
Abstract
Merkel cell carcinoma (MCC) is an aggressive cutaneous neuroendocrine (NE) carcinoma arising from integration of Merkel cell polyomavirus (MCPyV) DNA into a host cell or from ultraviolet light-induced genetic damage (proportions vary geographically). Tumors in the latter group include those with “pure” NE phenotype and those “combined” with other elements, most often squamous cell carcinoma (SCC). We performed comprehensive genomic profiling (CGP) of MCPyV+ and MCPyV− (pure and combined) tumors, to better understand their mutational profiles and shed light on their pathogenesis. Supplemental immunohistochemistry for Rb expression was also undertaken. After eliminating low quality samples, 37 tumors were successfully analyzed (14 MCPyV+, 8 pure MCPyV− and 15 combined MCPyV−). The SCC and NE components were sequenced separately in 5 combined tumors. Tumor mutational burden was lower in MCPyV+ tumors (mean 1.66 vs. 29.9/Mb, P < 0.0001). MCPyV− tumors featured frequent mutations in TP53 (95.6%), RB1 (87%), and NOTCH family genes (95.6%). No recurrently mutated genes were identified in MCPyV+ tumors. Mutational overlap in the NE and SCC components of combined tumors was substantial (‘similarity index’ >24% in 4/5 cases). Loss of Rb expression correlated with RB1 mutational (P < 0.0001) and MCPyV− status (P < 0.0001) in MCCs and it was observed more frequently in the SCC component of combined MCC than in a control group of conventional cutaneous SCC (P = 0.0002). Our results (i) support existing evidence that MCPyV+ and MCPyV− MCCs are pathogenetically distinct entities (ii) concur with earlier studies linking the NE and SCC components of combined MCCs via shared genetic profiles and (iii) lend credence to the proposal that an Rb-deficient subset of SCC’s is the source of phenotypically divergent combined MCCs.
Similar content being viewed by others


Introduction
Advances in the molecular characterization of Merkel cell carcinoma (MCC) have led to important insights on pathogenesis and possible therapeutic targets. First described in 1972 by Toker1, MCC is an uncommon cutaneous neuroendocrine carcinoma, frequently occurring on sun-exposed regions of elderly, Caucasian patients, and associated with a poor prognosis2,3,4,5. In 2008, the Merkel cell polyomavirus (MCPyV) was implicated in the pathogenesis of MCC6, and is estimated to drive development of most tumors (proportions vary geographically)4. Since then, viral status (MCPyV+ vs. MCPyV−) has served as a method to categorize MCC, leading to important discoveries regarding the distinct molecular pathogenesis of each subset of tumors. MCPyV− tumors, which are associated with a worse prognosis7,8,9,10,11, exhibit a UV-light induced mutational signature12,13,14, high tumor mutational burden (TMB)12,13,15,16,17, frequent mutations in TP53 and RB17,8,12,13,14,15,16,17,18, and frequent copy number variations (CNVs)18,19. In contrast, MCPyV+ tumors show few genetic abnormalities, consistent with a pathogenesis involving interactions between viral proteins and cell cycle regulatory pathways. The latter include retinoblastoma (Rb) and p53 proteins7,20,21. The cell of origin, which may differ between the two groups, has yet to be elucidated and is the subject of active research22,23,24,25.
In addition to polyomavirus status, MCC may also be classified morphologically into “pure” neuroendocrine and “combined” tumors with added non-neuroendocrine elements. Combined tumors, all of which are MCPyV−26,27, most commonly feature a component of in situ or invasive squamous cell carcinoma (SCC)28. These are estimated to comprise 5–20% of all MCC18,27,29. From a clinical perspective, categorization of MCCs on the basis of viral status has proven to be more important (prognostically) than that based on morphology, but the latter has value scientifically. In particular, it has the potential to shed light on the enigmatic cell (or cells) of origin of these tumors. Recent studies of combined MCCs have demonstrated a molecular relationship between Merkel cell carcinoma and associated SCC in situ, suggesting a pathogenetic relationship between the two, and proposing a keratinocytic origin of these tumors22,25. From a similar perspective, we have (i) characterized the molecular profiles of three subsets of MCC (pure MCPyV+, pure MCPyV− and combined MCPyV−) through comprehensive genomic profiling (CGP), (ii) performed comparative sequencing of segregated squamous and neuroendocrine elements of combined tumors and (iii) examined Rb protein expression in MCCs and related SCCs.
Materials and methods
This study was approved by the Nova Scotia Health Authority Research Ethics Board and performed in accordance with the Declaration of Helsinki. The requirement for obtaining individual patient consent was waived in accordance with institutional policies.
The study population represents a cohort of primary cutaneous Merkel cell carcinomas derived from the Maritime region of Canada (1993-2020). This has formed the basis of several prior publications18,26,28,30,31,32,33,34. The demographic, clinical, and pathological characteristic of the group have been well characterized. A total of 51 cases (21 MCPyV + , 13 pure MCPyV− and 17 combined MCPyV−) were selected. These mainly included excisional specimens and were chosen with a view to including adequate representation of different tumor subsets. The viral status of the cases had previously been determined by nuclear expression of viral large T antigen on immunohistochemistry (CM2B4 antibody; Santa Cruz Biotechnology Inc., Dallas, TX).
Tumor samples from formalin-fixed paraffin-embedded tissue were macrodissected from 2-5 unstained sections (10-20 µm) via scalpel blade, using a paired Hematoxylin & Eosin-stained slide to denote the area for dissection. In 12 combined tumors, an attempt was made to isolate the neuroendocrine and squamous (either in situ or invasive) components separately via selective macrodissection. Estimated tumor purity was a minimum of 30% for all cases, and >80% for the majority of cases. For combined tumors, only regions containing one histological component (NE or SCC) were dissected in order to minimize the risk of cross-contamination.
DNA extraction was performed using a commercial QIAamp DNA FFPE Tissue Kit (Qiagen; Hilden, Germany). Extracted DNA was quantified using a Nanodrop One instrument (ThermoFisher Scientific; Waltham, MA) and, subsequent to appropriate dilution as necessary, re-quantified using a Qubit fluorometer (ThermoFisher Scientific; Waltham, MA). NGS of extracted DNA was analyzed via the Illumina TSO500 hybrid capture DNA panel, performed on an Illumina NextSeq550 instrument (San Diego, CA), in accordance with the manufacturer’s instructions. The panel analyzes 523 cancer-related genes for small mutations, a subset of genes for copy number gains, as well as global parameters of tumor mutation burden (TMB) and microsatellite instability (MSI)35,36. Data were first analyzed using Illumina’s TSO500 LocalApp workflow for alignment and variant calling of mutations, defined for this study to include single nucleotide variants (SNVs), insertions/deletions (“indels”) and copy number variants (CNVs) (version 2.0.1.4). Additional copy number calling was performed using CNVKit37. SNVs and small indels were annotated using snpEff38 and vcfanno39 to add transcript and protein level annotations, population frequencies from gnomAD40, and information from the Catologue of Somatic Mutations in Cancer (COSMIC)41 and ClinVar42. This was used to filter variants based on known pathogenicity information, identify known COSMIC hotspots, and otherwise prioritize based on the impact on the protein. SNVs and small indels were filtered to remove variants with a population frequency >0.5%. We further selected for variants that were predicted to have an impact at the protein-coding level or that were present in ClinVar (pathogenic, likely-pathogenic, or unknown), or that had at least one entry in COSMIC. Analysis of CNVs detected via CNVKit included all copy number gains, as well as copy number losses in genes for which biallelic inactivation is predicted to be clinically relevant by the TARGET database version 3 (The Broad Institute)43. Mutational signatures were estimated using the R package DeconstructSigs44 and profiled using the COSMIC v2 signatures45,46,47. Details of phylogenetic analysis can be found in Supplement S5.
Cases with median exon coverage <100x (unique reads employed for all thresholds) were excluded from analysis. Intronic, synonymous, upstream and downstream gene, and 3ʹ and 5ʹ untranslated region variants were omitted, with the exception of upstream variants in TERT (i.e. TERT promoter mutations). Individual variants included in analysis were restricted to those with read depth ≥50x, with variant read count >10 for read depths 50–200x and variant allele frequency (VAF) > 5% for variant read depths >200x.
Thirty-three cases (9 MCPyV+, 10 pure MCPyV− and 14 combined MCPyV−) were sent to Mayo Clinic Laboratories (Rochester, MN) for Rb immunohistochemical (IHC) staining, which was performed in accordance with their institutional protocols. Appropriate staining of external control tissue was assessed at the referral laboratory and confirmed before interpretation. Tumor staining was also compared to internal control tissue to confirm intact expression in each case. Nuclear expression of Rb was assessed in MCC and SCC tumor cells, and qualitatively assessed as preserved vs. greatly reduced or absent (i.e. loss of Rb expression). For comparison, staining was also performed on a group of 13 cutaneous SCC (in situ and invasive, from various anatomical locations).
Data were summarized using descriptive statistics. Comparisons, where appropriate, were performed via two-tailed unpaired t test for continuous variables, and Fisher’s exact test for categorical variables. A threshold of P < 0.05 was employed for determining statistical significance.
Results
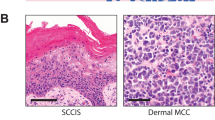
Following exclusion of cases with poor quality NGS metrics, 37 of 51 tumors remained for analysis (14 MCPyV+ , 8 pure MCPyV− and 15 combined MCPyV−). Examples of MCC with pure and combined morphology are displayed in Fig. 1. Clinical characteristics of these cases are displayed in Table 1, segregated by MCPyV status. There was no significant difference in mean age at presentation or in sex distribution between MCPyV+ and MCPyV− tumors. A higher proportion of MCPyV+ cases occurred on an extremity (P = 0.01) but, unlike in some other studies7,10,11, MCPyV− cases were not significantly more likely to occur on the head and neck in our cohort (P = 0.10). All 4 tumors on the trunk were MCPyV− (P = 0.28).

Pure Merkel cell carcinoma exhibits small cell-like neuroendocrine morphology (A. H&E, ×50; C. H&E, ×200). Combined Merkel cell carcinoma features areas of neuroendocrine and squamous cell carcinoma (B. H&E, ×50; D. H&E, ×200).
NGS analysis yielded a mean median exon coverage depth of 285 (standard deviation 98; minimum 102, maximum 796) (See Supplementary Table S1 for additional NGS quality metrics). Global parameters assessed via NGS analysis included TMB (Fig. 2), which was higher in MCPyV− tumors when compared to MCPyV+ cases (mean 29.9/Mb vs. 1.7/Mb; P < 0.0001). Additionally, MCPyV− tumors with combined morphology showed a higher TMB than those with pure morphology (mean 35.2/Mb vs. 20.0/Mb; P = 0.03). None of the tumors analyzed were MSI high.

Tumor mutation burden (TMB) observed in different subgroups of Merkel cell carcinoma, displayed as box-and-whisker plots with mean TMB denoted by an “x”: MCPyV+ (mean: 1.7/Mb, median: 1.6/Mb, range: 0–4.8/Mb, interquartile range (IQR): 0.8–2.6/Mb), all MCPyV− (mean: 29.9/Mb, median: 28.5/Mb, range: 0.8–71.2/Mb, IQR: 15.8–40.3/Mb), pure MCPyV− (mean: 20.0/Mb, median: 15.4/Mb, range: 0.8–40.9/Mb, IQR: 12.8–35.1/Mb), combined MCPyV− (mean: 35.2/Mb, median: 29.9/Mb, range: 10.2–71.2/Mb, IQR 27.5–48.2/Mb).
MCPyV+ tumors did not show a consistent pattern of mutations (Figs. 3 and 4). Genes with the highest frequency of mutations in this group included MET (3/14 cases), as well as ERCC family (4/14 cases) and NOTCH family (4/14 cases) genes. In contrast, MCPyV− tumors of pure and combined morphology demonstrated a similar complement of recurrent mutations. Genes most frequently demonstrating ≥1 mutation in MCPyV− tumors included TP53 (22/23; 95.6%), RB1 (20/23, 87.0%), and NOTCH family genes (NOTCH1-4) (22/23; 95.6%). Recurrent alterations were also observed in a number of other well-characterized tumor suppressor genes (TSGs, Figs. 3 and 4). Twenty of 23 MCPyV− tumors showed some evidence of UV mutational signature (COSMIC signature 7), versus 1/14 MCPyV+ tumors (P < 0.0001). A dominant UV signature, defined as >40% fit to that signature48, was present in 10/23 MCPyV− tumors, and absent in all MCPyV+ MCCs (P = 0.0056). There was no significant difference in the proportion of dominant UV signatures in pure (3/8) vs. combined (7/15) MCPyV− tumors (P = 0.48).

Genes frequently mutated in MCPyV+ MCC (A) and MCPyV− MCC (B). Mutational profiles of different subsets of MCC (C; P+ = Pure MCPyV+ , P− = Pure MCPyV−, C− = Combined MCPyV−). Combined cases with paired neuroendocrine and in situ (IS) or invasive squamous cell carcinoma (SCC) specimens are highlighted in gray, with a superscript ‘a’ denoting the squamous cell carcinoma component. Identical variants are present in both NE and SCC components in instances where shared variant types are reported, except where denoted by an “x”. Cases with dominant UV mutational signature (>40% COSMIC Signature 7) are highlighted in orange. *Indels restricted to in-frame insertions/deletions. **Truncation mutations include nonsense point mutations and frameshift insertions/deletions.

MCPyV+ Merkel cell carcinoma, which exhibits pure neuroendocrine morphology (A. H&E, ×50), was found to have retained expression of Rb in 8/9 cases evaluated (D. Rb IHC, ×50). Conversely, MCPyV− Merkel cell carcinoma combined with squamous cell carcinoma (B. H&E, ×100) demonstrated absence of Rb expression in both neuroendocrine and squamous elements in all 9 cases evaluated (E. Rb IHC, ×100). Conventional cutaneous invasive squamous cell carcinoma (C. H&E, ×100) exhibited retained expression of Rb in 11/13 cases (F. Rb IHC, ×100).
In five combined tumors, the neuroendocrine and squamous components were successfully sequenced separately (Fig. 3C). The clinicopathological features of these cases are documented in Table 2. All cases featured a component of SCCIS, and 3 cases also had elements of distinct invasive SCC and the presence of hybrid nests of neuroendocrine and squamous cells. Guided by feasibility/technical considerations, SCCIS components were selectively dissected for sequencing in four tumors (cases 33–36), and invasive SCC was differentially sequenced in one tumor (case 26). The frequency of overlapping mutations, or “similarity index”25, between the two components varied widely, from 3.1% (5/159 mutations; case 36) to 96.8% (90/93 mutations; case 33). The remaining tumors showed overlap in 24.3% (17/70), 66.7% (18/27), and 93.6% (59/63) of genetic variants, for median and mean similarity indices of 66.7 and 56.9%, respectively (Fig. 3C). Of note, in case 36, SCCIS and MCC were not observed in direct continuity and the possibility of a collision tumor was considered. Exclusion of case 36 from the analysis, given the likelihood of it representing two distinct tumors (see “Discussion”), results in median and mean similarity indices of 80.2 and 70.0%. Complete sequencing data is available in Supplementary S2, and VAF ranges for shared variants in each paired case are available in Supplementary S3. Phylogenetic analyses, performed for each tumor, can be viewed in Supplementary S4.
Results for immunohistochemical studies examining Rb protein expression in MCC are displayed in Table 3. Compared to MCPyV+ tumors, virus-negative MCC showed a significantly higher frequency of Rb loss (23/24 vs. 1/9, P < 0.0001). There was no significant difference in Rb expression between MCPyV− tumors with pure (loss in 9/10) vs. combined morphology (loss in 14/14). When compared to a control cohort of conventional cutaneous squamous cell carcinomas, the squamous elements of combined MCC were remarkable for significant Rb loss (9/9 vs. 2/13, P = 0.0002). In all MCCs, the presence of RB1 mutation correlated strongly with loss of Rb expression (P < 0.0001). Examples of Rb expression patterns in MCPyV+ MCC, combined MCPyV−MCC and conventional cutaneous SCC are presented in Fig. 4.
Discussion
The dichotomous nature of MCC, stratified by viral status, is well established. Studies have shown improved prognosis in MCPyV+ tumors7,8,9,10,11 and distinct patterns of molecular alterations that characterize MCPyV+ and MCPyV− tumors. While MCPyV+ MCC is associated with viral protein-mediated alteration of cell cycle regulation without a consistent mutational pattern7,20,21, MCPyV− tumors tend to exhibit a UV light-associated mutational signature12,13,14, high TMB12,13,15,16,17, TP53 and RB1 mutation7,8,12,13,14,15,16,17,18, and more frequent CNVs18,19. In each of these categories, our study results concur with demonstrated distinctions between MCPyV− and MCPyV+ MCCs.
Combined MCPyV− tumors (MCC intimately associated with SCCIS, invasive SCC or MCC with squamous differentiation) have been shown to share molecular alterations with their “pure” MCPyV− counterparts and are likely on the same spectrum of disease18. Hence study of combined tumors may shed light on the pathogenesis of virus-negative MCCs as a whole. In our investigations, MCPyV− MCC with combined morphology showed similar patterns of recurrent mutations to those in pure MCPyV− MCC, including frequent inactivating mutations in TP53 and RB1. While MCPyV− MCC also had much higher mean TMB than MCPyV+ MCC, as reported previously12,13,15,16,17,49 we now show that, within the MCPyV− family, combined tumors have higher TMB than those with pure NE morphology. Though speculative, this could signify an increased tendency for highly mutated combined tumors to derive from more highly mutated precursor lesions (e.g. in situ or invasive SCC), relative to pure MCPyV− MCC.
Of particular interest is the separate analysis of distinct morphological components in combined tumors. A study from our cohort in 2018 demonstrated overlapping mutational and CNV profiles in two paired samples from combined MCCs18. Two recent studies, examining four and seven paired samples (respectively) also revealed a molecular relationship between in situ squamous neoplasia and subjacent MCC22,25. The current study contributes to the limited data in this realm and includes evaluation of a second combined MCC with invasive, rather than in situ, SCC.
The squamous and NE elements of combined tumors were found to have a high degree of overlapping genetic alterations, with similarity indices greater than 24% in 4 of 5 cases. In the fifth case (36, Fig. 3C), SCCIS and MCC were not contiguous, and there was overlap of only 5 of 159 detected variants (all of uncertain significance) without shared mutations of RB1 or TP53. All 5 of the variants have been reported as single nucleotide polymorphisms (SNPs) with population allele frequencies of 0.01–0.1%40, and all had VAFs close to 0.5 in both the NE and SCCIS components (range 0.36–0.62). Hence, the histological and genomic findings together suggest that this is a case of SCCIS “colliding” with an unrelated MCC, the 5 overlapping variants representing (germline) SNPs. In the remaining paired cases, there was significant overlap of identical mutations (24.3–96.8%) with a minimum 17 overlapping variants. Each paired case had a shared variant in RB1, TP53, or both. These findings suggest a genuine ontogenetic relationship between the elements. The variation in overlap may be the result of differences with respect to the molecular stage of development at which the two components diverged (i.e. early development of a NE component in an SCC precursor lesion with subsequent accrual of abundant unique subclone/subtype-specific variants, vs. late divergence, which would leave less time for this phenomenon). This is supported by TMB data, with high similarity indices found in cases with similar level of TMB in NE and SCC components (cases 26 and 33, presumed late divergence). By contrast, case 34 has low similarity index (24.3%) and widely differing TMBs in the SCC (7.1/Mb) and NE (48.2/Mb) components (presumed early divergence).
Rb protein expression was studied in 14 combined tumors, including nine tumors in which SCC elements remained in the paraffin block. In all nine cases, Rb expression was absent in both the squamous and NE components of the tumor, in accordance with some15,22, but not all previous studies25. Similar loss of expression has been demonstrated in MCPyV− tumors with pure morphology7,8. Loss of Rb expression was uncommon in a comparison group of conventional cutaneous in situ and invasive SCC (2/13, 15%). This is consistent with the low rate of RB1 mutations (typically 4–15%) reported in cutaneous and non-cutaneous SCCs50. Although not considered a significant driver of cutaneous SCC, RB1 mutations have recently been observed in a distinct subset of SCC in situ (27%) and in a smaller group of aggressive invasive SCC (8%)51. Our findings with regard to RB1 mutation and loss of Rb protein expression in combined MCCs, concur with those recently described by Kervarrec et al.22. Those authors invoked RB1 inactivation as an early step in development of a Merkel cell phenotype mediated via induction of SOX222,52. In contrast, Harms et al. showed decreased Rb expression in the MCC components, but not in the squamous elements of combined MCCs despite the presence of RB1 variants in the latter25.
Despite our attempts to identify potential mutational trigger(s) of conversion from an SCC to a NE phenotype, no pattern of aberrations unique to the MCC component (as distinct from the SCC component) of combined tumors was identified. The findings of others have been similar in this regard22,25. Harms et al. did observe FBXW7 and SMARCA4 variants restricted to the MCC component in a subset of their cases25. In contrast, we observed FBXW7 variants in three of our independently sequenced combined MCC cases, in both the SCC and NE components of each case. Moreover, SMARCA4 mutations occurred in two such combined tumors, one featuring the variant in both tumor components, and the other only in the SCC component. Further studies on larger cohorts of combined tumors are required to rigorously address this question, but available data suggests that, in addition to a requisite group of mutations, epigenetic events and gene expression patterns may be involved in the development of a neuroendocrine phenotype25,53,54.
Evidence to date suggests that MCPyV− MCC arises from a keratinocytic precursor lesion, or from a tumor stem cell capable of squamous or neuroendocrine differentiation. The association of neuroendocrine carcinomas with epithelial elements in other organ systems (e.g. squamous cell or adenocarcinoma in Mixed Neuroendocrine-Nonneuroendocrine Neoplasms (MiNEN) of the gastro-entero-pancreatic tract) supports the concept of epithelial to neuroendocrine transition55. It seems likely that RB1 inactivation plays a pathogenetic role in the development of combined MCC. Hence, the subset of SCC in situ and aggressive invasive SCC with RB1 mutation51 may serve as precursors, with a propensity to transform to a neuroendocrine phenotype following acquisition of additional molecular aberrations. Additional studies on larger series of combined MCCs, including evaluation of gene expression patterns and epigenetic alterations, may clarify the source of these neoplasms.
Apart from the aforementioned genomic differences observed in MCPyV+ and MCPyV− MCCs12,13,15,16,17,49, additional comparisons between our genomic findings and those of others (e.g. relating to rates of mutation/hemizygous loss of commonly implicated TSGs, or of mutation/amplification of commonly implicated oncogenes between groups of MCCs) could be made, but are outside the scope of the current manuscript.
Limitations of our study include the small size of a single-center cohort of cases and resource restrictions. Prior studies resulting in depletion of tissue in archived paraffin blocks impaired our ability to sample dual components of many combined tumors. Lack of access to laser capture microdissection deterred more targeted comparative sequencing in combined MCCs with intimately admixed squamous and NE components. Lastly, the lack of parallel germline sequencing was an important limitation preventing subtraction of background variants/SNPs, leading to overinclusion of presumed somatic variants. These falsely implicate genes which are not involved in tumorigenesis and, in combined tumors, inflate similarity indices, as is believed to have happened for case 36 (“true” similarity index would likely be 0%, not 3.1%, if SNPs were identified and eliminated).
The strengths of this investigation include analysis of a well-characterized cohort of MCCs enriched by a subset of combined cases. The stratification of the tumors based on viral status and morphology complements prior studies addressing only one parameter or the other. Our comparative comprehensive profiling of five combined tumors represents a significant contribution to current limited knowledge of the relationship between the dual elements of these lesions and suggests that occasional “collision tumors” may be incorporated within the morphologically defined group of combined MCCs. Given the known demographic and geographic variability in the incidence and subtypes of MCC, our study sheds light on the features of a North American (Atlantic Canadian) subset of cases.
In conclusion, through CGP of a diverse cohort of MCCs we provide support for the biological dichotomy between MCPyV+ and MCPyV− MCC. Of importance, our findings support the concepts of (i) a pathogenetic relationship between MCPyV− MCCs with pure and combined morphology (ii) an ontogenetic relationship between neuroendocrine and squamous components of combined MCCs and (iii) an epithelial origin for combined (and possibly all) MCPyV− MCCs. A common molecular trigger, if one exists, for the development of a neuroendocrine phenotype in combined tumors has yet to be identified.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Toker, C. Trabecular carcinoma of the skin. Arch Dermatol 105, 107–10 (1972).
Harms, PW. Update on Merkel Cell Carcinoma. Clin Lab Med 37, 485–501 (2017).
Del Marmol, V, Lebbé, C. New perspectives in Merkel cell carcinoma. Curr Opin Oncol 31, 72-83 (2019).
Walsh, NM, Cerroni, L. Merkel cell carcinoma: A review. J Cutan Pathol 48, 411–21 (2021).
Becker, JC, Stang, A, DeCaprio, JA, Cerroni, L, Lebbé, C, Veness, M, et al. Merkel cell carcinoma. Nat Rev Dis Prim 3, 17077 (2017).
Feng, H, Shuda, M, Chang, Y, Moore, PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 319, 1096–100 (2008).
Sihto, H, Kukko, H, Koljonen, V, Sankila, R, Böhling, T, Joensuu, H. Merkel cell polyomavirus infection, large T antigen, retinoblastoma protein and outcome in Merkel cell carcinoma. Clin Cancer Res 17, 4806–13 (2011).
Higaki-Mori, H, Kuwamoto, S, Iwasaki, T, Kato, M, Murakami, I, Nagata, K, et al. Association of Merkel cell polyomavirus infection with clinicopathological differences in Merkel cell carcinoma. Hum Pathol 43, 2282–91 (2012).
Moshiri, AS, Doumani, R, Yelistratova, L, Blom, A, Lachance, K, Shinohara, MM, et al. Polyomavirus-Negative Merkel Cell Carcinoma: A More Aggressive Subtype Based on Analysis of 282 Cases Using Multimodal Tumor Virus Detection. J Invest Dermatol 137, 819–27 (2017).
Hoang, MP, Donizy, P, Wu, C-L, Kopczynski, J, Pieniazek, M, Miller, DM, et al. TdT Expression Is a Marker of Better Survival in Merkel Cell Carcinoma, and Expression of B-Cell Markers Is Associated With Merkel Cell Polyomavirus. Am J Clin Pathol 154, 38–47 (2020).
Harms, KL, Zhao, L, Johnson, B, Wang, X, Carskadon, S, Palanisamy, N, et al. Virus-positive Merkel Cell Carcinoma Is an Independent Prognostic Group with Distinct Predictive Biomarkers. Clin Cancer Res 27, 2494–504 (2021).
Harms, PW, Vats, P, Verhaegen, ME, Robinson, DR, Wu, Y-M, Dhanasekaran, SM, et al. The Distinctive Mutational Spectra of Polyomavirus-Negative Merkel Cell Carcinoma. Cancer Res 75, 3720–7 (2015).
Wong, SQ, Waldeck, K, Vergara, IA, Schröder, J, Madore, J, Wilmott, JS, et al. UV-Associated Mutations Underlie the Etiology of MCV-Negative Merkel Cell Carcinomas. Cancer Res 75, 5228–34 (2015).
Harms, PW, Collie, AMB, Hovelson, DH, Cani, AK, Verhaegen, ME, Patel, RM, et al. Next generation sequencing of Cytokeratin 20-negative Merkel cell carcinoma reveals ultraviolet-signature mutations and recurrent TP53 and RB1 inactivation. Mod Pathol 29, 240–8 (2016).
Pulitzer, MP, Brannon, AR, Berger, MF, Louis, P, Scott, SN, Jungbluth, AA, et al. Cutaneous squamous and neuroendocrine carcinoma: genetically and immunohistochemically different from Merkel cell carcinoma. Mod Pathol 28, 1023–32 (2015).
Goh, G, Walradt, T, Markarov, V, Blom, A, Riaz, N, Doumani, R, et al. Mutational landscape of MCPyV−positive and MCPyV−negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget 7, 3403–15 (2016).
Harms, KL, Lazo de la Vega, L, Hovelson, DH, Rahrig, S, Cani, AK, Liu, C-J, et al. Molecular Profiling of Multiple Primary Merkel Cell Carcinoma to Distinguish Genetically Distinct Tumors From Clonally Related Metastases. JAMA Dermatol 153, 505–12 (2017).
Carter, MD, Gaston, D, Huang, W-Y, Greer, WL, Pasternak, S, Ly, TY, et al. Genetic profiles of different subsets of Merkel cell carcinoma show links between combined and pure MCPyV−negative tumors. Hum Pathol 71, 117–25 (2018).
Paulson, KG, Lemos, BD, Feng, B, Jaimes, N, Peñas, PF, Bi, X, et al. Array-CGH reveals recurrent genomic changes in Merkel cell carcinoma including amplification of L-Myc. J Invest Dermatol 129, 1547–55 (2009).
Borchert, S, Czech-Sioli, M, Neumann, F, Schmidt, C, Wimmer, P, Dobner, T, et al. High-affinity Rb binding, p53 inhibition, subcellular localization, and transformation by wild-type or tumor-derived shortened Merkel cell polyomavirus large T antigens. J Virol 88, 3144–60 (2014).
Park, DE, Cheng, J, Berrios, C, Montero, J, Cortés-Cros, M, Ferretti, S, et al. Dual inhibition of MDM2 and MDM4 in virus-positive Merkel cell carcinoma enhances the p53 response. Proc Natl Acad Sci USA 116, 1027–32 (2019).
Kervarrec, T, Appenzeller, S, Samimi, M, Sarma, B, Sarosi, E-M, Berthon, P, et al. Merkel cell polyomavirus-negative -Merkel cell carcinoma originating from in situ squamous cell carcinoma: a keratinocytic tumor with neuroendocrine differentiation. J Invest Dermatol 142, 516-7 (2021).
Sunshine, JC, Jahchan, NS, Sage, J, Choi, J. Are there multiple cells of origin of Merkel cell carcinoma? Oncogene 37, 1409–16 (2018).
Kervarrec, T, Samimi, M, Guyétant, S, Sarma, B, Chéret, J, Blanchard, E, et al. Histogenesis of Merkel Cell Carcinoma: A Comprehensive Review. Front Oncol 9, 451 (2019).
Harms, PW, Verhaegen, ME, Hu, K, Hrycaj, SM, Chan, MP, Liu, C-J, et al. Genomic evidence suggests that cutaneous neuroendocrine carcinomas can arise from squamous dysplastic precursors. Mod Pathol 35, 506-14 (2021).
Ly, TY, Walsh, NM, Pasternak, S. The spectrum of Merkel cell polyomavirus expression in Merkel cell carcinoma, in a variety of cutaneous neoplasms, and in neuroendocrine carcinomas from different anatomical sites. Hum Pathol 43, 557–66 (2012).
Busam, KJ, Jungbluth, AA, Rekthman, N, Coit, D, Pulitzer, M, Bini, J, et al. Merkel cell polyomavirus expression in merkel cell carcinomas and its absence in combined tumors and pulmonary neuroendocrine carcinomas. Am J Surg Pathol 33, 1378–85 (2009).
Walsh, NM. Primary neuroendocrine (Merkel cell) carcinoma of the skin: morphologic diversity and implications thereof. Hum Pathol 32, 680–9 (2001).
Ogawa, T, Donizy, P, Wu, C-L, Cornejo, KM, Ryś, J, Hoang, MP. Morphologic Diversity of Merkel Cell Carcinoma. Am J Dermatopathol 42, 629–40 (2020).
Fleming, KE, Ly, TY, Pasternak, S, Godlewski, M, Doucette, S, Walsh, NM. Support for p63 expression as an adverse prognostic marker in Merkel cell carcinoma: report on a Canadian cohort. Hum Pathol 45, 952–60 (2014).
Lai, JH, Fleming, KE, Ly, TY, Pasternak, S, Godlewski, M, Doucette, S, et al. Pure versus combined Merkel cell carcinomas: immunohistochemical evaluation of cellular proteins (p53, Bcl-2, and c-kit) reveals significant overexpression of p53 in combined tumors. Hum Pathol 46, 1290–6 (2015).
Walsh, NM, Fleming, KE, Hanly, JG, Dakin Hache, K, Doucette, S, Ferrara, G, et al. A morphological and immunophenotypic map of the immune response in Merkel cell carcinoma. Hum Pathol 52, 190–6 (2016).
Pasternak, S, Carter, MD, Ly, TY, Doucette, S, Walsh, NM. Immunohistochemical profiles of different subsets of Merkel cell carcinoma. Hum Pathol 82, 232–8 (2018).
Walsh, NM, Saggini, A, Pasternak, S, Carter, MD, Fleming, K, Ly, TY, et al. p63 expression in Merkel cell carcinoma: comparative immunohistochemistry invokes TAp63 as the dominant isoform involved. Hum Pathol 97, 60–7 (2020).
Kroeze, LI, de Voer, RM, Kamping, EJ, von Rhein, D, Jansen, EAM, Hermsen, MJW, et al. Evaluation of a Hybrid Capture-Based Pan-Cancer Panel for Analysis of Treatment Stratifying Oncogenic Aberrations and Processes. J Mol Diagn 22, 757–69 (2020).
Sahajpal, NS, Mondal, AK, Ananth, S, Njau, A, Ahluwalia, P, Jones, K, et al. Clinical performance and utility of a comprehensive next-generation sequencing DNA panel for the simultaneous analysis of variants, TMB and MSI for myeloid neoplasms. PLoS One 15, e0240976 (2020).
Talevich, E, Shain, AH, Botton, T, Bastian, BC. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput Biol 12, e1004873 (2016).
Cingolani, P, Platts, A, Wang, LL, Coon, M, Nguyen, T, Wang, L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly (Austin) 6, 80–92 (2012).
Pedersen, BS, Layer, RM, Quinlan, AR. Vcfanno: fast, flexible annotation of genetic variants. Genome Biol 17, 118 (2016).
Karczewski, KJ, Francioli, LC, Tiao, G, Cummings, BB, Alföldi, J, Wang, Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–43 (2020).
Tate, JG, Bamford, S, Jubb, HC, Sondka, Z, Beare, DM, Bindal, N, et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res 47, D941–7 (2019).
Landrum, MJ, Lee, JM, Benson, M, Brown, GR, Chao, C, Chitipiralla, S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 46, D1062–7 (2018).
Allen, EMV, Wagle, N, Stojanov, P, Perrin, DL, Cibulskis, K, Marlow, S, et al. Whole-exome sequencing and clinical interpretation of FFPE tumor samples to guide precision cancer medicine. Nat Med 20, 682–8 (2014).
Rosenthal, R, McGranahan, N, Herrero, J, Taylor, BS, Swanton, C. deconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol 17, 31 (2016).
Alexandrov, LB, Stratton, MR. Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Curr Opin Genet Dev 24, 52–60 (2014).
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al.
Alexandrov, LB, Jones, PH, Wedge, DC, Sale, JE, Campbell, PJ, Nik-Zainal, S, et al. Clock-like mutational processes in human somatic cells. Nat Genet 47, 1402–7 (2015).
Zehir, A, Benayed, R, Shah, RH, Syed, A, Middha, S, Kim, HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 23, 703–13 (2017).
Knepper, TC, Montesion, M, Russell, JS, Sokol, ES, Frampton, GM, Miller, VA, et al. The Genomic Landscape of Merkel Cell Carcinoma and Clinicogenomic Biomarkers of Response to Immune Checkpoint Inhibitor Therapy. Clin Cancer Res 25, 5961–71 (2019).
Dotto, GP, Rustgi, AK. Squamous Cell Cancers: A Unified Perspective on Biology and Genetics. Cancer Cell 29, 622–37 (2016).
Lazo de la Vega, L, Bick, N, Hu, K, Rahrig, SE, Silva, CD, Matayoshi, S, et al. Invasive squamous cell carcinomas and precursor lesions on UV-exposed epithelia demonstrate concordant genomic complexity in driver genes. Mod Pathol 33, 2280–94 (2020).
Harold, A, Amako, Y, Hachisuka, J, Bai, Y, Li, MY, Kubat, L, et al. Conversion of Sox2-dependent Merkel cell carcinoma to a differentiated neuron-like phenotype by T antigen inhibition. PNAS 116, 20104–14 (2019).
Busam, KJ, Pulitzer, MP, Coit, DC, Arcila, M, Leng, D, Jungbluth, AA, et al. Reduced H3K27me3 expression in Merkel cell polyoma virus-positive tumors. Mod Pathol 30, 877–83 (2017).
Harms, KL, Chubb, H, Zhao, L, Fullen, DR, Bichakjian, CK, Johnson, TM, et al. Increased expression of EZH2 in Merkel cell carcinoma is associated with disease progression and poorer prognosis. Hum Pathol 67, 78–84 (2017).
La Rosa, S, Sessa, F, Uccella, S. Mixed neuroendocrine-nonneuroendocrine neoplasms (MiNENs): Unifying the Concept of a Heterogeneous Group of Neoplasms. Endocr Pathol 27, 284–311 (2016).
Acknowledgements
We gratefully acknowledge clerical and technical staff in the Division of Anatomical Pathology, Department of Pathology and Laboratory Medicine, Nova Scotia Health Authority (Central Zone) for their contributions to the project.
Funding
Financial support was provided by the Nova Scotia Health Authority Research Fund and the Department of Pathology and Laboratory Medicine Fund for Molecular Pathology housed at the QEII Foundation, Halifax, Nova Scotia, Canada.
Author information
Authors and Affiliations
Contributions
RCD, NMW, and MDC contributed to study concept and design. RCD, NMW, DG, TYL, SP, MN, and MDC contributed to the writing, review, and revision of the manuscript. RCD, DG, MN, SC, and MDC performed acquisition, analysis, and interpretation of data. RCD performed statistical analysis. DG and MN provided technical support.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study was approved by the Nova Scotia Health Authority Research Ethics Board (REB #1021594). The requirement for obtaining individual patient consent was waived in accordance with institutional policies. The study was performed in accordance with the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
DeCoste, R.C., Walsh, N.M., Gaston, D. et al. RB1-deficient squamous cell carcinoma: the proposed source of combined Merkel cell carcinoma. Mod Pathol 35, 1829–1836 (2022). https://doi.org/10.1038/s41379-022-01151-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-022-01151-2
