review and meta-analysis. JAMA Psychiatry. 2015;72:334–41.
Pizzagalli DA, Iosifescu D, Hallett LA, Ratner KG, Fava M. Reduced hedonic capacity in major depressive disorder: evidence from a probabilistic reward task. J Psychiatr Res. 2008;43:76–87.
Hasler G, Drevets WC, Manji HK, Charney DS. Discovering endophenotypes for major depression. Neuropsychopharmacology. 2004;29:1765–81.
Fava M. Diagnosis and definition of treatment-resistant depression. Biol Psychiatry. 2003;53:649–59.
Rush AJ, Gullion CM, Basco MR, Jarrett RB, Trivedi MH. The inventory of depressive symptomatology (IDS): psychometric properties. Psychol Med. 1996;26:477–86.
Trivedi MH, Fava M, Wisniewski SR, Thase ME, Quitkin F, Warden D, et al. Medication augmentation after the failure of SSRIs for depression. N Engl J Med. 2006;354:1243–52.
Kennedy N, Paykel ES. Residual symptoms at remission from depression: impact on long-term outcome. J Affect Disord. 2004;80:135–44.
Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163:1905–17.
Rush AJ, Trivedi MH, Wisniewski SR, Stewart JW, Nierenberg AA, Thase ME, et al. STAR*D Study Team. Bupropion-SR, sertraline, or venlafaxine-XR after failure of SSRIs for depression. N Engl J Med. 2006;354:1231–42.
McMakin DL, Olino TM, Porta G, Dietz LJ, Emslie G, Clarke G, et al. Anhedonia predicts poorer recovery among youth with selective serotonin reuptake inhibitor treatment-resistant depression. J Am Acad Child Adolesc Psychiatry. 2012;51:404–11.
Spijker J, Bijl RV, de Graaf R, Nolen WA. Determinants of poor 1-year outcome of DSM-III-R major depression in the general population: results of the Netherlands Mental Health Survey and Incidence Study (NEMESIS). Acta Psychiatr Scand. 2001;103:122–30.
Taylor DJ, Walters HM, Vittengl JR, Krebaum S, Jarrett RB. Which depressive symptoms remain after response to cognitive therapy of depression and predict relapse and recurrence? J Affect Disord. 2010;123:181–7.
Uher R, Perlis RH, Henigsberg N, Zobel A, Rietschel M, Mors O, et al. Depression symptom dimensions as predictors of antidepressant treatment outcome: replicable evidence for interest-activity symptoms. Psychol Med. 2012;42:967–80.
Chow TK, Bowie CR, Morton M, Lalovic A, McInerney SJ, Rizvi SJ. Contributors of functional impairment in major depressive disorder: a biopsychosocial approach. Current Behavioral Neuroscience Reports. 2022;9:59–72.
Carlezon WA Jr, Béguin C, Knoll AT, Cohen BM. Kappa-opioid ligands in the study and treatment of mood disorders. Pharmacol Ther. 2009;123:334–43.
Lutz PE, Kieffer BL. Opioid receptors: distinct roles in mood disorders. Trends Neurosci. 2013;36:195–206.
Naganawa M, Zheng MQ, Nabulsi N, Tomasi G, Henry S, Lin SF, et al. Kinetic modeling of (11)C-LY2795050, a novel antagonist radiotracer for PET imaging of the kappa opioid receptor in humans. J Cereb Blood Flow Metab. 2014;34:1818–25.
Naganawa M, Dickinson GL, Zheng MQ, Henry S, Vandenhende F, Witcher J, et al. Receptor occupancy of the κ-opioid antagonist LY2456302 measured with positron emission tomography and the novel radiotracer 11C-LY2795050. J Pharmacol Exp Ther. 2016;356:260–6.
Gray AM, Rawls SM, Shippenberg TS, McGinty JF. The kappa-opioid agonist, U−69593, decreases acute amphetamine-evoked behaviors and calcium-dependent dialysate levels of dopamine and glutamate in the ventral striatum. J Neurochem. 1999;73:1066–74.
Williams AV, Laman-Maharg A, Armstrong CV, Ramos-Maciel S, Minie VA, Trainor BC. Acute inhibition of kappa opioid receptors before stress blocks depression-like behaviors in California mice. Prog Neuropsychopharmacol Biol Psychiatry. 2018;86:166–74.
Rorick-Kehn LM, Witkin JM, Statnick MA, Eberle EL, McKinzie JH, Kahl SD, et al. LY2456302 is a novel, potent, orally-bioavailable small molecule kappa-selective antagonist with activity in animal models predictive of efficacy in mood and addictive disorders. Neuropharmacology. 2014;77:131–44.
Lowe SL, Wong CJ, Witcher J, Gonzales CR, Dickinson GL, Bell RL, et al. Safety, tolerability, and pharmacokinetic evaluation of single- and multiple-ascending doses of a novel kappa opioid receptor antagonist LY2456302 and drug interaction with ethanol in healthy subjects. J Clin Pharmacol. 2014;54:968–78.
Krystal AD, Pizzagalli DA, Smoski M, Mathew SJ, Nurnberger J Jr, Lisanby SH, et al. A randomized proof-of-mechanism trial applying the ‘fast-fail’ approach to evaluating κ-opioid antagonism as a treatment for anhedonia. Nat Med. 2020;26:760–8.
Pizzagalli DA, Smoski M, Ang YS, Whitton AE, Sanacora G, Mathew SJ, et al. Selective kappa-opioid antagonism ameliorates anhedonic behavior: evidence from the Fast-fail Trial in Mood and Anxiety Spectrum Disorders (FAST-MAS). Neuropsychopharmacology. 2020;45:1656–63.
Montgomery SA, Åsberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382–9.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders (DSM−5), 5th ed. Washington, DC: American Psychiatric Association; 2013.
Chandler GM, Iosifescu DV, Pollack MH, Targum SD, Fava M. RESEARCH: validation of the Massachusetts General Hospital Antidepressant Treatment History Questionnaire (ATRQ). CNS Neurosci Ther. 2010;16:322–5.
Guy W. ECDEU Assessment Manual for Psychopharmacology. Rockville, MD: US Department of Health, Education, and Welfare; 1976.
Franken IH, Rassin E, Muris P. The assessment of anhedonia in clinical and non-clinical populations: further validation of the Snaith-Hamilton Pleasure Scale (SHAPS). J Affect Disord. 2007;99:83–9.
Snaith RP, Hamilton M, Morley S, Humayan A, Hargreaves D, Trigwell P. A scale for the assessment of hedonic tone the Snaith-Hamilton Pleasure Scale. Br J Psychiatry. 1995;167:99–103.
Hamilton M. The diagnosis and rating of anxiety. In: Lader MM, editor. Studies of anxiety. Kent: Meedley Bros; 1969.
Shear MK, Vander Bilt J, Rucci P, Endicott J, Lydiard B, Otto MW, et al. Reliability and validity of a structured interview guide for the Hamilton Anxiety Rating Scale (SIGH-A). Depress Anxiety. 2001;13:166–78.
Bech P. Dose-response relationship of pregabalin in patients with generalized anxiety disorder. A pooled analysis of four placebo-controlled trials. Pharmacopsychiatry. 2007;40:163–8.
Fava M, Iosifescu DV, Pedrelli P, Baer L. Reliability and validity of the Massachusetts general hospital cognitive and physical functioning questionnaire. Psychother Psychosom. 2009;78:91–7.
Posner K, Brown GK, Stanley B, Brent DA, Yershova KV, Oquendo MA, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168:1266–77.
Fava M, Memisoglu A, Thase ME, Bodkin JA, Trivedi MH, de Somer M, et al. Opioid modulation with buprenorphine/samidorphan as adjunctive treatment for inadequate response to antidepressants: a randomized double-blind placebo-controlled trial. Am J Psychiatry. 2016;173:499–508.
Parker RD, Flint EP, Bosworth HB, Pieper CF, Steffens DC. A three-factor analytic model of the MADRS in geriatric depression. Int J Geriatr Psychiatry. 2003;18:73–7.
Borentain S, Gogate J, Williamson D, Carmody T, Trivedi M, Jamieson C, et al. Montgomery–Åsberg Depression Rating Scale factors in treatment-resistant depression at onset of treatment: derivation, replication, and change over time during treatment with esketamine. Int J Methods Psychiatr Res. 2022;31:e1927.
Gabryelewicz T, Styczynska M, Pfeffer A, Wasiak B, Barczak A, Luczywek E, et al. Prevalence of major and minor depression in elderly persons with mild cognitive impairment–MADRS factor analysis. Int J Geriatr Psychiatry. 2004;19:1168–72.
Ehrich E, Turncliff R, Du Y, Leigh-Pemberton R, Fernandez E, Jones R, et al. Evaluation of opioid modulation in major depressive disorder. Neuropsychopharmacology. 2015;40:1448–55.
Lindborg SR, Persinger CC, Sashegyi A, Mallinckrodt C, Ruberg SJ. Statistical refocusing in the design of phase II trials offers promise of increased R&D productivity. Nat Rev Drug Discov. 2014;13:638–40.
Rutherford BR, Sneed JR, Roose SP. Does study design influence outcome? The effects of placebo control and treatment duration in antidepressant trials. Psychother Psychosom. 2009;78:172–81.
Nestler EJ. Role of the brain’s reward circuitry in depression: transcriptional mechanisms. Int Rev Neurobiol. 2015;124:151–70.
Thase ME, Trivedi MH, Nelson JC, Fava M, Swanink R, Tran QV, et al. Examining the efficacy of adjunctive aripiprazole in major depressive disorder: a pooled analysis of 2 studies. Prim Care Companion J Clin Psychiatry. 2008;10:440–7.
Faries DE, Heiligenstein JH, Tollefson GD, Potter WZ. The double-blind variable placebo lead-in period: results from two antidepressant clinical trials. J Clin Psychopharmacol. 2001;21:561–8.
Melief EJ, Miyatake M, Carroll FI, Béguin C, Carlezon WA Jr, Cohen BM, et al. Duration of action of a broad range of selective κ-opioid receptor antagonists is positively correlated with c-Jun N-terminal kinase−1 activation. Mol Pharmacol. 2011;80:920–9.
National Library of Medicine. ClinicalTrials.gov identifier: NCT01913535. Proof-of-concept trial of CERC−501 therapy in treatment-resistant depression (RAPID KOR). Updated 02 July 2017. Available from: https://clinicaltrials.gov/study/NCT01913535. Accessed 18 March 2024.
Reed B, Butelman ER, Fry RS, Kimani R, Kreek MJ. Repeated administration of opra kappa (LY2456302), a novel, short-acting, selective kop-r antagonist, in persons with and without cocaine dependence. Neuropsychopharmacology. 2018;43:739–50.
Acknowledgements
We acknowledge Sandra Norris, PharmD of the Norris Communications Group LLC for medical writing assistance and Harry Ma, PhD (Janssen Global Services, LLC) for additional editorial support. We also acknowledge Adam Savitz, MD, for his contributions to protocol development, the statistical analysis plan, and interpretation of the study data during his employment by Janssen Research & Development, Dr. Savitz is currently employed by Alto Neuroscience. The authors thank the study participants and the following investigators for their participation in this study: Germany: Heike Benes, Bettina Bergtholdt; Moldova: Vitalie Lisnic; Russia: Alexey Agarkov, Olga Buhanovskaya, Alexander Golubev, Mikhail Ivanov, Dmitry Ivliev, Oxana Olevskaya, Alexander Parashchenko, Alexander Reznik, Vladislava Savitskaya, Alexander Shlafer, Alena Sidenkova; Ukraine: Vladyslav Demchenko, Yevgen Denysov, Iryna Kosenkova, Nataliya Maruta, Pavlo Palamarchuk, Gennadii Zilberblat; United Kingdom: Aliya Asher, Jeremy Dennison, Victoria Lynch, Tom Marland, Paul Westhead; United States – Arizona: Dena Petersen; Arkansas: Paul Wylie; California: Jesse Carr, Daniel Chueh, Stacey Layle, Paul Miller, Katrina Patrick; Florida: Carlos Danger, Jose Gamez, Cynthia Huffman, Morteza Nadjafi, Kelley Yokum; Georgia: Suneel Katragadda; Illinois: Yuhwen Chow, John Zajecka; Louisiana: Kashinath Yadalam; New Jersey: Olga Tchikindas; New York: Mark Dibuono, Inna Yuryev-Golger; North Carolina: James Barker; Ohio: Otto Dueno, Subhdeep Virk; Oklahoma: Courtney Nixon, Louise Thurman; Pennsylvania: Paul Gross; South Carolina: Robert Malcolm; Tennessee: Valerie Arnold; Utah: Barbara Rizzardi. Previous Presentations Oral presentation at the American College of Neuropsychopharmacology (ACNP) 61st annual meeting, December 4−7, 2022, Phoenix, AZ; presented as Pharmaceutical Pipeline Presentation and poster at the American Society of Clinical Psychopharmacology (ASCP) 2023, May 30-June 2, 2023, Miami FL.
Funding
Janssen Research and Development, LLC, Titusville, NJ, USA provided funding for this study.
Author information
Authors and Affiliations
Contributions
Mark E. Schmidt: Protocol development; study oversight; manuscript conceptualization (co-lead); manuscript writing – original draft (co-lead), formal analysis (supporting), review and editing (equal), Iva Kezic: Methodology (lead); protocol development and study oversight; formal analysis (lead); manuscript writing – review and editing (equal), Vanina Popova: Manuscript writing – original draft (co-lead), formal analysis (supporting), review and editing (equal), Rama Melkote: Formal analysis; manuscript writing – review and editing (equal), Peter Van Der Ark: Protocol development and study oversight; manuscript writing – review and editing (equal), Darrel J. Pemberton: Manuscript writing – review and editing (equal), Guy Mareels: Manuscript writing – review and editing (equal), Carla M. Canuso: Manuscript writing – review and editing (equal), Maurizio Fava: Protocol development and review; manuscript writing – review and editing (equal), Wayne C. Drevets: Study conceptualization and inception; protocol development; manuscript writing – review and editing (equal), All authors meet ICMJE criteria and all those who fulfilled those criteria are listed as authors. Authors had full access to all of the data, were involved in writing and/or revising the manuscript, and had final responsibility for the decision to submit for publication.
Corresponding author
Ethics declarations
Competing interests
MES, IK, VP, RM, DJP, GM, CMC, and WCD are employees of Janssen Research & Development, LLC and may hold stock or stock options in Johnson & Johnson. PVDA was an employee of Janssen Research & Development, LLC at the time this study was conducted. Maurizio Fava reports the following disclosures related to the last 3 years: Research Support Abbvie; Acadia Pharmaceuticals; Aditum Bio Management Company, LLC; Allergan, Alkermes, Inc.; Altimate Health Corporation; Alto Neuroscience, Inc.; Ancora Bio, Inc.; Angelini S.p.A; Aptinyx; Arbor Pharmaceuticals LLC; Avanir Pharmaceuticals Inc.; Axsome; Benckiser Pharmaceuticals, Inc.; BioClinica, Inc.; Biogen; BioHaven; BioShin Limited; Cambridge Science Corporation; Centrexion Therapeutics Corporation; Cerecor; Cybin IRL Limited; Damona Pharmaceuticals; EmbarkNeuro; Eliem Therapeutics LTD; Gate Neurosciences, Inc.; GenOmind, LLC; Gentelon, LLC; Gerbera Therapeutics, Inc.; Happify; Johnson & Johnson; Lundbeck Inc.; Marinus Pharmaceuticals; Medpace, Inc.; Methylation Sciences, Inc.; Millennium Pharmaceutics, Inc.; Minerva Neurosciences; Neuralstem; Neurocrine Biosciences, Inc.; NeuroRX Inc.; Novaremed; Novartis; Otsuka; Pfizer; Premiere Research International; Praxis Precision Medicines; Protagenic Therapeutics, Inc.; Relmada Therapeutics Inc.; Reckitt; Shenox Pharmaceuticals; Stanley Medical Research Institute (SMRI); Taisho; Takeda; University of Michigan; Vistagen; WinSanTor, Inc.; Xenon Pharmaceuticals, Inc.; National Institute of Drug Abuse (NIDA); National Institutes of Health (NIH); National Institute of Mental Health (NIMH); and PCORI. Dr. Fava has not done any personal consulting. Any consulting he has done has been on behalf of Massachusetts General Hospital, except for Revival Therapeutics and Sensorium Therapeutics. Speaking/Publishing Lecture given at Global Medical Education, Inc. Mood Disorders Summit, November 2020. Stock/Other Financial Options Equity Holdings: Compellis; Neuromity; Psy Therapeutics; Revival Therapeutics; Sensorium Therapeutics. Royalty/patent, other income: Patents for Sequential Parallel Comparison Design (SPCD), licensed by MGH to Pharmaceutical Product Development, LLC (PPD) (US_7840419, US_7647235, US_7983936, US_8145504, US_8145505); and patent application for a combination of Ketamine plus Scopolamine in Major Depressive Disorder (MDD), licensed by MGH to Biohaven. Patents for pharmacogenomics of Depression Treatment with Folate (US_9546401, US_9540691). Copyright for the MGH Cognitive & Physical Functioning Questionnaire (CPFQ), Sexual Functioning Inventory (SFI), Antidepressant Treatment Response Questionnaire (ATRQ), Discontinuation-Emergent Signs & Symptoms (DESS), Symptoms of Depression Questionnaire (SDQ), and SAFER; Belvior; Lippincott, Williams & Wilkins; Wolkers Kluwer; World Scientific Publishing Co. Pte. Ltd.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schmidt, M.E., Kezic, I., Popova, V. et al. Efficacy and safety of aticaprant, a kappa receptor antagonist, adjunctive to oral SSRI/SNRI antidepressant in major depressive disorder: results of a phase 2 randomized, double-blind, placebo-controlled study. Neuropsychopharmacol. (2024). https://doi.org/10.1038/s41386-024-01862-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41386-024-01862-x
