
Abstract
The WHO (2021) Classification classified a group of pediatric-type high-grade gliomas as IDH wildtype, H3 wildtype but as of currently, they are characterized only by negative molecular features of IDH and H3. We recruited 35 cases of pediatric IDH wildtype and H3 wildtype hemispheric glioblastomas. We evaluated them with genome-wide methylation profiling, targeted sequencing, RNAseq, TERT promoter sequencing, and FISH. The median survival of the cohort was 27.6 months. With Capper et al.’s36 methylation groups as a map, the cases were found to be epigenetically heterogeneous and were clustered in proximity or overlay of methylation groups PXA-like (n = 8), LGG-like (n = 10), GBM_MYCN (n = 9), GBM_midline (n = 5), and GBM_RTKIII (n = 3). Histology of the tumors in these groups was not different from regular glioblastomas. Methylation groups were not associated with OS. We were unable to identify groups specifically characterized by EGFR or PDGFRA amplification as proposed by other authors. EGFR, PDGFRA, and MYCN amplifications were not correlated with OS. 4/9 cases of the GBM_MYCN cluster did not show MYCN amplification; the group was also enriched for EGFR amplification (4/9 cases) and the two biomarkers overlapped in two cases. Overall, PDGFRA amplification was found in only four cases and they were not restricted to any groups. Cases in proximity to GBM_midline were all hemispheric and showed loss of H3K27me3 staining. Fusion genes ALK/NTRK/ROS1/MET characteristic of infantile glioblastomas were not identified in 17 cases successfully sequenced. BRAF V600E was only found in the PXA group but CDKN2A deletion could be found in other methylation groups. PXA-like cases did not show PXA histological features similar to findings by other authors. No case showed TERT promoter mutation. Mutations of mismatch repair (MMR) genes were poor prognosticators in single (p ≤ 0.001) but not in multivariate analyses (p = 0.229). MGMT had no survival significance in this cohort. Of the other common biomarkers, only TP53 and ATRX mutations were significant poor prognosticators and only TP53 mutation was significant after multivariate analyses (p = 0.024). We conclude that IDH wildtype, H3 wildtype pediatric hemispheric glioblastomas are molecularly heterogeneous and in routine practice, TP53, ATRX, and MMR status could profitably be screened for risk stratification in laboratories without ready access to methylation profiling.
Similar content being viewed by others


Introduction
Brain tumor is the commonest solid cancer in children and glioblastoma accounts for about 6% of all brain tumors in children1,2. Pediatric glioblastoma has an annual incidence of 1.78 per 100,000 population according to CBTRUS and is the commonest cause of cancer-related death in children3. There is now a lot of literature confirming that pediatric gliomas have different molecular pathological features from adult glioblastomas, in spite of histological similarity. In the new WHO 2021 CNS Tumor Classification, pediatric gliomas are classified separately from adult gliomas and are divided into low-grade and high-grade groups4. Pediatric-type high-grade gliomas are now classified by the new classification into Diffuse midline glioma, H3K27-altered; Diffuse hemispheric glioma, H3 G34-mutant; Diffuse pediatric-type high-grade glioma, H3 wildtype and IDH wildtype; and Infant-type hemispheric glioma [4, https://tumourclassification.iarc.who.int].
It is well known that pediatric gliomas are mostly isocitrate dehydrogenase wildtype. IDH-mutant pediatric gliomas are mostly found in older children or adolescents5,6,7. It is also well known that K27 mutation and G34 mutation of the H3 genes (HIST1H3B and H3F3A) are found in gliomas in the pediatric age group; the former group was mostly found in midline tumors and the latter being found in hemispheric tumors and in an older age group8,9,10,11. For glioblastoma located in the midline, other than the classical H3K27M-altered midline glioma, distinct groups including bithalamic gliomas in children with EGFR amplification or other alterations12,13, or tumors characterized by molecular changes such as mutations of ACVR114 and activation of EZHIP15,16 have been reported. The group of infantile hemispheric gliomas, generally taken as occurring between 1 and 3 years of age, are also characterized by specific fusion genes of ALK/NTRK/ROS1/MET14,17,18 and seem to have a better prognosis than those of the older children19.
The exact incidence of IDH wildtype (wt), H3 wildtype (wt) pediatric-type high-grade glioma is not known but in a clinical trial of non-brainstem pediatric high-grade gliomas in children aged 3–18 years, about 40% were H3 wildtype20. Gliomas arising within the context of constitutional mismatch repair deficiency (cMMRD), Lynch syndrome, Li Fraumeni syndrome or previous cranial irradiation, should be recognized as potential aetiologies for H3wt pediatric high-grade gliomas. In spite of advances in surgical and medical treatments, pediatric glioblastoma remains incurable. Median survival remains as 9–15 months overall for pediatric glioblastomas21,22, five-year survival is less than 20%21,22 and trials of drugs or targeted agents used against adult glioblastomas have not shown a survival benefit23.
H3wt, IDHwt pediatric high-grade gliomas are currently defined by negative mutations for IDH and H3 (H3F3A and HIST1H3B). They are not characterized by specific molecular or histological features. There have only been a few genomic studies of IDHwt and H3wt pediatric glioblastomas. Korshunov et al. studied a series of 87 cases with methylation profiling and proposed they should be divided into three groups enriched by MYCN amplification (pedGBM_MYCN), PDGFRA amplification (pedGBM_RTK1), and EGFR amplification (pedGBM_RTK2)24. In this study, tumors of the pedGBM_RTK2 group were associated with a favorable overall survival (OS), tumors of the pedGBM_RTK1 groups were associated with an intermediate OS, and tumors of the pedGBM_MYCN tumors had the worst clinical outcome. Next-generation sequencing (NGS) however was not performed in this study. So the question may arise whether in routine practice for those laboratories without day-to-day access to methylation profiling, one needs to test for EGFR, PDGFRA, and MYCN amplifications. In another study, Mackay et al. conducted a comprehensive meta-analysis of pediatric high-grade gliomas19. According to their supplementary data, a total of 43 IDHwt, H3wt glioblastomas located in the midline and the hemispheres were examined by methylation and NGS. The authors further examined 38 cases of pediatric glioblastomas from a clinical trial which were IDHwt, H3wt, and BRAFwt and concluded that they were genetically heterogeneous20.
In this study, we aimed to provide a comprehensive molecular landscape for pediatric IDHwt, H3wt glioblastoma by whole genome methylation profiling and NGS including both targeted sequencing and RNAseq and looked for their clinical significance. We examined in this study only hemispheric tumors as pediatric midline tumors have unique molecular pathologic changes like H3K27 alterations, EZHIP activation or EGFR amplification which were extensively studied13,15.
Materials and methods
Tumor samples
We searched cases diagnosed as hemispheric glioblastoma with the age range 1–18 inclusive in four institutions, Prince of Wales Hospital, Chinese University of Hong Kong (1990–2016), Huashan Hospital, Fudan University, Shanghai (2012–2018), First Affiliated Hospital of Zhengzhou University, Zhengzhou (2012–2018) and Children’s Hospital, Guangzhou (2017). The brackets showed the years when the archives were searched. The reason for the differences in years related to when electronic medical records were fully available in the institutions and only one year ‘s archive was available at Guangzhou Children’s Hospital as the neurosurgical unit was new there. Tumors which were diagnosed as anaplastic pleomorphic xanthoastrocytomas (PXA) at the time were not included in this study as their molecular pathology has been extensively covered in the literature25,26,27. High-grade pediatric gliomas located in the midline were also not recruited as they have been extensively studied12,13,15,16. Seventy-one glioblastomas were thus retrieved and genotyped for hotspot mutations of IDH1 and IDH2 and H3.3 (H3F3A) and H3.1 (HIST1H3B). Twenty cases were excluded from this study due to the presence of IDH1/2, H3.3, or H3.1 mutations. Out of the remaining 51 cases, 39 cases were subjected to methylation profiling. 12 cases were failed in quality control test. Methylation results suggested diagnoses other than glioblastomas in four cases which were subsequently confirmed and they were removed from further studies. So, a total of 35 cases of pediatric IDHwt, H3wt hemispheric glioblastomas were recruited for detailed analysis.
Histology of all selected cases was reviewed (HC and HKN) to confirm histological features of glioblastoma as per WHO 2016 Classification, namely mitoses, cellular atypia, microvascular proliferation, and necrosis1 The approach to gliomas in all three hospitals was maximal safe resection in the first instance. Ethics approvals were obtained from The Joint Chinese University of Hong Kong—New Territories East Cluster Clinical Research Ethics Committee, the Ethics Committees of Huashan Hospital, Shanghai, The First Affiliated Hospital of Zhengzhou University, Zhengzhou and the Children’s Hospital, Guangzhou. Data on patient demographics and therapeutic treatment was retrieved from institutional paper and electronic records. Survival data, for both OS and progression-free survival (PFS), were obtained from follow-up clinic visits and direct contact with patients or close relatives via phone as per our previous studies28,29,30.
IDH1, IDH2, H3.3 (H3F3A), H3.1 (HIST1H3B), and TERT promoter mutation analysis
Sanger sequencing was employed to confirm that all cases carried the wildtype for IDH1, IDH2, H3.3, and H3.131,32,33. Similarly, Sanger sequencing was done to detect hotspot mutations (C228T and C250T) at the promoter of the gene TERT (telomerase reverse transcriptase)34,35. In brief, formalin-fixed paraffin-embedded (FFPE) sections were dewaxed in xylene and rehydrated in graded alcohol. Tissues were then collected and immersed into Tris-EDTA buffer supplemented with proteinase K. The lysates were incubated at 56 °C overnight followed by heat inactivation at 95 °C for 5 min. DNA from the lysate was amplified using forward primer, reverse primer, KAPA Robust HotStart ReadyMix (Sigma)/ KAPA HiFi HotStart ReadyMix (Sigma). PCR products were visualized on electrophoresis gel, cleaned with a spin column-based PCR product purification kit (iNtRON Biotechnology) and sequenced with BigDye Terminator Cycle Sequencing kit v1.1 (Life Technologies). Primer sequencing and PCR conditions can be found in previous publications31,33,35.
Illumina infinium methylation EPIC BeadChip array
FFPE sections were sent to Sinotech Genomics Co., Ltd, Shanghai, where the DNA was extracted, bisulfite modified, and subjected to DNA-methylation profiling by EPIC Illumina Infinium Human (850k) array. Background correction, global dye-bias normalization, and calculation of DNA-methylation level were performed according to the previous publications36. The most variable probes were used to generate t-SNE plot (t-distributed stochastic neighbor embedding) with Rtsne R package v0.1336. MGMT promoter methylation status was determined by R package MGMT-STP2737.
To evaluate copy number variations (CNVs), probe-level signal intensities were subjected to background correction and dye-bias normalization. Probes targeting the sex chromosomes, containing single-nucleotide polymorphisms, or mapping to multiple locations in the human genome were excluded to avoid inaccurate assessment. Two sets of 50 control samples derived from male and female donors were used for normalization36. CNV analysis was performed using the ‘conumee’ R package in Bioconductor (http://www.bioconductor.org/packages/release/bioc/html/conumee.html). A log2 ratio ±0.35 was used as the cutoff of amplification/loss and a log2 ratio −0.415 was used as the cutoff for homozygous loss. GISTIC v2.0 analysis was conducted to identify significantly recurrent copy number amplification and deletions at arm-level and focal-level, defined as affected regions spanning less than 50% of a chromosome arm.
Targeted sequencing
DNA was obtained from FFPE sections using GeneRead DNA FFPE kit (Qiagen). The quality and quantity of extracted DNA were evaluated by QIAseq DNA QuantiMIZE Assay (Qiagen). Samples that passed quality control were subjected to library preparation with a custom QIAseq Targeted DNA Panel, covering either the coding exons or hotspot mutations of 74 genes altered in gliomas and other CNS tumors. The DNA libraries were then further assessed for quality and quantity before the libraries were sequenced with MiSeq v3 (Illumina).
Paired-end reads were aligned to the hg19 (GRCh37) build of the human reference genome with BWA-MEM algorithm on GeneGlobe platform (Qiagen). Variants were called using smCounter238 and annotated using wANNOVAR39. Variants with the following criteria were excluded: not passing quality filters, with variant allele fractions of ≤5%, with variant allele counts of ≤5, or with minor allele frequencies of >1% in overall human population or East Asians or documented in public databases (1000 Genomes, ExAc, gnomAD exome and genome databases).
Targeted RNA sequencing
Total RNA was extracted from FFPE sections using RNeasy FFPE kit (Qiagen). RNA passing quality control examination was converted into cDNA, and libraries were then generated using TruSight RNA Pan-Cancer Target Genes (Illumina), which designed to detect 1385 cancer-related genes. Paired-end reads were aligned to human genome assembly GRCh37 (hg19) and fusion genes were called using the STAR aligner and STAR fusion caller40.
Immunohistochemistry for H3K27me3 expression
H3K27me3 was detected according to the previous study41. In brief, 4 µm thickness FFPE tissue sections were dewaxed in xylene and rehydrated in graded alcohol. Immunohistochemical staining was done in BenchMark ULTRA automated tissue staining system (Ventana Medical Systems). Sections were incubated with the rabbit polyclonal anti-H3K27me3 antibody (1:100, C36B11, Cell Signaling). Immunostaining was detected with the OptiView DAB IHC Detection Kit (Ventana Medical Systems). Non-tumor cells such as endothelial cells and immune cells served as internal positive controls. Sections were scored positive when more than 80% tumor nuclei were stained positive for H3K27me342.
Fluorescence in-situ hybridization (FISH) analysis
MYCN amplification, EGFR amplification, and PDGFRA amplification were evaluated by FISH on selective cases to confirm amplification as revealed by methylation array. The BAC clone (RP11-355H10) containing the genomic sequences of chromosome 2p24 and the centromere probe (CEP 2 (D2Z1), Vysis) were used for MYCN amplification detection. The BAC clone (CTD-2199A14) and the centromere probe (CEP7, Vysis) were used for EGFR amplification detection. Probes of PDGFRA (CTD-2054G11 and RP11-231C18) and the centromere probe (CEP 4, Vysis) were employed to detected PDGFRA amplification. The target probe for EGFR and PDGFRA were labeled with Spectrum Orange and the reference probe was labeled with Spectrum Green. In the case of MYCN amplification, the target probe was labeled with Spectrum Green and the reference probe was labeled with Spectrum Orange.
In brief, FFPE sections were dewaxed by xylene, heated in 1 M sodium thiocyanate for 10 min at 80 °C and digested with pepsin at 37 °C for 20–30 min. Sections were then rinsed in molecular biology grade water to stop the digestion and dehydrated by heating. Locus-specific probe was then denatured at 80 °C for 10 min followed by incubation on slides for 16 h at 37 °C. On the next day, sections were washed twice in 1.5 M Urea/2× saline sodium citrate at 50 °C for 10 min, and stained with Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (Vector Laboratories). Fluorescent signals were visualized under a fluorescent microscope (Carl Zeiss).
At least 100 non-overlapping signals were counted and analyzed in each case. Amplifications were considered when clusters were appeared or >5% tumor cells displayed a ratio of target to reference signal >243.
Statistical analysis
Statistical analysis was performed on IBM SPSS software. OS was defined as the period of time between operation and death or the last follow-up. PFS was defined as the time between operation and recurrence as revealed by neuroradiological finding. Chi-square or Fisher’s test was applied to determine correlation between molecular alterations and clinical parameters and between different molecular alterations. Survival curves were evaluated by the Kaplan–Meier (KM) method, and log-rank test was done to compare survival distribution between groups. Multivariate analysis was performed by Cox proportional hazards model. P < 0.05 (two-sided) was considered statistically significant.
Results
Clinical features of the cohort
All tumors were wildtype for IDH1/2, H3.1 (HIST1H3B), and H3.3 (H3F3A) by Sanger sequencing. The clinical features of the 35 cases in this cohort are shown in Table 1. Male and female ratio was 1.06: 1. The mean and median ages at diagnosis were 12.2 and 13 years old (range 1–18), respectively. Only one patient was below three years old. Most tumors were located in the frontal lobes (n = 15). Ten cases were found in the temporal lobes and 8 and 2 cases were found in the parietal and occipital lobes respectively. Data of OS and PFS were available in 34 patients (97.1%) and 23 patients (65.7%) respectively. 27 patients had total resection. Radiotherapy and chemotherapy were given to 22 and 21 patients respectively. The mean and median OS of this cohort were 40.6 and 27.6 months, respectively. These appeared to be longer than those of adult IDHwt glioblastomas (TCGA data). Univariate Cox proportional hazard analyses were performed, and none of the clinical parameters, including sex, age, resection, chemotherapy and radiotherapy, was associated with OS and PFS (Supplementary Table 1).
Pediatric hemispheric IDHwt, H3wt glioblastomas were epigenetically heterogeneous
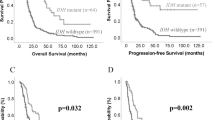
Using the topmost differentially methylated probes based on the Capper et al. publication36, we conducted t-SNE dimensionality reduction analysis. As shown in Fig. 1 and Table 1, overall, cases in the cohort were epigenetically heterogeneous, and were clustered in proximity or overlay of methylation clusters PXA (n = 8), LGG groups (n = 10), GBM_MYCN (n = 9), GBM_midline (n = 5), and GBM_RTKIII (n = 3). Cases assigned to GBM_MYCN, GBM_RTKIII directly overlaid the reference cases. Cases assigned to GBM_midline and PXA clustered well but the cases were only in proximity to the reference groups. Ten cases were not clustered well and could only be clustered in proximity to three LGG groups: LGG_DNT, LGG_GG, and LGG_SEGA. We named these case hereafter in this paper as “LGG-like” and “PXA-like” respectively for convenience. There was no correlation between methylation groups and clinical variations (Table 1). Survival analysis revealed that methylation classes had no impact on OS and PFS (Fig. 2A–D) though there was a trend for a better prognosis with the LGG/PXA-like cases. We have attempted using other means of clustering, e.g., k-means test, but the results were not satisfactory (data not shown).

Reference cases are indicated as colored circles. Each reference methylation cluster is indicated by a single color. Tumors in this study cohort are indicated as red triangles. PXA tumors were renamed to “PXA-like”, and tumors clustered in proximity to the reference groups LGG_GG, LGG_DNT, and LGG_SEGA were classified into “LGG-like” tumors.

Methylation groups have no impact on (A, C) OS and (B, D) PFS.
Histologically, we were unable to find typical features of PXA (pleomorphic xanthoastrocytoma) or epitheloid glioblastomas in the cases assigned to PXA-like (Supplementary Fig. 1). It should be noted that similar assignments of histologically non-PXA pediatric gliomas to the methylation group PXA were noted by other authors8,36,44,45,46 and that the Capper et al. classification (version 11b4) currently did not separate between regular PXA and anaplastic PXA. Similarly, cases assigned to LGG-like (low-grade gliomas) groups, as depicted in Supplementary Fig. 2 showed histologically usual features of glioblastomas similar to the experience of other authors8. All five cases assigned to GBM_midline were actually hemispheric as all cases in the whole cohort were hemispheric (Supplementary Fig. 3). Histologically, cases assigned to GBM_MYCN were also regular glioblastomas without features of embryonal tumors or “PNET” (Supplementary Fig. 4). Interestingly, all five cases assigned to GBM_midline showed loss of H3K27me3 staining with immunohistochemistry (Supplementary Fig. 5).
Copy number variations and MGMT methylation
CNVs together with the clinical and molecular details in this cohort is depicted in Fig. 3. CNVs in our cohort were derived from DNA-methylation profiles according to previous study36. We employed GISTIC analysis to identify statistically significant recurrent amplifications and losses at arm-level and focal-level. At arm-level, we found in the whole cohort significant amplification at chromosomes 7p, 7q, 20p, and 20q and significant loss at chromosome 10q (Supplementary Fig. 6; Supplementary Table 2; q < 0.05). At focal level, we identified significant amplification at chromosome region 7p11.2 where EGFR is located and losses at chromosome regions 15q11.2 and 19q13.42 (Supplementary Fig. 7; q < 0.05). Whole chromosome 7 gain/amplification and whole chromosome 10 loss were found in 8 (22.9%) and 4 (11.4%) cases respectively. No case showed a combined whole chromosome 7+/10− as per adult IDHwt glioblastomas47. Chromosome 7 or 10 alterations were not associated with clinical outcomes (Supplementary Table 2). Chromosome 1p (n = 3) and 1q (n = 3) amplifications were associated with poorer OS. Similarly, loss of chromosome 9p was correlated with a shorter OS (Supplementary Table 2) but the numbers for all these abnormalities were small.

OncoPrint illustrates the distribution of molecular alterations in pediatric hemispheric glioblastomas. Mutations are shown as green. Amplifications and losses are shown as red and blue respectively.
Next, we determined the prevalence of copy number changes at gene level as per Shirahata et al.48. EGFR, PDGFRA and MYCN amplifications were observed in 25.7%, 11.4% and 17.1%, respectively (Supplementary Table 3). EGFR, PDGFRA, and MYCN amplifications were validated by FISH in 9/9, 3/4, and 3/6 of amplified tumors respectively (Supplementary Fig. 8). The remaining cases could not be further evaluated due to a lack of tissues. CDK4 amplification (n = 11) and CDKN2B loss (n = 11) were common and present in around one-third of our hemispheric tumors (Supplementary Table 3). Other common alterations included CCND2 amplification, MET amplification and CDKN2A loss (Supplementary Table 3). CDKN2A loss was found in all methylation groups and was not unique to cases assigned to PXA-like (Fig. 3). CNVs at gene level were not correlated with clinical outcomes (Supplementary Table 3) including CDKN2A/B loss. MGMT methylation was identified only in five cases (14.3%), a frequency lower than the regular adult glioblastomas49 and it had no prognostic significance (Supplementary Fig. 9).
Association between CNVs and methylation classes
As expected, MYCN amplification was significantly associated with the GBM_MYCN group (p = 0.008; Supplementary Table 4). MYCN amplification was confirmed by FISH in three cases with sufficient tissue materials. EGFR amplification (4/9 cases) was also common in GBM_MYCN and two cases in this group harbored both molecular alterations (Fig. 3).
EGFR amplification was found in cases assigned to LGG-like group (3/10 cases) and GBM_midline cluster (1/5 cases). In the whole cohort, PDGFRA amplification was found in only four cases (11.4%) and they were found in different methylation groups. So we were unable to identify methylation groups specifically characterized by EGFR or PDGFRA amplification as proposed by other authors24. EGFR, MYCN and PDGFRA amplifications were all not correlated with clinical outcomes (Supplementary Table 3).
Amplification of chromosome 20p was associated with GBM_MYCN class (p = 0.019; Supplementary Table 5) where it was found in >50% of the cases. Similarly, amplification of chromosome 20q was associated with GBM_MYCN group (p = 0.019) and chromosome 15q loss was associated with GBM_midline group (p = 0.006).
TP53, ATRX and MMR mutations were poor prognosticators
We then conducted target sequencing to reveal the mutational profiling of 74 genes relevant to gliomas (Supplementary Table 6). The list of mutations identified in this cohort is shown in Supplementary Table 7. The mutational landscape of this study is also depicted in Fig. 3. None of our cases carried TERT promoter mutation.
We detected on average 14.94 ± 29.55 mutations (range 1–121) per sample. The majority of cases (27/35; 77%) showed few mutations (≤10 mutations); however, 8 tumors had more than 10 mutations with three tumors carrying >100 mutations. All three of these cases carried mutations in the mutations of the mismatch repair (MMR) genes (MSH2, MSH6, MLH1 and PMS2). Tumors carrying mutations of the MMR genes had significantly higher number of mutations (p = 0.001; 39.70 ± 48.10 vs 5.04 ± 3.41 mutations/sample). Overall, 10 cases showed MMR protein genes. We evaluated carefully the history of the 10 cases and could not find any history of any other cancers or tumors. We then separated tumors into two groups based on mutation counts. Samples with mutation count ≥15 were classified into “high mutation count” and those <15 were classified into “low mutation count”. We detected a trend towards shorter OS in high mutation samples (p = 0.068; Supplementary Fig. 10). We performed immunohistochemistry on selected cases with enough tissue materials to confirm MMR mutations (Supplementary Fig. 11).
The gene with most frequent mutations in this study was TP53 (n = 17; 48.6%; Supplementary Table 8). Other frequent mutations included NF1 (34.3%), PDGFRA (28.6%) and ROS1 (25.7%). NF1 mutation found across all methylation groups. We evaluated carefully the clinical history of all cases with NF1 mutations but could not find any case with documented clinical manifestation of the NF1 syndrome. All five BRAF V600E mutations were found in cases assigned to PXA-like group (p = 0.001).
For clinical correlation, TP53 mutation was significantly associated with shorter OS (p = 0.001; Fig. 4A) and PFS (p = 0.040; Fig. 4B). ATRX mutation was also a negative prognosticator and associated with shorter OS (p = 0.022; Fig. 5A) and PFS (p = 0.047; Fig. 5B). MMR genes were the poor prognosticators for OS (p = 0.007; Fig. 6A) and PFS (p = 0.039; Fig. 6B) but significance was lost in multivariate analyses (Table 2). In multivariate analysis after adjusting age, sex, extent of resection, chemotherapy, and radiotherapy, TP53 mutation remained as an independent molecular prognosticator for OS (p = 0.024) and showed a trend towards significance for PFS (Table 2). When TP53 and ATRX mutation status was combined, tumors carrying ATRX and/or TP53 mutations also had a significant shorter OS and PFS (Supplementary Fig. 12A, B).

Pediatric glioblastomas harboring TP53 mutations had a significant shorter (A) OS and (B) PFS.

(A) OS and (B) PFS curves for ATRX mutation. ATRX mutation was associated with a shorter OS and PFS.

IDHwt, H3wt pediatric hemispheric glioblastomas with mutations in the MMR genes (MSH2, MSH6, MLH1 and PMS2) had a significantly shorter (A) OS and (B) PFS.
Fusion genes found in infantile glioblastomas were absent
We carried out RNAseq designed to detect fusions of 1385 cancer-related genes. Seventeen of 35 samples passed the QC to proceed library preparation. Two fusion genes were detected. A MN1-PATZ1 fusion gene predicted to be in-frame was found in an 8-year-old girl with the tumor classified as GBM_MYCN. This fusion was previously also reported in gliomas and other brain tumors50,51. The patient was still alive with a survival of 65.8 months. The other predicted to be in-frame fusion gene was detected in a 16-year-old boy, and it was a fusion between the genes ZC3H7B and ARFGAP3. The patient was alive with a survival of 26.5 months. Fusion genes ALK/NTRK/ROS1/MET characteristic of infantile glioblastomas were not identified17.
Discussion
Korshunov et al. found that some cases diagnosed as pediatric glioblastomas epigenetically fell into PXA or low-grade glioma groups though histologically they might not look like PXA or LGG8. As indicated in the supplementary data of this study, 40 pediatric glioblastomas were thus classified and they were IDHwt and H3wt. In a follow-up paper, after excluding those cases which were classified epigenetically as PXA or low-grade gliomas, this group of authors determined DNA-methylation profiles in 87 IDHwt, H3wt pediatric glioblastomas24. They classified them into three methylation groups by amplifications of MYCN, PDGFRA, and EGFR (which the authors termed pedGBM_MYCN, pedGBM_RTK1, and pedGBM_RTK2, respectively). They found the group enriched by EGFR amplification had the better prognosis, the group enriched by MYCN amplification the worse prognosis and the group by PDGFRA amplification an intermediate prognosis. In our study, we used the commonly used Capper plot for epigenetic clustering for brain tumors and clustered this cohort of IDHwt, H3wt hemispheric glioblastomas into PXA-like, LGG-like, GBM_MYCN, GBM_midline, and GBM_RTKIII groups using Capper et al.’s terminology (version 11b4). The clustering was not perfect as described under Results.
In our cohort, EGFR amplification was found in GBM_MYCN cluster (4/9) and also in LGG-like (3/10 cases), and GBM_midline cluster (1/5 cases). MYCN and EGFR amplifications overlapped in 2 cases. Four out of 9 cases of GBM_MYCN did not show MYCN amplification. PDGFRA amplification was only found in 4/35 cases (11.4%) and they were found in different methylation groups. None of MYCN, EGFR or PDGFRA amplification was a prognosticator in our cohort. So overall we were unable to identify distinct methylation groups characterized specifically by EGFR or PDGFRA amplification using the Capper et al. plot. We appreciate that there are other means of epigenetic clustering but Capper et al.’s plot is the one most commonly referenced and many of its methylation groups have been written into WHO 2021 Classification as desirable diagnostic criteria4.
In this cohort, there was no significant survival difference among the clusters but LGG/PXA-like cases had a trend towards a better prognosis than the other groups. The lack of survival difference could be attributed to the relative smaller sample size of PXA-like and LGG-like in our cohort (18 cases). One should also be aware that methylation array is not available to most pathology laboratories in the world due to cost and lack of expertise in interpretation, e.g., tSNE and this may restrict their use in routine diagnoses. BRAF V600E was specific to the PXA-like tumors in this cohort and there might be consideration for its regular testing, which is available in many pathology laboratories, as target therapy may be considered52. The PXA-like cases did not have the typical histological appearance of PXA and were not epitheloid glioblastomas and histologically were indistinguishable from regular glioblastomas. But it is well known in the literature that epigenetic PXA is not equivalent to histological PXA8,36,44,45,46 and the Capper plot does not have a group of anaplastic PXA. Similarly, the LGG-like cases in our cohort showed regular features of glioblastomas and may not be distinguishable by histology. A similar experience was also reported by other authors8,19.
In this cohort, 5/9 cases the GBM_MYCN showed MYCN amplification and one case of MYCN amplification was found in GBM_midline. Mackay et al. examined 38 cases of IDHwt, BRAFwt, H3wt pediatric glioblastomas from a clinical trial20. The commonest methylation group in their cohort was GBM_MYCN but only one of their group MYCN showed MYCN amplification. MYCN is also known to be upregulated in H3.3 G34R/V pediatric gliomas and therefore caution should be exercised in the interpretation of MYCN status in pediatric gliomas53. Interestingly, pediatric glioblastomas with MYCN amplification has also been associated with “CNS-PNET” in the literature44,54 and adult glioblastoma with features of PNET are known to show MYCN amplification55. However, we found that our cases with MYCN amplification showed histological features of regular glioblastoma instead of embryonal tumors.
GBM_midline may actually be defining a group of tumors with similar epigenetic rather than clinical features. In our cohort, none of the tumors was located midline. Similar findings of a non-midline location of pediatric gliomas clustered to this group were reported by Mackay et al.19. Consideration of an alternate name for this methylation cluster should be made for better communication with clinicians and patients. Interestingly, our GBM_midline tumors, although not midline-located, shared with midline gliomas the interesting property of loss of H3K27me356. Also similar to the truly midline pediatric glioblastomas, this group of GBM_midline tumors was enriched with TP53 mutation (5/5 cases)56,57,58,59. GBM-RTKIII is similar to the cytogenetic abnormalities of adult GBM. The prognosis is poor similar to the GBM_midline and GBM_MYCN but the number is too small (n = 3) for further analyses.
Gliomas may exhibit MMR genes and loss of their staining immunohistochemically60,61,62. These tumors may be associated with Lynch syndrome in the older population, and in the pediatric patients, consideration should also be given to cMMRD syndrome63 which we were unable to find any hint in the history of the 10 cases with mutations of the MMR genes. Interestingly, mutations of MMR in our cohort were associated with a poor prognosis though this was not significant in multivariate analyses. They were also associated with a genomic pattern suggestive of a hypermutation state. In adults, MMR deficient IDH-mutant astrocytomas also have a poor prognosis64,65.
Fusion genes of ALK/NTRK/ROS1/MET, where potential target therapy may be available were found in infantile glioblastomas17,18,66. They were not identified in this cohort of which only one patient was an infant. It should be noted that we were only able to successfully perform RNA sequencing in 17 cases of the cohort. RNAseq was not conducted in previous studies on IDHwt, H3wt pediatric glioblastomas.
In conclusion, we showed in this study that pediatric IDHwt, H3wt glioblastomas are very heterogeneous as regards to their molecular and epigenetic landscapes. TP53 and ATRX mutations and MMR mutations were associated with adverse outcomes and TP53 was the only biomarker with multivariate significance. As methylation array is not universally available to most pathology centers due to the cost incurred and the clinical significance of the methylation groups is still unclear, we propose that it will be more useful to risk-stratify pediatric hemispheric IDHwt, H3wt glioblastomas by TP53, MMR, and ATRX mutations. In regular diagnostic practice, immunohistochemistry has been shown to be a reliable method for screening for TP53, MMR, and ATRX mutations by other authors63,67,68.
Data availability
The datasets generated during the current study are available at http://www.acp.cuhk.edu.hk/hkng/.
References
Louis, D. N. et al. Glioblastoma, IDH-wildtype. In WHO Classification of Tumours of the Central Nervous System (eds Louis, D. N. et al.) 28–45 (IRAC, Lyon, 2016).
Ostrom, Q. T. et al. Alex’s Lemonade stand foundation infant and childhood primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol. 16, x1–x36 (2015).
Ostrom, Q. T. et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro Oncol. 20, iv1–iv86 (2018).
Louis, D. N. et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 23, 1231–1251 (2021).
Antonelli, M. et al. Prognostic significance of histological grading, p53 status, YKL-40 expression, and IDH1 mutations in pediatric high-grade gliomas. J. Neurooncol. 99, 209–215 (2010).
Balss, J. et al. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 116, 597–602 (2008).
Ryall, S. et al. Integrated molecular and clinical analysis of 1,000 pediatric low-grade gliomas. Cancer Cell. 37, 569–583.e565 (2020).
Korshunov, A. et al. Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol. 129, 669–678 (2015).
Korshunov, A. et al. Histologically distinct neuroepithelial tumors with histone 3 G34 mutation are molecularly similar and comprise a single nosologic entity. Acta Neuropathol. 131, 137–146 (2016).
Lim, K. Y. et al. H3 G34-mutant high-grade glioma. Brain Tumor Pathol. 38, 4–13 (2021).
Picart, T. et al. Characteristics of diffuse hemispheric gliomas, H3 G34-mutant in adults. Neurooncol. Adv. 3, vdab061 (2021).
Mondal, G. et al. Pediatric bithalamic gliomas have a distinct epigenetic signature and frequent EGFR exon 20 insertions resulting in potential sensitivity to targeted kinase inhibition. Acta Neuropathol. 139, 1071–1088 (2020).
Sievers, P. et al. A subset of pediatric-type thalamic gliomas share a distinct DNA methylation profile, H3K27me3 loss and frequent alteration of EGFR. Neuro Oncol. 23, 34–43 (2021).
Wu, G. et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 46, 444–450 (2014).
Castel, D. et al. Histone H3 wild-type DIPG/DMG overexpressing EZHIP extend the spectrum diffuse midline gliomas with PRC2 inhibition beyond H3-K27M mutation. Acta Neuropathol. 139, 1109–1113 (2020).
Pratt, D. et al. Diffuse intrinsic pontine glioma-like tumor with EZHIP expression and molecular features of PFA ependymoma. Acta Neuropathol. Commun. 8, 37 (2020).
Guerreiro Stucklin, A. S. et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat. Commun. 10, 4343 (2019).
Clarke, M. et al. Infant high-grade gliomas comprise multiple subgroups characterized by novel targetable gene fusions and favorable outcomes. Cancer Discov. 10, 942–963 (2020).
Mackay, A. et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 32, 520–537.e5 (2017).
Mackay, A. et al. Molecular, pathological, radiological, and immune profiling of non-brainstem pediatric high-grade glioma from the HERBY phase II randomized trial. Cancer Cell 33, 829–842 (2018).
Adams, H. et al. Evaluating extent of resection in pediatric glioblastoma: a multiple propensity score-adjusted population-based analysis. Childs Nerv. Syst. 32, 493–503 (2016).
Gottardo, N. G. & Gajjar, A. Chemotherapy for malignant brain tumors of childhood. J. Child Neurol. 23, 1149–1159 (2008).
Jones, C. et al. Pediatric high-grade glioma: biologically and clinically in need of new thinking. Neuro Oncol. 19, 153–161 (2017).
Korshunov, A. et al. H3-/IDH-wild type pediatric glioblastoma is comprised of molecularly and prognostically distinct subtypes with associated oncogenic drivers. Acta Neuropathol. 134, 507–516 (2017).
Hosono, J. et al. Role of a promoter mutation in TERT in malignant transformation of pleomorphic Xanthoastrocytoma. World Neurosurg. 126, 624–630 (2019).
Phillips, J. J. et al. The genetic landscape of anaplastic pleomorphic xanthoastrocytoma. Brain Pathol. 29, 85–96 (2019).
Vaubel, R. et al. Biology and grading of pleomorphic xanthoastrocytoma-what have we learned about it? Brain Pathol. 31, 20–32 (2021).
Wong, Q. H. et al. Molecular landscape of IDH-mutant primary astrocytoma Grade IV/glioblastomas. Mod. Pathol. 34, 1245–1260 (2021).
Wong, G. C. et al. Clinical and mutational profiles of adult medulloblastoma groups. Acta Neuropathol. Commun. 8, 191 (2020).
Li, K. K. et al. Identification of subsets of IDH-mutant glioblastomas with distinct epigenetic and copy number alterations and stratified clinical risks. Neurooncol. Adv. 1, vdz015 (2019).
Yang, R. R. et al. Pediatric low-grade gliomas can be molecularly stratified for risk. Acta Neuropathol. 136, 641–655 (2018).
Zhang, Z. et al. Glioma groups classified by IDH and TERT promoter mutations remain stable among primary and recurrent gliomas. Neuro Oncol. 19, 1008–1010 (2017).
Zhang, R. Q. et al. Biomarker-based prognostic stratification of young adult glioblastoma. Oncotarget. 7, 5030–5041 (2016).
Aibaidula, A. et al. Adult IDH wild-type lower-grade gliomas should be further stratified. Neuro Oncol. 19, 1327–1337 (2017).
Chan, A. K. et al. TERT promoter mutations contribute to subset prognostication of lower-grade gliomas. Mod Pathol. 28, 177–186 (2015).
Capper, D. et al. DNA methylation-based classification of central nervous system tumours. Nature. 555, 469–474 (2018).
Bady, P., Delorenzi, M. & Hegi, M. E. Sensitivity analysis of the MGMT-STP27 model and impact of genetic and epigenetic context to predict the MGMT methylation status in gliomas and other tumors. J. Mol. Diagn. 18, 350–361 (2016).
Xu, C. et al. smCounter2: an accurate low-frequency variant caller for targeted sequencing data with unique molecular identifiers. Bioinformatics 35, 1299–1309 (2019).
Yang, H. & Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 10, 1556–1566 (2015).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29, 15–21 (2013).
Venneti, S. et al. Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of Zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathol. 23, 558–564 (2013).
Panwalkar, P. et al. Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol. 134, 705–714 (2017).
Horbinski, C., Miller, R. & Perry, A. Gone FISHing: clinical lessons learned in brain tumor molecular diagnostics over the last decade. Brain Pathol. 21, 57–73 (2011).
Sturm, D. et al. New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell. 164, 1060–1072 (2016).
Boisseau, W. et al. Molecular profiling reclassifies adult astroblastoma into known and clinically distinct tumor entities with frequent mitogen-activated protein kinase pathway alterations. Oncologist. 24, 1584–1592 (2019).
Lehman, N. L. et al. Genomic analysis demonstrates that histologically-defined astroblastomas are molecularly heterogeneous and that tumors with MN1 rearrangement exhibit the most favorable prognosis. Acta Neuropathol. Commun. 7, 42 (2019).
Stichel, D. et al. Distribution of EGFR amplification, combined chromosome 7 gain and chromosome 10 loss, and TERT promoter mutation in brain tumors and their potential for the reclassification of IDHwt astrocytoma to glioblastoma. Acta Neuropathol. 136, 793–803 (2018).
Shirahata, M. et al. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol. 136, 153–166 (2018).
Brennan, C. W. et al. The somatic genomic landscape of glioblastoma. Cell. 155, 462–477 (2013).
Burel-Vandenbos, F. et al. A polyphenotypic malignant paediatric brain tumour presenting a MN1-PATZ1 fusion, no epigenetic similarities with CNS High-Grade Neuroepithelial Tumour with MN1 Alteration (CNS HGNET-MN1) and related to PATZ1-fused sarcomas. Neuropathol. Appl. Neurobiol. 46, 506–509 (2020).
Zschernack, V. et al. Supratentorial ependymoma in childhood: more than just RELA or YAP. Acta Neuropathol. 141, 455–466 (2021).
Nobre, L. et al. Outcomes of BRAF V600E pediatric gliomas treated with targeted BRAF inhibition. JCO Precis. Oncol. 4, PO.19.00298 (2020).
Bjerke, L. et al. Histone H3.3. mutations drive pediatric glioblastoma through upregulation of MYCN. Cancer Discov. 3, 512–519 (2013).
Korshunov, A. et al. Molecular analysis of pediatric CNS-PNET revealed nosologic heterogeneity and potent diagnostic markers for CNS neuroblastoma with FOXR2-activation. Acta Neuropathol. Commun. 9, 20 (2021).
Perry, A. et al. Malignant gliomas with primitive neuroectodermal tumor-like components: a clinicopathologic and genetic study of 53 cases. Brain Pathol. 19, 81–90 (2009).
Dufour, C. et al. Identification of prognostic markers in diffuse midline gliomas H3K27M-mutant. Brain Pathol. 30, 179–190 (2020).
Solomon, D. A. et al. Diffuse midline gliomas with histone H3-K27M mutation: a series of 47 cases assessing the spectrum of morphologic variation and associated genetic alterations. Brain Pathol. 26, 569–580 (2016).
Buczkowicz, P. et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 46, 451–456 (2014).
Castel, D. et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 130, 815–827 (2015).
Caccese, M. et al. Mismatch-repair protein expression in high-grade gliomas: a large retrospective multicenter study. Int. J. Mol. Sci. 21, 6716 (2020).
Johnson, A. et al. Comprehensive genomic profiling of 282 pediatric low- and high-grade gliomas reveals genomic drivers, tumor mutational burden, and hypermutation signatures. Oncologist. 22, 1478–1490 (2017).
Indraccolo, S. et al. Genetic, epigenetic, and immunologic profiling of MMR-Deficient Relapsed Glioblastoma. Clin. Cancer Res. 25, 1828–1837 (2019).
Kim, B., Tabori, U. & Hawkins, C. An update on the CNS manifestations of brain tumor polyposis syndromes. Acta Neuropathol. 139, 703–715 (2020).
Suwala, A. K. et al. Primary mismatch repair deficient IDH-mutant astrocytoma (PMMRDIA) is a distinct type with a poor prognosis. Acta Neuropathol. 141, 85–100 (2021).
Yang, R. R. et al. Mismatch repair proteins PMS2 and MLH1 can further refine molecular stratification of IDH-mutant lower grade astrocytomas. Clin. Neurol. Neurosurg. 208, 106882 (2021).
Perreault, S. et al. Canadian consensus for biomarker testing and treatment of TRK Fusion Cancer in Pediatric Patients. Curr. Oncol. 28, 346–366 (2021).
Takami, H. et al. Revisiting TP53 mutations and immunohistochemistry–a comparative study in 157 diffuse gliomas. Brain Pathol. 25, 256–265 (2015).
Tanboon, J., Williams, E. A. & Louis, D. N. The diagnostic use of immunohistochemical surrogates for signature molecular genetic alterations in gliomas. J. Neuropathol. Exp. Neurol. 75, 4–18 (2016).
Author contributions
LH, ZFS, KKL, ZYZ, FCL, YM, and HKN designed the study. LH, WWW, RRY, HC, XZL, DTC, WCL, and ZYZ participated in data acquisition. LH, KKL, JSK, and HKN analyzed the methylation and NGS data. HKN was the major contributor in manuscript writing. All authors read and approved the final manuscript.
Funding
This study was supported by the Health and Medical Research Fund (HMRF), the Food and Health Bureau of Hong Kong (reference number: 07180736); Children Cancer Foundation; National Natural Science Foundation of China (reference number 82072020, 81702465 and U1804172 and U1904148); and, Shanghai Municipal Science and Technology Major Project, China (reference number 2018SHZDZX01).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This study was approved by The Joint Chinese University of Hong Kong - New Territories East Cluster Clinical Research Ethics Committee, the Ethics Committees of Huashan Hospital, Shanghai, The First Affiliated Hospital of Zhengzhou University, Zhengzhou and the Children’s Hospital, Guangzhou.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Hong, L., Shi, ZF., Li, K.KW. et al. Molecular landscape of pediatric type IDH wildtype, H3 wildtype hemispheric glioblastomas. Lab Invest 102, 731–740 (2022). https://doi.org/10.1038/s41374-022-00769-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41374-022-00769-9
