
Abstract
Cancer is one of the leading causes of death worldwide. During the last few decades prognosis has improved dramatically and patients are living longer and suffering long-term cardiovascular consequences of chemotherapeutic agents. Cardiovascular disease is a leading cause of morbidity and mortality in cancer survivors second only to recurrent cancer. In some types of cancer, cardiovascular disease is a more common cause of death than the cancer itself. This has led to a new sub-specialty of cardiology coined cardio-oncology to manage this specific population. Hypertension is one of the most common cardiovascular disease seen in this cohort. The aetiology of hypertension in cardio-oncology is complex and multifactorial based on the type of chemotherapy, type of malignancy and intrinsic patient factors such as age and pre-existing comorbidities. A variety of different oncological treatments have been implicated in causing hypertension. The effect can be transient whilst undergoing treatment or can be delayed occurring decades after treatment. A tailored management plan is recommended given the plethora of agents and their differing underlying mechanisms and speed of this mechanism in causing hypertension. Management by a multidisciplinary team consisting of oncology, general practice and cardiology is advised. There are currently no trials comparing antihypertensives in this specific cohort of patients. In the absence of evidence demonstrating otherwise, hypertension in cardio-oncology should be managed utilising the same treatment guidelines for the general population.
Similar content being viewed by others

Introduction
Cancer is one of the leading causes of death worldwide. In the United States (US) cancer is the second most common cause of death with an estimated toll of 606,520 in 2020. Advances in oncological treatment have led to improved survival of patients with a year on year decline in mortality between 1991 and 2017 in the US [1]. The increase in cancer survivors has been accompanied by increasing cardiovascular disease (CVD) morbidity and mortality due to downstream side effects of treatment and an increasing age of patients due to improved survival. The overall societal burden of CVD in oncology is likely to increase with an increasing aging population and an overall lifetime risk of nearly 40% of developing cancer in the US.
Cardio-oncology is a relatively new clinical field focusing on the diagnosis, prevention and treatment of the cardiovascular consequences of cancer and its treatment. Approximately 75% of cancer survivors have chronic health problems [2]. CVD is the leading cause of morbidity and mortality in this population second only to recurrent malignancy [2]. The risk of CVD in cancer survivors is 800% higher than that of the general population [2]. The relative risk of coronary artery disease and heart failure is over 1000% more in cancer survivors as compared with their cancer free sibling [2]. Cancer treatments in general share various detrimental effects in common, especially upregulation of cardiovascular risk factors [3]. This can lead to both short- and long-term cardiovascular complications. The increasing recognition of this resulted in the building of the world’s first cardio-oncology unit in the MD Anderson Center in the US in 2000. Following this, the international cardio-oncology society was born in 2009 [4].
Although the field of cardio-oncology has received increasing attention in recent years, many aspects of both radiation-induced and cancer drug-induced CVD are still to be fully elucidated. As the field advances, insights into mechanisms of cardiovascular toxicities and hypertension have become more evident. There are currently ten main CVD stigmata of cancer treatments, these are (Fig. 1):
-
Left ventricular systolic dysfunction
-
Arterial hypertension
-
Pulmonary hypertension
-
Valvular disease
-
Cardiac arrhythmias
-
Thromboembolic disease
-
Peripheral vascular disease
-
Stroke
-
Coronary artery disease
-
Pericardial disease

Cancer treatment and its stigmata on the cardiovascular system.
Essential hypertension is a leading cause of morbidity and mortality worldwide. The relationship between blood pressure (BP) and CVD is continuous, but for the sake of pragmatism hypertension is defined by the European Society of Cardiology (ESC) as a BP ≥ 140/90 [5]. The overall prevalence of hypertension in adults is around 30–45% and this increases with increasing age, with a prevalence of >60% in people aged >60 years [6].
Hypertension has been reported to be the most common comorbidity in cancer patients [7]. Hypertension is a well-recognised cause of CVD morbidity and mortality and is implicated in strokes, coronary heart disease, peripheral arterial disease, heart failure and renal disease.
The relationship between hypertension and cancer is multifaceted. Risk factors for hypertension can be risk factors for specific tumours e.g. smoking and lung cancer. Improving cancer survival is leading to an older population at more risk of developing hypertension. In cancer patients, specific treatments and some cancers lead to the development of hypertension. The prevalence of hypertension in patients is therefore difficult to estimate given the heterogeneity present. However, retrospective data in patients without a prior diagnosis of hypertension have demonstrated that around 33% of patients will develop hypertension [8]. This is particularly more profound in patients treated with angiogenesis inhibitors, where rates of hypertension close to 70% have been reported with some therapies [9]. The overall lack of data in prevalence suggests widespread under-diagnosis and undertreatment [10].
Table 1 grades the severity of hypertension occurring secondary to anticancer therapies based on the latest version of US Common Terminology Criteria for Adverse Events [11]. This rating system categorises “any unfavourable symptom, sign or disease associated with the use of a medical treatment or procedure that may or may not be considered related to or caused by the medical treatment or procedure”. This rating system is often utilised in analysis of hypertension secondary to oncology treatment.
The focus of this article is to review the literature with regards to the development and management of hypertension in cardio-oncology. Specifically, the article will focus on hypertension as a consequence of cancer therapy rather than hypertension induced by cancer.
Aetiology of hypertension in relation to oncology treatments
Table 2 demonstrates biologically plausible pathways for various agents in causing hypertension.
Angiogenesis inhibitors
Angiogenesis is an essential process in the natural course of cancer, as it mediates tumour growth and metastasis. Angiogenesis inhibitors generally exert their effect via inhibition of a component of the vascular endothelial growth factor (VEGF) signalling pathway via two main mechanisms. The first directly inhibit VEGF ligand’s ability to bind to its target receptor and includes bevacizumab and ramucirumab. These medications are classified as VEGF inhibitors. The second class inhibit tyrosine kinases which would be activated by the VEGF ligand–receptor interactions and includes agents such as sunitinib and sorafenib. These medications are classified as tyrosine kinase inhibitors (TKI).
Angiogenesis inhibitors are a well-recognised cause of CVD including hypertension. All commercially available angiogenesis inhibitors have been implicated in the development of hypertension to varying degrees [12]. VEGF signalling inhibitor-induced elevation in BP appears to be not an adverse event of the therapy, but rather a mechanism-dependent on-target toxicity [13]. In a metanalysis of 77 studies of 11 different angiogenesis inhibitors and 30,593 patients the likelihood of new hypertension had an odds ratio of 5.28 (95% Confidence interval of 4.53–6.15) [14]. Table 3 demonstrates the prevalence of hypertension with a variety of different angiogenesis inhibitors are reported in meta-analysis’.
Drug-related hypertension may occur from initiation of therapy to up to a year after treatment onset [15]. The incidence of hypertension seems to depend on multiple variables, such as type of drug, dose and schedule utilised. Patient characteristics further play a part with those with pre-existing hypertension at greater risk, along with the elderly (age > 60 years) and more obese patients (body mass index ≥ 25) [16]. There is some evidence that severity is dose-dependent and appears to be transient with normal BP values restored after discontinuation [17].
The pathophysiology of new or worsening hypertension is unclear; but is likely to be multifactorial with multiple biologically plausible theories hypothesised. There is evidence that VEGF activation induces the expression of nitric oxide synthase in endothelial cells, which promotes vasodilation. The inhibition of this pathway thus suppresses the nitric oxide pathway and induces hypertension via vasoconstriction. Furthermore, there is evidence in increasing levels of endothelin-1, the most potent vasoconstrictor known, which further contributes. Plasma endothelin-1 levels are elevated twofold to threefold in patients treated with VEGF inhibitors [18]. This appears to be dose-dependent and may explain the dose-dependent rise in BP [19]. In addition, VEGF is expressed in endothelial cells and in the kidneys and is known to play an important role in cellular proliferation and homoeostasis in both sites. It has also been shown that there are parallel losses of capillary circulation in both tumour and non-tumour tissue. It is thought that these factors, in combination with systemic thrombotic microangiopathy, contribute to the resulting development of hypertension [20].
Mitotic inhibitors
Mitotic inhibitors inhibit mitosis via disruption of microtubules. Vinca alkaloids such as vincristine have been suggested to cause hypertension [21]. The mechanism for this is unclear and the strength of the association is confounded by the fact that these agents are often utilised in combination with other drugs [22]. There is some evidence that vinca alkaloids result in mitosis-mediated inhibition of endothelial cell proliferation and endothelial cell caspase-mediated apoptosis. It is unclear if this contributes to hypertension [23].
Antimetabolite therapy
Antimetabolite therapy interferes with deoxyribonucleic acid production and inhibit cell division. There is some evidence to suggest that gemcitabine can induce hypertension in the context of thrombotic microangiopathy [24]. Reports from a case series of 29 patients also demonstrate that gemcitabine induced or worsened hypertension in 26 of them likely secondary to nephrotoxicity [25].
Alkylating and alkylating-like agents
Alkylating agents stop the proliferation of neoplasia via attaching an alkyl group to DNA causing subsequent damage. Alkylating-like agents act similarly to alkylating agents; however, they lack the alkyl group. These agents were historically and originally utilised as mustard gas during the second world war. However, alkylating agents such as cyclophosphamide, chlorambucil and busulfan and alkylating-like agents such as cisplatin have since found widespread medical utility and represent the oldest class of anticancer medications. In a case series of multiple alkylating agents, 15/18 patients developed new hypertension [26]. The mechanism(s) for this is unclear. Alkylating-like agents have been implicated in causing hypertension which is thought to be secondary to underlying nephrotoxicity [27]. Cisplatin treatment has been correlated with a dose-dependent increase in hypertension in testicular cancer survivors [28].
Anthracyclines
Anthracyclines such as doxorubicin and daunorubicin work by interfering with DNA metabolism. The dose-dependent left ventricular dysfunction effects of anthracyclines are well-recognised. It is however less clear if they cause hypertension. There is evidence that hypertension has a synergistic effect with anthracycline-induced cardiotoxicity, producing substantially higher risks of heart failure [29]. The finding that pre-existing hypertension predisposes to higher rates of heart failure following treatment with anthracyclines has been seen in multiple other studies [30, 31]. This raises the importance of appropriate management of hypertension in this cohort of patients.
Calcineurin inhibitors
Calcineurin inhibitors are potent immunosuppressive drugs which can be utilised in oncology often as adjuvant therapy. Calcineurin is a calcium- and calmodulin-dependent serine–threonine phosphatase important for function of T-helper cells [32]. Examples include cyclosporin and tacrolimus. These have been linked to hypertension by a multifactorial combination of renal artery vasoconstriction [33] and activation of the renin–angiotensin system [34].
Proteasome inhibitors
Proteasome inhibition may prevent degradation of pro-apoptotic factors such as the p53 protein, permitting activation of programmed cell death in neoplastic cells dependent upon suppression of pro-apoptotic pathways [35]. Examples include carfilzomib and bortezomib. A 2014 retrospective analysis of 2509 patients treated with bortezomib were noted to have a non-significant trend towards hypertension compared with those not treated with bortezomib [36]. It is possible that this mechanism of hypertension is mediated via diminished NO bioavailability and subsequent vasoconstriction [37].
Radiotherapy
There is some evidence that radiotherapy to the head and neck can result in hypertension [38]. However, there is also evidence to suggest that radiotherapy for head and neck cancers results in a permanent reduction in BP [39]. It is likely the mechanism for both hypertension and hypotension is baroceptor failure. The mechanism responsible for hypertension in survivors of testicular cancer following radiotherapy is however less clear [28]. The pattern of hypertension post radiotherapy is also seen in those undergoing thorax and abdominal radiation [40].
Steroids
Steroid therapy is utilised in a variety of chemotherapy regimens and for treating symptoms of cancer. It has long been recognised that steroids induce hypertension and that this is dose dependent [41]. Mineralocorticoid hypertension is thought to be mainly secondary to sodium retention, whilst glucocorticoid hypertension is believed to result from altered vascular reactivity [41].
Diagnosis
Hypertension is predominantly an asymptomatic condition that is best detected by frequent and careful screening of at-risk groups such as cancer patients. Some chemotherapeutic agents cause hypertension in the first few cycles and others are more likely to cause hypertension 10 years after diagnosis. Therefore, cancer patients require both frequent and early assessment and a long-term approach. Patients undergoing chemotherapy warrant close monitoring of their BP throughout the course of treatment. A 2016 position paper by the ESC This can be done via weekly visits to their general practice clinic or via home monitoring following patient education [42].
The diagnosis of hypertension is based on a persistent BP ≥ 140/90. The BP should be checked in both arms (unless contraindicated by lymphoedema or indwelling venous line). In cancer patients it is important to assess for the presence of temporary interfering substances that could be causative in a persistently elevated BP such as pain and high dose steroids. It is recommended to utilise ambulatory BP monitoring over a 3–6-day period as a means of diagnosis rather than spot testing to avoid over-diagnosis due to “white coat” hypertension. Ambulatory monitoring is typically performed every 15–30 min during the day and every 30–60 min at night. Diagnosis via ambulatory monitoring is the gold standard due to a stronger association with cardiovascular outcomes, reflecting the hypertension ‘load’ over the 24 h. The diagnostic threshold in ambulatory monitoring is often lowered to an average BP ≥ 135/85 [5]. It is likely that BP monitoring in oncology clinics is lacking leading to under-diagnosis. Telehealth monitoring of BP has demonstrated useful utility [43]. It is likely that telehealth will become more common following the worldwide pandemic of COVID-19 [44].
The role of telehealth monitoring of BP is likely to take an increasingly larger role in the future given the trend towards remote reviews expediated by the COVID-19 crisis.
Management
Effectively lowering BP reduces morbidity and mortality from congestive heart failure, myocardial infarction, stroke and renal insufficiency. The risk of these adverse outcomes is proportional to the level and duration of the BP [45]. A multidisciplinary team is required as the aetiology of hypertension is complex and can arise before/during/after treatment. Treatment is often best guided by a combination of physicians trained in oncology, cardiology and primary care. Traditional recommendations regarding lifestyle changes including physical exercise, weight reduction, dietary change and sodium restriction, though potentially beneficial and advisable, may be unachievable for some patients in the setting of advanced malignancy. Therefore, pharmacological therapy tends to dominate in this cohort. However, with increasing early diagnosis of malignancy and improving prognosis there is a large cohort in whom conservative measures are the most important starting step. This highlights the importance of individualising treatment for each patient.
In keeping with the complex aetiology there is no universal approach to pharmacological choices. In essentially hypertension, combination therapy is usually required to control BP adequately. In hypertension induced by oncology there is little data indicating how responsive patients are to therapy, but it is probable that combination therapy will be required. In an increasing proportion of cardio-oncology patients, life expectancy is good and therefore the primary coal of treatment is to prevent the long-term stigmata of elevated BP. In some patients, life expectancy can be much more limited and therefore, the goals of treatment tend to differ depending on this context. Treatment remains important to prevent acute complications of hypertension even in those with the most limited prognosis. An individualised treatment approach is advised if there is evidence of an underlying aetiology and treatment should take note of patient’s comorbid conditions such as chronic kidney disease, diabetes and heart failure. Treatment is likely to result in some adverse effects including but not limited due an increased risk of renal dysfunction, falls, medication related side effects, cognitive impairment, urinary incontinence and functional decline [46].
All patients should undergo a formal evaluation and documentation of pre-treatment risk for CVD. BP values and proteinuria should be assessed before initiation of treatment, and if hypertension is present (BP ≥ 140/90) antihypertensive treatment should be started first [42]. The purpose of this assessment is to identify patients at high risk for chemotherapy-induced hypertension, especially if VEGF inhibitors are being considered. There is evidence that pre-existing hypertension in cancer patients confers worse prognosis with increasing mortality [46]. The main goal of treatment is to reduce BP to less than 140/90. This target is based on existing guidelines for the treatment of hypertension in all patients [5, 47]. There are no individualised targets for oncology patients per se.
In higher risk patients, particularly those with diabetes or chronic kidney disease, stricter targets should be utilised aiming for 130/80. In patients already taking antihypertensive medication adherence to treatment should be initially verified and compliance ensured. In patient’s adherent to medication whose BP is still above their target range, a second agent should be introduced. Hypertension should not preclude initiation of chemotherapy unless the risk of hypertension is deemed too high by the treating physician. No studies have compared the efficacy of different antihypertensive agents in treating chemotherapy-induced hypertension. Therefore, in the absence of evidence or a compelling secondary indication, angiotensin converting enzyme inhibitors, Angiotensin receptor blockers and dihydropyridine calcium channel blockers (CCB) are all considered viable first line therapy. Addition of a second agent is preferred to increasing the dose if BP remains high as this has been shown to be more effective [48]. Diuretics and second-generation beta blockers (especially when a secondary indication exists) are considered viable second line treatment options. Caution is advised with diuretic use as they may cause electrolyte depletion and consequent QT prolongation. This may be worsened in the setting of chemotherapy agents that commonly cause diarrhoea and potential dehydration.
Hypertension during VEGF treatment
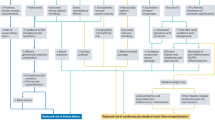
New hypertension during treatment with VEGF inhibitors is so prevalent that this topic warrants its own discussion regarding its management. Figure 2 demonstrates an algorithm for BP management during treatment with VEGF inhibitors. In this cohort it recognised that the majority of increase in BP occurs during the first cycle [49]. Treatment should be initiated when hypertension develops, or diastolic BP increases by 20 mm Hg. This is based on expert recommendations by the Cardiovascular Toxicities Panel of the National Cancer Institute [50]. There are conflicting data regarding which class of medications is more effective in reduction on BP. Currently there is no formal evidence to suggest any agent is more efficacious and therefore none is currently recommended over another. The main recommendation is to avoid non-dihydropyridine CCBs such as verapamil and diltiazem. This is based on the fact that non-dihydropyridine CCBs inhibit cytochrome P450 3A4, the enzyme that metabolises VEGF inhibitors, leading to potentially high VEGF inhibitor plasma levels, which may aggravate VEGF inhibitor-induced hypertension [51]. Temporary cessation of VEGF inhibitors has been demonstrated to be useful when hypertension is proving difficult to control with normal agents [52]. In general, the hypertensive effect of VEGF inhibitors dissipates after cessation of the agent. This necessitates monitoring of BP following completion of a treatment course with withdrawal of antihypertensive medications when BP returns to baseline. In general, prognosis is good, treatment-induced hypertension secondary to angiogenesis inhibitors is commonly low-grade, and easily correctable with standard antihypertensive medications.

ACEi, Ace inhibitor; ARB, Angiotensin receptor blocker; CCB, calcium channel blocker; VEGF, Vascular endothelial growth factor.
Hypertension in the context of angiogenesis inhibitors has even been suggested as a possible biomarker of clinical efficacy. A retrospective analysis of nearly 5000 patients with renal cell carcinoma demonstrated that treatment associated hypertension with sunitinib was significantly and independently associated with improved clinical outcomes. Furthermore, the utilisation of antihypertensives to control high BP has no effect on treatment outcome, suggesting that there is no contraindication to managing hypertension properly in patients treated with angiogenesis inhibitors [53].
Several treatments have been proposed based on the pathophysiology of hypertension in patients treated with angiogenesis inhibitors. This includes endothelin-1 receptor blockers, increasing NO bioavailability and salt restriction. To date there have been no trials with endothelin-1 receptor blockers. There have been case reports on the efficacy of NO donors for the treatment of VEGF-induced hypertension [54]. There is also one trial looking at the value of a low salt (<4 g/day) diet on the VEGF-induced hypertension due to finish on in 2020 ((Dutch trial register NTR7556).
Conclusion
Hypertension and CVD are common in patients undergoing treatment for cancer. This likely contributes to increasing CVD morbidity and mortality as compared with the general population. The estimated prevalence of hypertension in cancer patients is expected to increase as the prognosis of cancer improves and more patients survive to experience long-term consequences of chemotherapy. All cancer patients should undergo a pre-chemotherapy risk assessment to identify and appropriately manage hypertension. These patients should be monitored closely during their chemotherapy (especially if angiogenesis inhibitors are utilised) and post treatment for the development of hypertension.
There is a paucity of data with regards to the management of hypertension in cardio-oncology. High quality trials are required to generate evidence-based guidance for clinicians on the best management strategies in this population. In the absence of evidence to the contrary, hypertension in cardio-oncology should be treated utilising the same medications used in the general population. Key areas to investigate would be randomised treatment trials in patients with hypertension post chemotherapy to create evidenced based treatment plans and the data studies looking at the effect of reducing hypertension in preventing cardiovascular events in cancer patients.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30.
Oeffinger KC, Mertens AC, Sklar CA, Kawashima T, Hudson MM, Meadows AT, et al. Chronic health conditions in adult survivors of childhood cancer. N. Engl J Med. 2006;355:1572–82.
Ewer MS, Ewer SM. Cardiotoxicity of anticancer treatments. Nat Rev Cardiol. 2015;12:547–58.
Lenihan DJ, Cardinale D, Cipolla CM. The compelling need for a cardiology and oncology partnership and the birth of the International CardiOncology Society. Prog Cardiovasc Dis. 2010;53:88–93.
Williams B, Mancia G, Spiering W, Agabiti Rosei E, Azizi M, Burnier M, et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension: The Task Force for the management of arterial hypertension of the European Society of Cardiology and the European Society of Hypertension: The Task Force for the management of arterial hypertension of the European Society of Cardiology and the European Society of Hypertension. J Hypertens. 2018;36:1953–2041.
Chow CK, Teo KK, Rangarajan S, Islam S, Gupta R, Avezum A, et al. Prevalence, awareness, treatment, and control of hypertension in rural and urban communities in high-, middle-, and low-income countries. JAMA. 2013;310:959–68.
Piccirillo JF, Tierney RM, Costas I, Grove L, Spitznagel EL. Prognostic importance of comorbidity in a hospital-based cancer registry. JAMA. 2004;291:2441–7.
Fraeman KH, Nordstrom BL, Luo W, Landis SH, Shantakumar S. Incidence of New-Onset Hypertension in Cancer Patients: A Retrospective Cohort Study. Int J Hypertens. 2013;2013:379252.
Schlumberger M, Tahara M, Wirth LJ, Robinson B, Brose MS, Elisei R, et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N. Engl J Med. 2015;372:621–30.
Małyszko J, Małyszko M, Kozlowski L, Kozlowska K. Hypertension in malignancy—an underappreciated problem. Oncotarget 2018;9:20855–71.
Common Terminology Criteria for Adverse Events (CTCAE)—CTCAE_v5_Quick_Reference_8.5x11.pdf 2020 Version 5. https://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf.
El-Kenawi AE, El-Remessy AB. Angiogenesis inhibitors in cancer therapy: mechanistic perspective on classification and treatment rationales. Br J Pharm. 2013;170:712–29.
Simons M, Eichmann A. On-target” cardiac effects of anticancer drugs: lessons from new biology. J Am Coll Cardiol. 2012;60:626–7.
Abdel-Qadir H, Ethier JL, Lee DS, Thavendiranathan P, Amir E. Cardiovascular toxicity of angiogenesis inhibitors in treatment of malignancy: a systematic review and meta-analysis. Cancer Treat Rev. 2017;53:120–7.
Escalante CP, Zalpour A. Vascular endothelial growth factor inhibitor-induced hypertension: basics for primary care providers. Cardiol Res Pr. 2011;2011:816897.
Hamnvik OP, Choueiri TK, Turchin A, McKay RR, Goyal L, Davis M, et al. Clinical risk factors for the development of hypertension in patients treated with inhibitors of the VEGF signaling pathway. Cancer 2015;121:311–9.
Corr BR, Breed C, Sheeder J, Weisdack S, Behbakht K. Bevacizumab induced hypertension in gynecologic cancer: Does it resolve after completion of therapy? Gynecol Oncol Rep. 2016;17:65–8.
Kappers MH, van Esch JH, Sluiter W, Sleijfer S, Danser AH, van den Meiracker AH. Hypertension induced by the tyrosine kinase inhibitor sunitinib is associated with increased circulating endothelin-1 levels. Hypertension. 2010;56:675–81.
Lankhorst S, Baelde HJ, Kappers MH, Smedts FM, Hansen A, Clahsen-van Groningen MC, et al. Greater sensitivity of blood pressure than renal toxicity to tyrosine kinase receptor inhibition with sunitinib. Hypertension. 2015;66:543–9.
Robinson ES, Khankin EV, Karumanchi SA, Humphreys BD. Hypertension induced by VEGF signaling pathway inhibition: mechanisms and potential use as a biomarker. Semin Nephrol. 2010;30:591–601.
Bokemeyer C, Berger CC, Kuczyk MA, Schmoll HJ. Evaluation of long-term toxicity after chemotherapy for testicular cancer. J Clin Oncol. 1996;14:2923–32.
Stoter G, Koopman A, Vendrik CP, Struyvenberg A, Sleyfer DT, Willemse PH, et al. Ten-year survival and late sequelae in testicular cancer patients treated with cisplatin, vinblastine, and bleomycin. J Clin Oncol. 1989;7:1099–104.
Soultati A, Mountzios G, Avgerinou C, Papaxoinis G, Pectasides D, Dimopoulos MA, et al. Endothelial vascular toxicity from chemotherapeutic agents: preclinical evidence and clinical implications. Cancer Treat Rev. 2012;38:473–83.
Izzedine H, Isnard-Bagnis C, Launay-Vacher V, Mercadal L, Tostivint I, Rixe O, et al. Gemcitabine-induced thrombotic microangiopathy: a systematic review. Nephrol Dial Transpl. 2006;21:3038–45.
Glezerman I, Kris MG, Miller V, Seshan S, Flombaum CD. Gemcitabine nephrotoxicity and hemolytic uremic syndrome: report of 29 cases from a single institution. Clin Nephrol 2009;71:130–9.
Graves SW, Eder JP, Schryber SM, Sharma K, Brena A, Antman KH, et al. Endogenous digoxin-like immunoreactive factor and digitalis-like factor associated with the hypertension of patients receiving multiple alkylating agents as part of autologous bone marrow transplantation. Clin Sci. 1989;77:501–7.
Kooijmans EC, Bökenkamp A, Tjahjadi NS, Tettero JM, van Dulmen-den Broeder E, van der Pal HJ, et al. Early and late adverse renal effects after potentially nephrotoxic treatment for childhood cancer. Cochrane Database Syst Rev. 2019;3:CD008944.
Sagstuen H, Aass N, Fosså SD, Dahl O, Klepp O, Wist EA, et al. Blood pressure and body mass index in long-term survivors of testicular cancer. J Clin Oncol. 2005;23:4980–90.
Hershman DL, McBride RB, Eisenberger A, Tsai WY, Grann VR, Jacobson JS. Doxorubicin, cardiac risk factors, and cardiac toxicity in elderly patients with diffuse B-cell non-Hodgkin’s lymphoma. J Clin Oncol. 2008;26:3159–65.
Mantarro S, Rossi M, Bonifazi M, D’Amico R, Blandizzi C, La Vecchia C, et al. Risk of severe cardiotoxicity following treatment with trastuzumab: a meta-analysis of randomized and cohort studies of 29,000 women with breast cancer. Intern Emerg Med. 2016;11:123–40.
Russo G, Cioffi G, Gori S, Tuccia F, Boccardi L, Khoury G, et al. Role of hypertension on new onset congestive heart failure in patients receiving trastuzumab therapy for breast cancer. J Cardiovasc Med. 2014;15:141–6.
Hoorn EJ, Walsh SB, McCormick JA, Zietse R, Unwin RJ, Ellison DH. Pathogenesis of calcineurin inhibitor–induced hypertension. J Nephrol. 2012;25:269–75.
Kaye D, Thompson J, Jennings G, Esler M. Cyclosporine therapy after cardiac transplantation causes hypertension and renal vasoconstriction without sympathetic activation. Circulation 1993;88:1101–9.
Lassila M. Interaction of cyclosporine A and the renin-angiotensin system; new perspectives. Curr Drug Metab. 2002;3:61–71.
Kubiczkova L, Pour L, Sedlarikova L, Hajek R, Sevcikova S. Proteasome inhibitors—molecular basis and current perspectives in multiple myeloma. J Cell Mol Med. 2014;18:947–61.
Laubach JP, Moslehi JJ, Francis SA, San Miguel JF, Sonneveld P, Orlowski RZ, et al. A retrospective analysis of 3954 patients in phase 2/3 trials of bortezomib for the treatment of multiple myeloma: towards providing a benchmark for the cardiac safety profile of proteasome inhibition in multiple myeloma. Br J Haematol. 2017;178:547–60.
Chari A, Hajje D. Case series discussion of cardiac and vascular events following carfilzomib treatment: possible mechanism, screening, and monitoring. BMC Cancer. 2014;14:915.
Timmers HJ, Wieling W, Karemaker JM, Lenders JW. Baroreflex failure: a neglected type of secondary hypertension. Neth J Med. 2004;62:151–5.
Leibowitz A, Grossman E, Berkovitch A, Levartovski M, Appel S, Sharabi Y, et al. The effect of head and neck radiotherapy on blood pressure and orthostatic hypotension in patients with head and neck tumors. Am J Hypertens. 2018;31:235–9.
Meacham LR, Chow EJ, Ness KK, Kamdar KY, Chen Y, Yasui Y, et al. Cardiovascular risk factors in adult survivors of pediatric cancer-a report from the childhood cancer survivor study. Cancer Epidemiol Biomark Prev. 2010;19:170–81.
Mantero F, Boscaro M. Glucocorticoid-dependent hypertension. J Steroid Biochem Mol Biol. 1992;43:409–13.
Zamorano JL, Lancellotti P, Rodriguez Muñoz D, Aboyans V, Asteggiano R, Galderisi M, et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur Heart J. 2016;37:2768–801.
Omboni S, Gazzola T, Carabelli G, Parati G. Clinical usefulness and cost effectiveness of home blood pressure telemonitoring: meta-analysis of randomized controlled studies. J Hypertens. 2013;31:455–67. discussion 67–8
Hollander JE, Carr BG. Virtually Perfect? Telemedicine for Covid-19. N. Engl J Med. 2020;382:1679–81.
Lewington S, Clarke R, Qizilbash N, Peto R, Collins R, Collaboration PS. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet. 2002;360:1903–13.
Menditto E, Gimeno Miguel A, Moreno Juste A, Poblador Plou B, Aza Pascual-Salcedo M, Orlando V, et al. Patterns of multimorbidity and polypharmacy in young and adult population: systematic associations among chronic diseases and drugs using factor analysis. PLoS One. 2019;14:e0210701.
Whelton PK, Carey RM, Aronow WS, Casey DE, Collins KJ, Dennison Himmelfarb C, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: executive summary: a report of the American college of cardiology/american heart association task force on clinical practice guidelines. Circulation. 2018;138:e426–e83.
MacDonald TM, Williams B, Webb DJ, Morant S, Caulfield M, Cruickshank JK, et al. Combination therapy is superior to sequential monotherapy for the initial treatment of hypertension: a double-blind randomized controlled trial. J Am Heart Assoc. 2017;6:e006986.
Azizi M, Chedid A, Oudard S. Home blood-pressure monitoring in patients receiving sunitinib. N. Engl J Med. 2008;358:95–7.
Maitland ML, Bakris GL, Black HR, Chen HX, Durand JB, Elliott WJ, et al. Initial assessment, surveillance, and management of blood pressure in patients receiving vascular endothelial growth factor signaling pathway inhibitors. J Natl Cancer Inst. 2010;102:596–604.
Humphreys BD, Atkins MB. Rapid development of hypertension by sorafenib: toxicity or target? Clin Cancer Res. 2009;15:5947–9.
Jain M, Townsend RR. Chemotherapy agents and hypertension: a focus on angiogenesis blockade. Curr Hypertens Rep. 2007;9:320–8.
Rini BI, Cohen DP, Lu DR, Chen I, Hariharan S, Gore ME, et al. Hypertension as a biomarker of efficacy in patients with metastatic renal cell carcinoma treated with sunitinib. J Natl Cancer Inst. 2011;103:763–73.
Kruzliak P, Kovacova G, Pechanova O. Therapeutic potential of nitric oxide donors in the prevention and treatment of angiogenesis-inhibitor-induced hypertension. Angiogenesis. 2013;16:289–95.
Daher IN, Yeh ET. Vascular complications of selected cancer therapies. Nat Clin Pr Cardiovasc Med. 2008;5:797–805.
Doll DC, List AF, Greco FA, Hainsworth JD, Hande KR, Johnson DH. Acute vascular ischemic events after cisplatin-based combination chemotherapy for germ-cell tumors of the testis. Ann Intern Med. 1986;105:48–51.
Damiano S, Scanni R, Ciarcia R, Florio S, Capasso G. Regulation of sodium transporters in the kidney during cyclosporine treatment. J Nephrol 2010;23:S191–8.
Qi WX, Shen Z, Tang LN, Yao Y. Risk of hypertension in cancer patients treated with aflibercept: a systematic review and meta-analysis. Clin Drug Investig. 2014;34:231–40.
Zhu X, Stergiopoulos K, Wu S. Risk of hypertension and renal dysfunction with an angiogenesis inhibitor sunitinib: systematic review and meta-analysis. Acta Oncol. 2009;48:9–17.
Qi WX, Lin F, Sun YJ, Tang LN, He AN, Yao Y, et al. Incidence and risk of hypertension with pazopanib in patients with cancer: a meta-analysis. Cancer Chemother Pharm. 2013;71:431–9.
Funakoshi T, Latif A, Galsky MD. Risk of hypertension in cancer patients treated with sorafenib: an updated systematic review and meta-analysis. J Hum Hypertens. 2013;27:601–11.
Qi WX, He AN, Shen Z, Yao Y. Incidence and risk of hypertension with a novel multi-targeted kinase inhibitor axitinib in cancer patients: a systematic review and meta-analysis. Br J Clin Pharm. 2013;76:348–57.
Qi WX, Shen Z, Lin F, Sun Y, Min D, Tang LN, et al. Incidence and risk of hypertension with vandetanib in cancer patients: a systematic review and meta-analysis of clinical trials. Br J Clin Pharm. 2013;75:919–30.
Wang Z, Xu J, Nie W, Huang G, Tang J, Guan X. Risk of hypertension with regorafenib in cancer patients: a systematic review and meta-analysis. Eur J Clin Pharm. 2014;70:225–31.
Wang J, Wang Z, Zhao Y. Incidence and risk of hypertension with ramucirumab in cancer patients: a meta-analysis of published studies. Clin Drug Investig. 2015;35:221–8.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
HE has nothing to declare. RD has nothing to declare. GYHL reports consultancy and speaker fees from Bayer, Bayer/Janssen, BMS/Pfizer, Biotronik, Medtronic, Boehringer Ingelheim, Microlife, Roche, and Daiichi-Sankyo outside the submitted work. No fees received personally. DW has received speaker and consultancy fees from Boston scientific and medtronic.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Essa, H., Dobson, R., Wright, D. et al. Hypertension management in cardio-oncology. J Hum Hypertens 34, 673–681 (2020). https://doi.org/10.1038/s41371-020-0391-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41371-020-0391-8
This article is cited by
-
Chronic diseases spectrum and multimorbidity in elderly inpatients based on a 12-year epidemiological survey in China
BMC Public Health (2024)
-
Beta-blocker adjunct therapy as a prospective anti-metastatic with cardio-oncologic regulation
Clinical & Experimental Metastasis (2024)
-
Hypertension in Cardio-Oncology Clinic: an update on etiology, assessment, and management
Cardio-Oncology (2023)
-
Chapter 4: CKD treatment in cancer survivors, from Clinical Practice Guidelines for the Management of Kidney Injury During Anticancer Drug Therapy 2022
International Journal of Clinical Oncology (2023)
