
Abstract
Tumor purity, or the relative contribution of tumor cells out of all cells in a pathological specimen, influences mutation identification and clinical interpretation of cancer panel next generation sequencing results. Here, we describe a method of calculating tumor purity using pathologist-guided copy number analysis from sequencing data. Molecular calculation of tumor purity showed strong linear correlation with purity derived from driver KRAS or BRAF variant allele fractions in colorectal cancers (R2 = 0.79) compared to histological estimation in the same set of colorectal cancers (R2 = 0.01) and in a broader dataset of cancers with various diagnoses (R2 = 0.35). We used calculated tumor purity to quantitate ERBB2 copy number in breast carcinomas with equivocal immunohistochemical staining and demonstrated strong correlation with fluorescence in situ hybridization (R2 = 0.88). Finally, we used calculated tumor purity to infer the germline status of variants in breast and ovarian carcinomas with concurrent germline testing. Tumor-only next generation sequencing correctly predicted the somatic versus germline nature of 26 of 26 (100%) pathogenic TP53, BRCA1 and BRCA2 variants. In this article, we describe a framework for calculating tumor purity from cancer next generation sequencing data. Accurate tumor purity assessment can be assimilated into interpretation pipelines to derive clinically useful information from cancer genomic panels.
Similar content being viewed by others


Introduction
Over the past decade, massively parallel next generation sequencing (NGS) of cancer specimens has been rapidly adopted to become standard of care in many oncology practices. NGS has replaced single gene assays in many molecular pathology laboratories1. As of 2017, 76% of oncologists in the United States in a nationally representative survey reported using NGS tests to guide treatment decisions for patients with advanced disease, to determine eligibility for clinical trials, and to prescribe off-label therapy2.
Cancer panel NGS assays have been validated in multiple academic and commercial laboratories3,4,5,6, and these comprehensive assays are capable of detecting many types of genetic alterations within cancer genomes, including sequence alterations (nucleotide substitutions, insertions and deletions), structural variants and copy number alterations7. Identifying different types of alterations usually requires multiple informatics tools8,9. As a result, single nucleotide variants and copy number alterations are commonly assessed independently in validation and in the clinical report. Clinical practices vary among laboratories in the reporting of higher complexity findings, including tumor purity, variant allele fraction, copy number analysis and clinical variant interpretation10.
Pathological evaluation of tumor tissue is an important pre-analytical consideration for cancer molecular testing. Most cancer NGS assays are validated to detect somatic mutations at variant allele fraction of as low as 5–10% and generally require tissue specimens containing at least 20% tumor nuclei. While most laboratories conduct histological tumor purity assessment, there is a need for standardization in clinical practice11. Tumor purity can also be used to guide analysis of somatic copy number variation and is necessary if quantitative copy number analysis is performed. It has been shown previously that computational assessment of tumor purity can help refine NGS analysis, including such parameters as germline mutation inference and tumor mutational burden12,13. While several tools exist to calculate tumor purity based on the sequencing data, these generally rely on paired tumor-normal samples or require whole genome or whole exome sequencing14,15,16,17.
In this article, we describe methods to analyze panel sequencing data and achieve a more comprehensive analytical and clinical interpretation of an unpaired tumor specimen. We demonstrate that NGS data can be used to quantitate tumor purity, which can in turn be used to improve quantitative copy number analysis and to infer the somatic or germline status of pathogenic variants.
Materials and methods
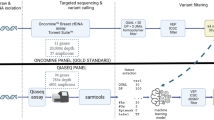
Cancer next generation sequencing
Next generation sequencing (NGS) was performed using OncoPanel, a hybrid-capture based targeted sequencing assay5. Pre-analytical histologic examination was performed for all cases prior to sequencing. A tumor-enriched region of interest was outlined on a hematoxylin and eosin-stained slide, and a histological estimation of tumor purity was recorded. Sequencing required at least 20% tumor nuclei in the region of interest. Deoxyribonucleic acid was isolated after macro-dissection of the corresponding tissue from unstained slides. A panel of 447 cancer-associated genes was enriched using solution-based hybrid capture (Agilent SureSelect; Agilent Technologies; Santa Clara, CA). Massively parallel sequencing was performed using Illumina HiSeq2500 (Illumina, Inc., San Diego, CA). Single nucleotide variants were detected using Mutect version 2. Insertion and deletion variants were detected using GATK version 4. Copy number analysis was visualized with RobustCNV version 2.0.1. Germline polymorphism variant allele fractions were visualized using a laboratory developed tool.
Tumor purity calculation
Relative copy number variation was calculated and visualized using RobustCNV. Briefly, the relative read contribution of each gene exon in the tumor specimen, or the proportion of reads mapping to the exon compared to all reads, was compared to relative read contribution of the same exon in a non-neoplastic control. To improve copy number detection, systematic bias and GC bias correction was implemented to increase the signal to noise ratio via a two-step process. In step one, systematic bias was removed by fitting a robust regression model with iteratively re-weighted least squares, as implemented in the MASS package in R. The model was then used to estimate expected values for each gene exon. The log2(observed/predicted) value was then calculated for each gene exon. In step two GC bias was removed through Local Polynomial Regression (loess) where the previously normalized values were fitted against GC content. The resulting model was then used to generate a set of predicted values which were subtracted from the observed values. Relative read contributions from each gene exon was plotted in log2 scale, with relative copy number gains displayed as values above 0, and relative copy number losses displayed as values below 0. Manual copy number calls were made by human reviewers via an interactive user interface portal. In validation, copy number detection by NGS achieved 86% sensitivity and 98% specificity compared to array comparative genomic hybridization5.
The cancer panel incidentally captured germline polymorphisms, which were detected by MuTect and plotted for evaluation concurrent with copy number changes. Pathologist copy number interpretation was performed akin to analysis of microarray data in accordance with technical standards18,19.
The tumor purity was calculated by the following formula, where T represents tumor purity, X represents the median log2 ratio of a one-copy (haploid) state, and Y represents the median log2 ratio of a two-copy (diploid) state. These reference states were selected by the pathologist and correspond to a whole chromosome or chromosome region or arm.
Absolute copy number estimation was performed as follows, where A represents absolute copy number, Z represents the median log2 ratio of gene of interest, Y represents the median log2 ratio of the diploid copy number state, and T represents calculated tumor purity.
Examples of pathologist-guided copy number estimation and tumor purity calculation are provided in Supplemental File 1.
To infer the germline status of pathogenic variants in BRCA1, BRCA2 and TP53, expected variant allele fractions were modeled for both germline and somatic scenarios at the observed copy number state (either one copy deletion or copy number neutral loss of heterozygosity). A probability density function was constructed for the expected number of mutant reads given the observed target coverage with normal approximation of the binomial distribution. The variant was inferred to be germline if the probability of the observed allele fraction in the expected germline scenario is greater than that in the expected somatic scenario.
Case selection
All sequencing results were reviewed by a board-certified molecular pathologist (F.D.). Cases were excluded if there were no identifiable copy number changes due to low tumor purity or have highly complex copy number changes precluding determination of one-copy and two-copy states.
To validate tumor purity calculations, 12 colorectal adenocarcinomas with driver oncogenic mutations in KRAS or BRAF were identified20. Analysis was limited to cases with mutations at non-amplified diploid loci, and the driver mutations were assumed to represent de novo mutations involving all tumor cells and absent in non-neoplastic cells. For KRAS or BRAF mutations involving one of two tumor alleles, tumor purity was estimated as follows, where T represent tumor purity and X represents driver mutation variant allele fraction, and compared to tumor purity calculation based on copy number variants:
Analysis of absolute copy number quantitation included 19 invasive breast carcinomas with equivocal (2+) expression of ERBB2 (HER2) by immunohistochemistry. ERBB2 fluorescence in situ hybridization (FISH) was performed in accordance to the 2018 American Society of Clinical Oncology/College of American Pathologists (ASCO/CAP) guidelines21. By FISH, tumors with ERBB2 copy number ≥4.0 and ERBB2/CEP17 ratio ≥2.0 were considered positive. Tumors with ERBB2 copy number <4.0 and ERBB2/CEP17 ratio <2.0 were considered negative. By NGS, tumors with ERBB2 copy number ≥4.0 and ERBB2/NF1 ratio ≥2.0 were considered positive. Tumors with ERBB2 copy number <4.0 and ERBB2/NF1 ratio <2.0 were considered negative.
To infer germline status from tumor sequencing, nine cases of invasive breast carcinoma and six cases of high grade serous carcinoma of the ovary harboring BRCA1 or BRCA2 mutations were identified. Each BRCA1, BRCA2, and TP53 variant was classified based on tumor-only NGS data as germline or somatic. All 15 patients received concurrent germline testing for BRCA1, BRCA2 and TP53 as a component of clinical care via a targeted sequencing panel. Clinical germline testing results were collected by chart review. The tumor-only NGS classification was compared to germline panel testing results for each variant.
Results
Calculation of tumor purity from NGS data
We first calculated tumor purity for a clinical dataset of tumors sequenced by NGS. We identified 265 NGS cases consecutively reported by a single pathologist. Of these 265 cases, 135 (51%) were excluded due to failed sequencing (n = 4, 2%), having a flat copy number profile indicating tumor purity below level of detection of copy number alterations (n = 85, 32%), or having complex number profile precluding reliable identification of the diploid copy number state (n = 46, 17%). Cases with flat copy number profiles are enriched for specimens of borderline adequacy, defined as a histological estimate of 30% or fewer tumor nuclei. 43 of 85 (51%) cases with flat copy number profiles were of borderline adequacy compared to 24 of 130 (18%) cases that underwent successful tumor purity analysis (Fisher’s exact p < 0.001). Cases with complex copy number profiles had a similar rate of borderline adequacy (11 of 46, 24%) compared to cases that underwent successful tumor purity analysis (Fisher’s exact p = 0.52).
TP53 variants were identified by sequencing in 142 cases in the cohort. Specimens excluded due to a flat copy number profile had a mean TP53 variant allele fraction of 18% (standard deviation 12%), which was significantly lower than the mean TP53 variant allele fraction observed in specimens that were successfully analyzed for tumor purity (mean TP53 allele fraction 52%, standard deviation 19%, t-test p < 0.001). The mean TP53 variant allele fraction for cases excluded due to complex copy number changes was 48% (standard deviation 20%), which was not significantly different from that of cases that were successfully analyzed (t-test p = 0.35). These findings demonstrated that specimens with low tumor purity were likely to exhibit flat copy number profiles, limiting the utility of methodologies dependent on copy number changes in this scenario.
The successfully analyzed dataset included 130 tumors representing the spectrum of clinical specimens sequenced at our institution (Fig. 1A). Calculated tumor purity was compared to estimated tumor purity by histology, and these variables showed a weak positive association (R2 = 0.35, 95% Confidence Interval (CI) = 0.47–0.69, Fig. 1B). Calculated tumor purity was within 10% of histologic estimation in 45% (59 of 130) of tumors and within 20% histologic estimation in 74% (96 of 130) of tumors (Fig. 1C). In this cohort, histology review estimated higher tumor purity compared to the calculated tumor purity in 57% of cases. Of note, the actual value may be higher since specimens with lowest tumor purity with flat copy number profiles were excluded from this calculation.

A Pie chart of primary sites for 130 tumors, representing the spectrum of clinical solid tumor specimens sequenced at the institution. B Tumor purity based on histologic estimation versus tumor purity based on calculation by NGS. C Difference in tumor purity based on histology and sequencing in cancer specimens.
To further validate our method of tumor purity calculation, we analyzed the calculated tumor purity based on copy number alterations in 12 colorectal adenocarcinomas with driver oncogenic mutations in KRAS or BRAF (Table 1). Pathogenic pathway-activating somatic mutations in the RAS-MAPK pathway genes have been strongly implicated as causative events of colorectal tumor evolution22,23,24. Calculated tumor purity based on copy number alterations was compared to tumor purity based on driver mutation allele fraction, demonstrating a strong positive correlation (R2 = 0.79, 95% CI = 0.64–0.97, Fig. 2A). In contrast, there was poor correlation between histologic estimation and calculated tumor purity by copy number alterations (R2 = 0.01, 95% CI = −0.51–0.63, Fig. 2B) or between histologic estimation and calculated tumor purity by driver mutations (R2 = 0.05, 95% CI = −0.41–0.70).

A Calculated tumor purity in colorectal cancers based on copy number changes (X-axis) versus RAS driver mutation allele fraction (Y-axis) shows linear correlation (R2 = 0.79). B Calculated tumor purity (X-axis) versus histologic estimation (Y-axis) shows poor correlation (R2 = 0.01).
Finally, we assessed interobserver variability between two experienced reviewers (D.K.M. and F.D.) across 12 specimens. The calculated tumor purity between the two observers was linearly correlated (R2 = 0.83, 95% CI 0.72–0.98, Fig. 3). The median absolute difference in tumor purity between the two observers was 3.5%.

The calculated tumor purity between the two observers was linearly correlated (R2 = 0.83).
Absolute copy number quantitation
To demonstrate the clinical utility of calculating tumor purity, we used calculated tumor purity to quantitate unknown copy number states. We selected the clinically relevant target gene ERBB2, which is amplified in a subset of breast carcinoma and serves as both a prognostic biomarker and a target for ERBB2 inhibitor therapies25. Current clinical guidelines according to the 2018 ASCO/CAP call for ERBB2 gene evaluation by a combination of immunohistochemistry and FISH21. Carcinomas that are equivocal (2+) for ERBB2 protein expression by immunohistochemistry represent a diagnostic dilemma, as these tumors demonstrate a broad range of copy number states in ERBB226.
We evaluated 19 invasive breast carcinomas with equivocal (2+) ERBB2 expression by immunohistochemistry, including 12 cases that were positive for ERBB2 amplification and 7 that were negative for ERBB2 amplification by FISH (Table 2). First, we quantitated absolute ERBB2 copy number by NGS and compared this to the absolute copy number assessment by manual counting of FISH specimens. ERBB2 copy numbers by NGS and FISH were strongly correlated (R2 = 0.88, 95% CI = 0.84–0.98, Fig. 4A). Since clinical guidelines evaluate the ratio of ERBB2 copy number to a chromosome 17 centromeric probe, we approximated this ratio by assessing the copy number ratio of ERBB2 to NF1, the gene in our NGS panel on chromosome 17q that is closest to the centromere. A comparison of the ratios of ERBB2 to the chromosome 17 control (ERBB2 to CEP17 ratio by FISH or ERBB2 to NF1 copy number by NGS) showed that the ratios determined by FISH and NGS were linearly correlated (R2 = 0.76, 95% CI = 0.67–0.95, Fig. 4B).

A Absolute ERBB2 copy number by fluorescence in situ hybridization (FISH) versus NGS (linear regression R2 = 0.88). B Comparison of ERBB2/CEP17 copy number ratio by FISH versus ERBB2/NF2 copy number ratio by NGS (linear regression R2 = 0.76). C NGS data plots of log2(read ratio) for chromosome 17 from three representative invasive breast carcinoma cases; each dot represents read ratio data point from target genes included in the NGS panel; “C” denotes location of centromere and shaded boxes labeled NF1 and ERBB2 denote the read ratios corresponding to these target genes. Top: focal ERBB2 amplification, positive by FISH and NGS. Middle: ERBB2 gain as part of chromosome 17q gain, negative by FISH and NGS. Bottom: ERBB2 gain as part of complex chromosome 17q copy number changes, positive by FISH and NGS.
We next adapted the ASCO/CAP algorithm to determine categorical amplification status for our cohort of 19 tumors and found 95% concordance (18 of 19 cases) between NGS and FISH. In the one case of discordance, ERBB2 amplification was found to be positive by FISH (ERBB2 to CEP17 ratio 6.7/3.3 = 2.1) but negative by NGS (ERBB2 to NF1 ratio 2.0/2.1 = 0.9). Although we cannot completely explain this discordance, this specimen consisted of a breast carcinoma metastasis in a patient with a known prior specimen that was negative for ERBB2 amplification, and the discordance could be related to tumor heterogeneity. Another possible explanation may be the presence of a hyperdiploid genomic copy number state that could not be deduced from the NGS copy number profile. An additional benefit of ERBB2 analysis by NGS is the ability to visualize ERBB2 copy number with respect to the rest of chromosome 17. Examples of ERBB2 focal amplification, ERBB2 gain as a part of chromosome 17q arm-level gain, and ERBB2 gain as a part of complex copy number alterations involving chromosome 17q are shown in Fig. 4C.
Inference of germline alterations using tumor-only sequencing
With an ability to calculate tumor purity, we hypothesized that tumor-only sequencing data could be used to infer the germline status of variants. In most tumor predisposition syndromes, non-neoplastic cells harbor a pathogenic variant in one of two germline alleles, and the wild type allele is lost in neoplastic cells during tumorigenesis, commonly by gene deletion or copy number neutral loss of heterozygosity. The observed variant allele fraction observed in tumor-only sequencing data can be used to infer whether non-neoplastic cells in the specimen harbor the variant.
To test our hypothesis, we evaluated 9 invasive breast carcinomas and 6 ovarian high grade serous carcinomas with that harbored variants in BRCA1 or BRCA2 (n=15). Eleven of these cases also harbored concurrent sporadic mutations in TP53, which were used as additional somatic variants in our analysis. BRCA1, BRCA2 and TP53 germline status were previously assessed by clinical germline panel testing. In total, the cohort included 26 mutations with known germline status (12 germline, 14 somatic) (Table 2).
The calculated tumor purity of breast and ovarian carcinomas in the dataset ranged from 28 to 87%. Overall, the observed variant allele fraction was correlated with calculated tumor purity (R2 = 0.53, 95% CI = 0.47–0.87), and demonstrated close agreement with predicted values for the corresponding germline and somatic status (Fig. 5A, B). Based on comparison of the expected tumor purity and observed variant allele fraction, 26 of 26 (100%) variants were correctly classified as germline or somatic based on tumor-only NGS data (Table 3).

A Observed variant allele fraction compared to expected variant allele fraction for known germline and somatic variants with single copy loss. B Observed variant allele fraction compared to expected variant allele fraction for known somatic and germline variants with copy number neutral loss of heterozygosity. Observed variant allele fractions (circles) and expected variant allele fractions (lines) are shown in red for somatic mutations and blue for germline mutations.
Discussion
The histopathological examination of tumor tissue is a key pre-analytical quality control measure for cancer NGS testing. Inadequate specimens with tumor purity below the molecular limit of detection can be rejected prior to sequencing, saving labor, time and cost. An accurate assessment of tumor purity is useful in downstream analysis and interpretation, including the clinical identification of artifactual or contaminating sequences and the detection of somatic mutations and copy number changes27. Despite the importance of histological review, the pathologist’s estimation of tumor purity is relatively inaccurate compared to manually counting tumor cells, which may impact the interpretation of molecular test results28. Multiple factors can contribute to inaccuracies in tumor purity estimation. Technical factors include changes of tumor purity in deeper levels of the tissue block, particularly in small tissue samples or specimens focally involved by tumor cells. Pathologists may be biased to accept specimens to expand potential treatment options for patients with advanced cancer. Pathologists may also be biased from clinical training to fixate on positive events or diagnostic cells relative to the non-neoplastic background29,30. A comparison of pathologist estimation of PD-L1 immunohistochemistry shows relative overestimation of percent of cells with PD-L1 expression compared to automated algorithms31. Due to limitations in visual interpretation, calculation of tumor purity based on molecular data may be useful to guide the subsequent clinical interpretation of molecular results.
Establishing the accurate calculation of tumor purity enables us to utilize this value to expand the downstream information yielded in our NGS pipeline. Our first demonstration of this principle is seen with the application of calculated tumor purity to quantitate gene amplification. We describe the reliable quantitation of ERBB2 copy number in a cohort of breast carcinomas with equivocal (2+) ERBB2 expression by immunohistochemistry. Previous studies have demonstrated high rates of concordance between NGS and FISH approaches to assessing ERBB2 amplification across the full spectrum of breast and other carcinomas and within equivocal or clinically challenging subsets26,32,33,34,35,36. Compared to prior studies, the current method approximates clinical FISH guidelines with quantitative estimations of absolute ERBB2 copy number and the ratio of ERBB2 to the centromeric region of chromosome 17. Future work with larger datasets may enable validation of NGS cut-offs further adjudicate cancers with equivocal findings33.
A potential limitation of our approach is the use of the chromosome 17q gene NF1 as a surrogate centromere marker, with a risk that this gene can be affected by chromosomal instability compared to a centromeric control. However, NGS has the advantage of providing copy state information over multiple genes on chromosome 17 that can distinguish focal from broad events, which cannot be determined based on standard FISH approaches37. Breast carcinomas exhibiting ERBB2 absolute copy number gain but without amplification relative to chromosome 17 have been shown to not benefit from targeted therapy38. Similar findings can be seen in carcinomas with polysomy 17 or 17q gain39. NGS can distinguish between examples of true ERBB2-specific amplification, compared with 17q gain or whole chromosome instability (Fig. 4C). Larger cohort studies will be useful to optimize calculation of centromere chromosome 17 controls and to standardize metrics for distinguishing focal ERBB2 amplification from broader copy number alterations.
A second utility of tumor purity calculation is the ability to successfully infer germline status of variants. Sequencing of paired non-tumor tissue detects pathogenic germline mutations in 3–16% of patients with cancer3,32,40,41. Current institutional guidelines including consent protocols for many sequencing platforms do not specifically address germline status, due to the assumption that germline information cannot be ascertained from tumor only sequencing data42. However, our findings show that most germline BRCA1 and BRCA2 variants can be accurately categorized as germline events from tumor sequencing. While our approach addresses the most common biological scenarios (loss of the wild type allele by deletion or copy number neutral loss of heterozygosity), more complex scenarios, such as greater than two copies of a variant or somatic loss of a germline variant, can be modeled by the same principles. Expected allele fractions for somatic and germline variants at multiple copy number states and tumor purities are provided in Supplemental File 2. A limitation of germline inference is that the expected variant allele fraction of germline versus somatic events converge as tumor purity approaches 100% and may not be informative in specimens with very high tumor purity.
A limitation to this study is an overall high failure rate. 32% of specimens failed due to a flat copy number profiles, indicative of low tumor purity. This high rate of failure in our study is related to the study population of unselected clinical specimens. Compared to research samples used to populate large datasets, clinical specimens in anatomic pathology laboratories have a range of tumor purity estimations depending on tumor biology, growth pattern, and the diagnostic procedure performed. Accurate assessment on low tumor purity specimens remains a clinical challenge in molecular diagnostics. Although not addressed in our current clinical sequencing platform, molecular technologies optimized to detect copy number changes in limited specimens and improved informatics algorithms to reduce artifact in copy number analysis could expand application of quantitative methods in limited clinical specimens.
An additional 17% of specimens failed analysis due to genomic complexity, where a diploid baseline could not be established. This observation reflects the complexity of interpreting cancer genomes, where copy number changes are generated via diverse mechanisms and complex events like genome doubling occur frequently43. While our analysis is based on purely analytical features of NGS data to set baseline copy number states, incorporation of additional biological and clinical information, such as expected copy number changes for tumor type, may provide tumor purity estimations for more samples. Alternatively, assessment by an orthogonal method with absolute copy number quantitation, such as FISH analysis of a limited subset of gene targets, would be expected to provide baseline ploidy for most cancer specimens.
The ability to determine germline status in tumor specimens for a variety of pathogenic variants has broad implications for clinical care. For example, <20% of patients with breast or ovarian cancer meeting eligibility criteria for germline evaluation undergo genetic testing44. With appropriate patient consent, germline alterations could effectively be incorporated into analytical pipelines to yield clinically relevant information, while being masked within reporting schemes according to patient preference45,46.
Tumor-only sequencing uses population databases to filter germline events; however, studies have shown that population databases are insufficient to remove rare private variants from cancer sequencing panels47. These considerations are important in the calculation of tumor mutational burden, an emerging biomarker that predicts response to immune checkpoint inhibitor therapy. The consideration of variant allele fraction in the context of tumor purity may assist in filtering incidental germline event and improving accuracy for tumor mutational burden calculation, especially for non-Caucasian individuals who are underrepresented in population databases.
In summary, we have demonstrated that NGS data can be used to quantitate tumor purity, which can be integrated with analytical algorithms to improve quantitative copy number analysis and infer the germline status of variants without paired normal sequencing. Cancer NGS interpretation requires molecular pathologists to integrate complex data and make clinical recommendations. This paper demonstrates that we can derive more clinically useful information from existing sequencing data with little additional cost, which has potential to expand the utility of cancer NGS and better integrate histopathological and molecular data into patient care.
Data availability
All data used and analyzed in the current study are available in the published paper and Supplemental Files.
References
Nagarajan, R., Bartley, A.N., Bridge, J.A., Jennings, L.J., Kamel-Reid, S., Kim A., et al. A window into clinical next-generation sequencing–based oncology testing practices. Arch. Pathol. Lab. Med. 141, 1679–1685 (2017).
Freedman, A.N., Klabunde, C.N., Wiant, K., Enewold, L., Gray, S.W., Filipski, K.K., et al. Use of next-generation sequencing tests to guide cancer treatment: results from a nationally representative survey of oncologists in the United States. JCO Precis. Oncol. 2 (2018) https://doi.org/10.1200/PO.18.00169.
Cheng, D.T., Prasad, M., Chekaluk, Y., Benayed, R., Sadowska, J., Zehir, A., et al. Comprehensive detection of germline variants by MSK-IMPACT, a clinical diagnostic platform for solid tumor molecular oncology and concurrent cancer predisposition testing. BMC Med. Genom. 10, 1–9 (2017).
Frampton, G.M., Fichtenholtz, A., Otto, G.A., Wang, K., Downing, S.R., He, J., et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 31, 1023–31 (2013).
Garcia, E.P., Minkovsky, A., Jia, Y., Ducar, M.D., Shivdasani, P., Gong, X., et al. Validation of OncoPanel: a targeted next-generation sequencing assay for the detection of somatic variants in cancer. Arch. Pathol. Lab. Med. 141, 751–758 (2017).
Hirsch, B., Endris, V., Lassmann, S., Weichert, W., Pfarr, N., Schirmacher, P., et al. Multicenter validation of cancer gene panel-based next-generation sequencing for translational research and molecular diagnostics. Virchows Arch. 472, 557–565 (2018).
Horak, P., Fröhling, S., Glimm, H. Integrating next-generation sequencing into clinical oncology: strategies, promises and pitfalls. ESMO Open 1, e000094–e000094 (2016).
Wang, Q., Jia, P., Li, F., Chen, H., Ji, H., Hucks, D., et al. Detecting somatic point mutations in cancer genome sequencing data: a comparison of mutation callers. Genome Med. 5, 91 (2013).
Zhang, L., Bai, W., Yuan, N., Du, Z. Comprehensively benchmarking applications for detecting copy number variation. PLOS Comput. Biol. 15, e1007069 (2019).
Sirohi, D., Schmidt, R.L., Aisner, D.L., Behdad, A., Betz, B.L., Brown, N., et al. Multi-institutional evaluation of interrater agreement of variant classification based on the 2017 Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists Standards and Guidelines for the interpretation and reporting of sequence variants in cancer. J. Mol. Diagn. 22, 284–293 (2020).
Devereaux, K.A., Souers, R.J., Graham, R.P., Portier, B.P., Surrey, L.F., Yemelyanova, A., et al. Neoplastic cellularity assessment in molecular testing. Arch. Pathol. Lab. Med. (2022) https://doi.org/10.5858/arpa.2021-0166-CP.
Hong, T.H., Cha, H., Shim, J.H., Lee, B., Chung, J., Lee, C., et al. Clinical advantage of targeted sequencing for unbiased tumor mutational burden estimation in samples with low tumor purity. J. Immunother. Cancer 8, 1–11 (2020).
Smith, K.S., Yadav, V.K., Pei, S., Pollyea, D.A., Jordan, C.T., De, S. SomVarIUS: somatic variant identification from unpaired tissue samples. Bioinformatics 32, 808–813 (2016).
Carter, S.L., Cibulskis, K., Helman, E., McKenna, A., Shen, H., Zack, T., et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 30, 413–21 (2012).
Favero, F., Joshi, T., Marquard, A.M., Birkbak, N.J., Krzystanek, M., Li, Q., et al. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann. Oncol. 26, 64–70 (2015).
Riester, M., Singh, A.P., Brannon, A.R., Yu, K., Campbell, C.D., Chiang, D.Y., et al. PureCN: copy number calling and SNV classification using targeted short read sequencing. Source Code Biol. Med. 11, 13 (2016).
Shen, R., Seshan, V.E. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 44, 1–9 (2016).
Cooley, L.D., Lebo, M., Li, M.M., Slovak, M.L., Wolff, D.J. American College of Medical Genetics and Genomics technical standards and guidelines: microarray analysis for chromosome abnormalities in neoplastic disorders. Genet. Med. 15, 484–494 (2013).
Mikhail, F.M., Biegel, J.A., Cooley, L.D., Dubuc, A.M., Hirsch, B., Horner, V.L., et al. Technical laboratory standards for interpretation and reporting of acquired copy-number abnormalities and copy-neutral loss of heterozygosity in neoplastic disorders: a joint consensus recommendation from the American College of Medical Genetics and Genomics. Genet. Med. 21, 1903–1915 (2019).
Landrum, M.J., Lee, J.M., Riley, G.R., Jang, W., Rubinstein, W.S., Church, D.M., et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 42, D980–D985 (2014).
Wolff, A.C., Hammond, M.E.H., Allison, K.H., Harvey, B.E., Mangu, P.B., Bartlett, J.M.S., et al. Human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline focused update. J. Clin. Oncol. 36, 2105–2122 (2018).
Costigan, D.C., Dong, F. The extended spectrum of RAS-MAPK pathway mutations in colorectal cancer. Genes Chromosom. Cancer 59, 152–159 (2020).
Jinesh, G.G., Sambandam, V., Vijayaraghavan, S., Balaji, K., Mukherjee, S. Molecular genetics and cellular events of K-Ras-driven tumorigenesis. Oncogene 37, 839–846 (2018).
Rajagopalan, H., Bardelli, A., Lengauer, C., Kinzler, K.W., Vogelstein, B., Velculescu, V.E. RAF/RAS oncogenes and mismatch-repair status. Nature 418, 934 (2002).
Stocker, A., Hilbers, M.L., Gauthier, C., Grogg, J., Kullak-Ublick, G.A., Seifert, B., et al. HER2/CEP17 ratios and clinical outcome in HER2-positive early breast cancer undergoing trastuzumab-containing therapy. PLoS ONE 11, 1–10 (2016).
Hoda, R.S., Bowman, A.S., Zehir, A., Razavi, P., Brogi, E., Ladanyi, M., et al. Next-generation assessment of human epidermal growth factor receptor 2 gene (ERBB2) amplification status in invasive breast carcinoma: a focus on Group 4 by use of the 2018 American Society of Clinical Oncology/College of American Pathologists HER2 testing guideline. Histopathology 78, 498–507 (2021).
Li, Y.Y., Schmidt, R.J., Manning, D.K., Jia, Y., Dong, F. Contamination assessment for cancer next-generation sequencing: method development and clinical implementation. Arch. Pathol. Lab. Med. 146, 227–232 (2022).
Smits, A.J.J., Kummer, J.A., De Bruin, P.C., Bol, M., Van Den Tweel, J.G., Seldenrijk, K.A., et al. The estimation of tumor cell percentage for molecular testing by pathologists is not accurate. Mod. Pathol. 27, 168–174 (2014).
Brunyé, T.T., Carney, P.A., Allison, K.H., Shapiro, L.G., Weaver, D.L., Elmore, J.G. Eye movements as an index of pathologist visual expertise: a pilot study. PLoS ONE 9, (2014).
Rangrej, S.B., Sivaswamy, J., Srivastava, P. Scan, dwell, decide: strategies for detecting abnormalities in diabetic retinopathy. PLoS ONE 13, 1–18 (2018).
Widmaier, M., Wiestler, T., Walker, J., Barker, C., Scott, M.L., Sekhavati, F., et al. Comparison of continuous measures across diagnostic PD-L1 assays in non-small cell lung cancer using automated image analysis. Mod. Pathol. 33, 380–390 (2020).
Dumbrava, E.I., Brusco, L., Daniels, M.S., Wathoo, C., Shaw, K.R. Expanded analysis of secondary germline findings from matched tumor/normal sequencing identifies additional clinically significant mutations. JCO Precis Oncol. 3 (2019) https://doi.org/10.1200/PO.18.00143.
Nakamura, K., Aimono, E., Oba, J., Hayashi, H., Tanishima, S., Hayashida, T., et al. Estimating copy number using next-generation sequencing to determine ERBB2 amplification status. Med. Oncol. 38, 1–8 (2021).
Pfarr, N., Penzel, R., Endris, V., Lier, C., Flechtenmacher, C., Volckmar, A-L., et al. Targeted next-generation sequencing enables reliable detection of HER2 (ERBB2) status in breast cancer and provides ancillary information of clinical relevance. Genes Chromosom. Cancer 56, 255–265 (2017).
Robinson, C.L., Harrison, B.T., Ligon, A.H., Dong, F., Maffeis, V., Matulonis, U., et al. Detection of ERBB2 amplification in uterine serous carcinoma by next-generation sequencing: an approach highly concordant with standard assays. Mod. Pathol. 34, 603–612 (2021).
Ross, D.S., Zehir, A., Cheng, D.T., Benayed, R., Nafa, K., Hechtman, J.F., et al. Next-generation assessment of human growth factor receptor 2 (ERBB2) amplification status: clinical validation in the context of a hybrid capture-based, comprehensive solid tumor genomic profiling assay. J. Mol. Diagn. 19, 244–254 (2017).
Haskell, G.T., Liu, Y.J., Chen, H., Chen, B., Meyer, R.G., Yuhas, J.A., et al. Integrated analysis of HER2 copy number by cytogenomic microarray in breast cancers with nonclassical in situ hybridization results. Am. J. Clin. Pathol. 149, 135–147 (2018).
Perez, E.A., Reinholz, M.M., Hillman, D.W., Tenner, K.S., Schroeder, M.J., Davidson, N.E., et al. HER2 and chromosome 17 effect on patient outcome in the N9831 adjuvant trastuzumab trial. J. Clin. Oncol. 28, 4307–4315 (2010).
Ahn, S., Woo, J.W., Lee, K., Park, S.Y. HER2 status in breast cancer: changes in guidelines and complicating factors for interpretation. J. Pathol. Transl. Med. 54, 34–44 (2020).
Schrader, K.A., Cheng, D.T., Joseph, V., Prasad, M., Walsh, M., Zehir, A., et al. Germline variants in targeted tumor sequencing using matched normal DNA. JAMA Oncol. 2, 104–111 (2016).
Seifert, B.A., O’Daniel, J.M., Amin, K., Marchuk, D.S., Patel, N.M., Parker, J.S., et al. Germline analysis from tumor-germline sequencing dyads to identify clinically actionable secondary findings. Clin. Cancer Res. 22, 4087–4094 (2016).
Reid, S., Pal, T. Update on multi-gene panel testing and communication of genetic test results. Breast J. 26, 1513–1519 (2020).
Zack, T.I., Schumacher, S.E., Carter, S.L., Cherniack, A.D., Saksena, G., Tabak, B., et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 45, 1134–40 (2013).
Childers, C.P., Childers, K.K., Maggard-Gibbons, M., Macinko, J. National estimates of genetic testing in women with a history of breast or ovarian cancer. J. Clin. Oncol. 35, 3800–3806 (2017).
Watson, M.S. ACMG policy statement: Updated recommendations regarding analysis and reporting of secondary findings in clinical genome-scale sequencing. Genet. Med. 17, 68–69 (2015).
Green, R.C., Berg, J.S., Grody, W.W., Kalia, S.S., Korf, B.R., Martin, C.L., et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 15, 565–574 (2013).
Garofalo, A., Sholl, L., Reardon, B., Taylor-Weiner, A., Amin-Mansour, A., Miao, D., et al. The impact of tumor profiling approaches and genomic data strategies for cancer precision medicine. Genome Med. 8, 1–10 (2016).
Author information
Authors and Affiliations
Contributions
S.E.S. and F.D. performed study design, development of methodology, data collection, writing, and revision of the paper. D.K.M. and P.K.D. performed data collection and revision of the paper. All authors read and approved the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This project was approved by the Mass General Brigham Human Research Committee, the institutional review board for Brigham and Women’s Hospital, and was conducted in accordance with the Declaration of Helsinki. Informed consent was waived as this research was deemed no more than minimal risk.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Siegmund, S.E., Manning, D.K., Davineni, P.K. et al. Deriving tumor purity from cancer next generation sequencing data: applications for quantitative ERBB2 (HER2) copy number analysis and germline inference of BRCA1 and BRCA2 mutations. Mod Pathol 35, 1458–1467 (2022). https://doi.org/10.1038/s41379-022-01083-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-022-01083-x
