
Abstract
In addition to constituting the genetic material of an organism, DNA is a tracer for the recognition of foreign pathogens and a trigger of the innate immune system. cGAS functions as a sensor of double-stranded DNA fragments and initiates an immune response via the adaptor protein STING. The cGAS-STING pathway not only defends cells against various DNA-containing pathogens but also modulates many pathological processes caused by the immune response to the ectopic localization of self-DNA, such as cytosolic mitochondrial DNA (mtDNA) and extranuclear chromatin. In addition, macrophages can cause inflammation by forming a class of protein complexes called inflammasomes, and the activation of the NLRP3 inflammasome requires the release of oxidized mtDNA. In innate immunity related to inflammasomes, mtDNA release is mediated by macropores that are formed on the outer membrane of mitochondria via VDAC oligomerization. These macropores are specifically formed in response to mitochondrial stress and tissue damage, and the inhibition of VDAC oligomerization mitigates this inflammatory response. The rapidly expanding area of research on the mechanisms by which mtDNA is released and triggers inflammation has revealed new treatment strategies not only for inflammation but also, surprisingly, for neurodegenerative diseases such as amyotrophic lateral sclerosis.
Similar content being viewed by others

Introduction
DNA, the blueprint of life, contains the genetic information needed for all organisms to develop, survive, and reproduce. In addition to carrying genetic information, DNA signals the host immune system to pathogens, such as DNA viruses and bacteria, inducing its response1. Pathogen-derived nucleic acids, such as single-stranded (ss) or double-stranded (ds) DNA and RNA, and RNA–DNA hybrids are recognized by various sensors in host cells and trigger signaling pathways to elicit innate immune responses involving the production of type I interferons (IFNs) and proinflammatory cytokines2,3.
Host cells initiate innate immune responses upon recognition of various types of conserved pathogen structures called pathogen-associated molecular patterns (PAMPs), such as lipopolysaccharide (LPS), yeast zymosan, and bacterial flagellin, as well as pathogen-derived nucleic acids3. Recent studies have indicated that damage-associated molecular patterns (DAMPs), which are endogenous factors released from damaged, dead, or transformed cells, also activate innate immune responses3,4. Pattern recognition receptors (PRRs) are critical for sensing both PAMPs and DAMPs in the extracellular space, endosome, and cytoplasm3,5. Over the over the past two decades, various classes of PRRs have been identified, including Toll-like receptors (TLRs), c-type lectin-like receptors (CLRs), retinoic acid-inducible gene I–like receptors (RLRs), nucleotide-binding oligomerization domain-like receptors (NLRs), absent in melanoma 2 (AIM2)-like receptors (ALRs), and cytosolic DNA receptors3,4,5. In infected cells, pathogen-derived nucleic acids are commonly exposed in subcellular compartments, and each type of PRR recognizes them depending on the location and polynucleotide species of the nucleic acids3,5. For instance, the RIG-1 in the RLR family senses 5′-tri/diphosphorylated dsRNAs, the cytosolic replication intermediates of RNA viruses6,7. In the TLR family, TLR-3 and TLR-7/8 detect dsRNAs and ssRNAs, respectively, and TLR-9 recognizes CpG-hypomethylated DNA in endosomes4,7. AIM2 in the ALR family binds to cytosolic DNA to induce the formation of the inflammasome, which consists of multiprotein complexes, in myeloid cells5,8,9,10,11. Subsequently, inflammasome assembly leads to the maturation of pro-interleukin-1β (IL-1β) and pro-IL-18 through the activation of the protease caspase-110,11.
A number of proteins have been proposed to sense DNA in the cytosol: DNA-dependent activators of IRFs (DAI), RNA polymerase III, DExD/H box helicases (namely, DDX41), and LRR-binding FLII-interacting protein 1 (LRRFIP1)3,12,13. However, whether some of these proteins are DNA sensors are controversial. For example, PRRs have not been universally accepted as major DNA sensors because they recognize specific DNA sequences or function only in specific cell types3,13. In this review, we focus on cyclic GMP-AMP synthase (cGAS), which is a recently characterized cytosolic DNA sensor that is essential for the DNA-induced immune response, irrespective of cell type or DNA sequence3. Upon recognition of DNA, cGAS catalyzes the formation of cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) (cGAMP), which is a second messenger molecule that associates with and activates the adaptor protein stimulator of interferon genes (STING)3,14. Activated STING recruits TANK-binding kinase 1 (TBK1), which phosphorylates the transcription factor interferon regulatory factor 3 (IRF3)3,14,15,16. Phosphorylated IRF3 homodimerizes and then is translocated into the nucleus, where it increases the expression of type I IFNs, thereby leading to antiviral immune responses3,14,15. STING also upregulates inflammatory cytokines and chemokines by activating the kinase IKK, which phosphorylates and inactivates the IkB family of inhibitors of the transcription factor NF-kB7.
As eukaryotic cells evolved, DNA was compartmentalized into the cell nucleus and mitochondria, effectively separating the main genetic material from the cytosol3,4. However, DNA can be released from these organelles and accumulate in the cytosol under certain pathological conditions. Thus, the cGAS-STING pathway is activated by not only exogenous DNA derived from pathogens but also self-DNA, such as mtDNA and nuclear DNA3,4. In addition to their primary role of producing ATP via oxidative phosphorylation (OXPHOS), mitochondria are involved in a wide range of biological processes crucial for cell proliferation and survival4,11,17. Despite the symbiotic relationship between the cell and mitochondria, mtDNA is a potent stimulator of inflammatory responses when it is released into the cytosol or extracellular space11,17.
Much is known about how self-DNA is recognized as a DAMP by PRRs to induce the production of type I IFN and inflammatory cytokines through the cGAS-STING pathway, TLR-9, and inflammasome formation4,5,7. In this review, we focus on the current state of the understanding of the cGAS-STING pathway and how it mediates inflammation in response to ectopic mtDNA localization. In addition, we discuss how mtDNA is released from mitochondria and how this knowledge can be leveraged to treat diseases associated with ectopic mtDNA.
Discovery of cGAS-STING signaling
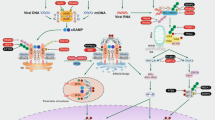
In screening performed to identify mediators of the type-I IFN response, several research groups independently identified a protein called STING that is activated by DNA and upregulates type-I IFN responses1,18,19. It was initially postulated that STING directly detects cytosolic DNA and triggers antiviral gene expression. Subsequently, it was found that the ligand of STING was a cyclic dinucleotide (CDN), not DNA. STING recognizes cyclic diadenosine monophosphate (c-dAMP) and cyclic diguanylate monophosphate (c-dGMP) in bacteria20, as well as 2’3’-cGAMP, which is produced by cGAS (also known as MB21D1 or C6orf150) upon its binding to DNA in mammalian cells12,21,22,23 (Fig. 1). STING is composed of a basic, unstructured N-terminal domain, a central nucleotidyltransferase (NTase, the catalytic sequence) domain, and a C-terminal Mab-21-homologous domain that binds to DNA via the positively charged Zn2+-ribbon/thumb motif3,24.

The host immune response is initiated by the recognition of cytosolic DNA, such as pathogen-derived nucleic acids (e.g., viruses and bacteria) and self-DNA (e.g., from dying cells, tumor cells or mtDNA). Upon recognition of cytosolic DNA, cGAS catalyzes the formation of cGAMP and thereby activates STING. STING then activates TBK1, which phosphorylates IRF3. Phosphorylated IRF3 activates the expression of type I IFNs in the nucleus. STING also activates NF-κB by phosphorylating the kinase IKK. NF-κB then activates the transcription of genes encoding proinflammatory cytokines.
Although cGAS has been mostly reported to be a cytoplasmic protein, it has also been found in the nucleus and on the plasma membrane3,15,25,26. cGAS interacts with a viral capsid sensor in the nucleus, and it has been proposed that nuclear or perinuclear localization of cGAS facilitates viral DNA detection during the integration and replication phases of viruses25. cGAS has also been reported to bind to the inner leaflet of the plasma membrane through its interaction with phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2)26. An obvious advantage of plasma membrane localization is microbe detection at the point of cell entry26.
Upon binding to DNA, cGAS undergoes a conformational change that leads to a rearranged catalytic pocket of the enzyme to allow ATP or GTP binding, thereby synthesizing the second messenger cGAMP3,12,15,23. cGAS binds cytosolic dsDNA without sequence specificity, but it binds only B-form dsDNA, not A-form dsDNA3. Although other types of nucleic acids, such dsRNA, can bind to cGAS, these species do not stimulate cGAS activity3,12. However, cGAS specificity is still controversial because some studies have suggested that cGAS senses synthetic RNA/DNA hybrids or unpaired DNA nucleotides flanking short base-paired DNA stretches derived from human immunodeficiency virus type-1 (HIV-1)13,27.
As suggested by its lack of sequence specificity, cGAS binds to DNA only on the sugar-phosphate backbone, without interacting with bases2,3. However, cGAS requires a minimum of 18 bp of dsDNA for binding, and dsDNA ≥40 bp can form a more stable “ladder network” with cGAS, further increasing the protein catalytic activity2,22,28. In addition, the presence of long DNA sequences leads to a dimeric structure (a 2:2 DNA:cGAS complex) that promotes the liquid phase condensation, further stabilizing the active dimeric state through multivalent interactions between nearby cGAS molecules2,29,30 (Fig. 2).

cGAS binds to dsDNA ≥18 bp and can form dimers when the DNA sequence is sufficiently long or is bent by other factors (e.g., TFAM). Liquid phase condensation leads to the stable ladder/net interaction of cGAS and enhances its catalytic activity.
Posttranslational modification of cGAS
A complex network of pathways modulates cGAS activity both before and after DNA binding. Protein kinase B (PKB), also known as Akt, phosphorylates human cGAS at the Ser305 residue (Ser291 in mouse cGAS) near the catalytic site of cGAS31, suppressing cGAMP synthesis31. Similarly, DNA-dependent protein kinase (DNA-PK) phosphorylates Thr68 and Ser213 and suppresses cGAMP synthesis and antiviral innate immunity32. When cells enter mitosis, aurora kinase B (AurB) hyperphosphorylates the N-terminus of cGAS to suppress cGAS activity, thereby preventing cGAS from sensing chromatin33. Protein phosphatase 6 catalytic subunit (PPP6c), which is a member of the serine/threonine protein phosphatase family, is constitutively associated with cGAS, dephosphorylating human cGAS at the Ser 435 residue (Ser420 in mouse cGAS) in resting cells34. This interaction prevents autoimmune reactions by retaining cGAS in the inactive state in the absence of ligands34. In addition to phosphorylation, glutamylation plays an essential role in the regulation of cGAS activity. The polyglutamylation of mouse cGAS at Glu272 by tubulin tyrosine ligase-like enzyme 6 (TTLL6) abolishes the cytosolic dsDNA-binding ability of cGAS, and the monoglutamylation of cGAS at Glu302 by TTLL4 suppresses cGAS catalytic activity35. Ubiquitination and SUMOylation are also reversible posttranslational modifications of cGAS. During the early phase of viral infection, tripartite motif-containing 38 (Trim38, an E3 ubiquitin ligase) stabilizes cGAS by preventing its Lys48 (K48)-linked ubiquitination and subsequent degradation36. In the late phase of a viral infection, SUMO-specific protease 2 (SENP2) deSUMOylates cGAS, leading to cGAS degradation through the ubiquitin-proteasomal or chaperone-mediated autophagy pathways36. Ring finger protein 185 (RNF185, an E3 ubiquitin ligase) interacts with cGAS during viral infections and specifically catalyzes the K27-linked polyubiquitination of cGAS at K137 and K384, enhancing the enzymatic activity of cGAS37. K48-linked ubiquitination at K414 promotes p62-mediated autophagic degradation of cGAS, whereas tripartite motif-containing 14 (Trim14) recruits ubiquitin-specific peptidase 14 (USP14) to cleave the K48-linked ubiquitin chains of cGAS at K414 and increase cGAS stability38.
Intercellular cGAMP transmission and signaling networks
Interestingly, cGAMP in the extracellular space can propagate the intercellular immune response. For example, direct administration of cGAMP to mice induced an innate immune response by upregulating IFN expression, which did not occur in STING-deficient mice39,40,41. On the other hand, ecto-nucleotide pyrophosphatase phosphodiesterase 1 (ENPP1) degraded extracellular cGAMP and blunted the host immune response42,43. Mutating histidine 362 led to the loss of its cGAMP-degradation activity and enhanced cellular resistance to HSV-1 infection but induced systemic inflammation by enhancing STING signaling44.
The extracellular function of cGAMP indicates that intracellular cGAMP can be released into the extracellular space through transmembrane carriers, such as channels or extracellular vesicles, and various nonspecific biological mechanisms may induce extracellular cGAMP activation of intracellular STING. A genome-wide CRISPR led to the identification of solute carrier family 19 member 1 (SLC19A1), which can import exogenous cGAMP in a cell- and species-dependent manner45,46. In a subsequent study, the purinergic P2X7 receptor (P2X7R) was identified as a novel transporter of extracellular cGAMP released from membrane- compromised dying tumor cells47,48,49. Another transporter of cGAMP is leucine-rich repeat-containing protein 8 (LRRC8)A/E, a subunit of the volume-controlled anion channel (VRAC), which has been shown to mediate both the efflux and influx of cGAMP and other CDNs49. Indeed, genetic ablation or chemical blockade of LRRC8A/E-containing VRACs reduced the intercellular transport of cGAMP and antiviral immune response49,50 (Fig. 3). It is possible that extracellular cGAMP can be sensed directly. One of the three alternatively spliced isoforms of STING is localized to the plasma membrane, and the CTD of the spliced STING isoform is secreted from the cell due to the lack of a transmembrane domain, in contrast in canonical STING51. This isoform is expressed with the same pattern as the canonical isoform of endoplasmic reticulum STING and activates the TBK1/IRF3/IFN pathway51. The intercellular transfer of cGAMP may not require its release into the extracellular space. Intracellular cGAMP can be delivered via intercellular connexin proteins that form gap junctions with adjacent cells, as well as via extracellular vesicles, such as exosomes, microparticles, and viral particles52,53,54,55.

Intracellular cGAMP can be transferred to neighboring or adjacent cells through gap junctions, formed by the connexin protein, or via plasma-membrane transporters, including SLC19A1, P2X7R, and LRRC8. Degradation of extracellular cGAMP by ENPP1 modulates extracellular cGAMP levels.
Activation of STING
STING (also named TMEM173, MITA, MPYA, and ERIS) is an endoplasmic reticulum (ER) membrane protein12,15,23. It is composed of an N-terminal transmembrane domain with four transmembrane helices, a cytosolic ligand-binding domain (LBD), and a C-terminal tail (CTT) with a phosphorylation motif and a TBK1-binding motif56,57. The crystal structure of STING shows that the LBD generates a V-shaped ligand pocket through dimerization with its cytoplasmic domain under steady-state conditions58,59,60. Upon binding to cGAMP, STING undergoes a substantial conformational change to tightly hold the ligand and then transforms into a tetrameric or higher-ordered oligomeric structure, which migrates to the ER–Golgi intermediate compartment (ERGIC) or the Golgi apparatus60,61,62. The oligomeric assembly recruits TBK1 to a highly conserved motif in the CTT of STING and leads to trans-phosphorylation of TBK114,63. Activated TBK1 phosphorylates the CTT, which is directly adjacent to a p-Leu-x-Ile-Ser (pLxIS, where p indicates phosphorylation and x denotes any amino acid) motif1,58,64. This modification generates a docking site for IRF3 and recruits IRF3 for phosphorylation by TBK17,14,63,64. Phosphorylated IRF3 homodimerizes and then is translocated into the nucleus to induce the expression of type I IFNs7,63,64 (Fig. 4).

Upon binding to cGAMP, STING undergoes a conformational change, and then, the dimers oligomerize. STING oligomerization recruits TBK1 and promotes autotransphosphorylation of TBK1 at the ERGIC. Activated TBK1 phosphorylates the CTT of STING to generate IRF3 docking sites and then phosphorylates the recruited IRF3.
Newly secreted IFNs bind to and activate heterodimeric receptor complexes consisting of IFNα receptor 1 (IFNAR1) and IFNAR265,66. Dimerized IFNAR1 and IFNAR2 trigger the autophosphorylation of the receptor-associated protein tyrosine kinase Janus kinase 1 (JAK1), which phosphorylates and activates signal transducer and activator of transcription 1 (STAT1) and STAT267. Phosphorylated STAT1 and STAT2 dimerize and then interact with IFN-regulatory factor 9 (IRF9) to form a trimolecular complex called IFN-stimulated gene factor 3 (ISGF3)68. ISGF3 is then translocated to the nucleus and binds to IFN-stimulated response elements (ISREs), thereby activating the transcription of IFN-stimulated genes (ISGs), which defend against infection through various mechanisms, such as by inhibiting viral replication and degrading viral nucleic acids68,69.
The role of STING in the host immune system is not limited to type I interferon signaling. STING also stimulates NF-κB-mediated transcription to increase the expression of inflammatory cytokines and chemokines70,71,72. The precise molecular mechanisms remain unclear, as conflicting interpretations of STING regulation of the transcriptional activity of NF-κB have been proposed71,72,73. It was first suggested that TBK1 is required for the activation of IKK, which promotes the degradation of IkB family proteins, which inhibit NF-κB71,72,73. However, subsequent studies have shown that activation of NF-κB by STING is not dependent on CTT, which is required to activate TBK74,75. Noncanonical NF-κB responses, such as those of p52–RELB, have also been reported to be regulated by STING70,76. Hence, understanding how STING activates NF-κB requires more investigation.
Regulation of STING activity through posttranslational modifications
The most common and well-characterized posttranslational modification of STING is ubiquitination. TRIM56 was identified as an interferon-inducible E3 ubiquitin ligase that promotes the K63-linked ubiquitination of STING77,78. TRIM56 is upregulated by dsDNA and ubiquitinates STING at K150 77, causing STING to dimerize and recruit TBK177. In contrast, RING finger 5 (RNF5, an E3 ubiquitin ligase) and RNF90 also ubiquitinate STING at K15079,80 but catalyze K48-linked ubiquitination of STING, which causes the degradation of STING through the proteasome-dependent pathway and thus reduces the cellular antiviral response79,80. Similar to TRIM56, TRIM32 catalyzes the K63-linked ubiquitination of STING at K20, 150, 224, and 236, which increases STING-induced IFN expression by recruiting TBK1 to STING81. Endoplasmic reticulum-localized autocrine motility factor receptor (AMFR, an E3 ubiquitin ligase) catalyzes the K27-linked polyubiquitination of STING at K137, K150, K224, and K236 in an insulin-induced gene 1 (INSIG1)-dependent manner in the presence of cytosolic DNA82. This modification causes STING and TBK1 to congregate at perinuclear microsomes as mediated through the Golgi apparatus and activates IRF382.
The counterpart to STING in the antiviral response against dsRNA viruses is mitochondrial antiviral-signaling protein (MAVS, an antiviral adaptor protein with activity against RNA viruses), and its regulation is quite complex. In uninfected cells, MAVS is maintained in an inactivated state by RNF115, which catalyzes the K48-linked ubiquitination of MAVS83. After viral infection, RNF115 disassociates from MAVS and catalyzes the K63-linked ubiquitination of STING at K20, K224, and K289, thereby promoting MAVS translocation to the ERGIC and the recruitment of TBK183. The E3 ubiquitin ligase RNF26 then catalyzes the K11-linked polyubiquitination of STING at K150, thereby protecting STING from K48-linked ubiquitination and degradation in the early phase of viral infection84. On the other hand, these modifications suppress antiviral activity by degrading IRF3 in an autophagy-dependent manner in the late phase of viral infection. Mitochondrial E3 ubiquitin-protein ligase 1 (MUL1, an E3 ubiquitin ligase) catalyzes the K63-linked ubiquitination of STING at K224, which is a required modification for IRF3 activation but not for NF-κB activation85. The K6-linked ubiquitination of STING at K19 by TRIM13, K48-linked ubiquitination of STING at K83, K151, K288, and K337 by TRIM29, and K48-linked ubiquitination of STING at K275 by TRIM30α promote proteasome-dependent STING degradation, thereby suppressing antiviral responses86,87,88. Thus, K11-, K27-, or K63-linked ubiquitination mostly enhances the STING-mediated antiviral response, whereas K6- or K48-linked ubiquitination suppresses this response.
The cause and immunostimulatory effect of mtDNA release
The endosymbiotic model, which indicates that mitochondria are derived from ancient aerobic bacteria, mitochondria and bacteria share several similar features, such as a circular genome and hypomethylated/unmethylated CpG dinucleotide motifs89. Owing to these similarities, mitochondrial components, such as mtDNA, are recognized as DAMPs by the host immune system upon release from mitochondria into the cytoplasmic or extracellular milieu. Recent extensive studies have shown that mtDNA is released under pathological conditions, such as oxidative stress, genotoxic stress, high levels of proinflammatory factors, viral infection, and mitochondrial dysfunction, subsequently activating the cGAS-STING pathway.
Oxidative stress
Numerous studies have shown that oxidative stress caused by excess reactive oxygen species (ROS) production is a major source of DNA damage4. Intracellular ROS are predominantly produced in the electron transport chain (ETC) in the mitochondrial matrix during oxidative phosphorylation4. Compared to nuclear DNA, mtDNA is more vulnerable to oxidative damage because mtDNA resides near the ETC and is not protected by histones4,10,11. As a result, mitochondrial oxidative stress disrupts mtDNA integrity, facilitating the release of oxidized mtDNA into the cytoplasm and eventually into the extracellular space, thereby triggering IFN and proinflammatory responses4,10,11,17.
Three prime exonuclease 1 (TREX1), which is localized to the ER in the basal state, degrades cytosolic DNA and prevents the activation of the cGAS-STING pathway90,91,92. Loss of TREX1 activity in humans and mice causes autoimmune diseases such as systemic lupus erythematosus (SLE) and Aicardi–Goutieres syndrome91,92. Oxidized DNA is resistant to TREX1-mediated degradation and therefore accumulates in the cytosol, leading to cGAS activation90. Therefore, one can infer that mtDNA released into the cytosol or extracellular space in response to oxidative damage induces particularly robust inflammatory responses. Indeed, in SLE, oxidized mtDNA-protein complexes are released from mitochondria into the cytoplasm in the neutrophils and are spontaneously extruded to the extracellular space93. The extrusion of oxidized mtDNA in turn activates plasmacytoid DCs and causes the production of anti–oxidizing mtDNA autoantibodies93. In a mouse model of SLE, mitochondrial oxidation induced neutrophil extracellular trap (NETosis), releasing oxidized mtDNA into the extracellular environment94. Consistent with this outcome, reducing mtROS via the mitochondrion-targeted antioxidant Mito-TEMPO ameliorated disease in a mouse model of SLE94.
Inflammasome activation
Oxidative damage to both mitochondria and mtDNA is essential for the activation of the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome, which triggers IL-1β secretion from macrophages10,95. Activation of the NLRP3 inflammasome requires a priming step stimulated by PAMPs (e.g., LPS) or inflammatory cytokines, followed by an activation step mediated through the assembly of components, such as the NLRP3 inflammasome, apoptosis-associated speck-like protein containing a CARD (ASC), and pro-caspase-110,11,95. The priming step increases the expression of inflammasome components, lowers mitochondrial membrane potential and increases mtROS levels and de novo synthesis of mtDNA10,11,95. Depletion of mtDNA with ethidium bromide treatment or knockdown of mtDNA polymerase-γ (POLγ) inhibited NLRP3 inflammasome activation in mouse bone marrow-derived macrophages (BMDMs)11. Deficient mitochondrial transcription factor A (TFAM), which regulates the replication and compaction of mtDNA, markedly reduced the mtDNA content and suppressed inflammasome activation in BMDMs11. TFAM ablation reduced both mtROS and ox-mtDNA in response to NLRP3 activators, although the expression levels of general inflammasome components, such as pro–IL-1β, pro-caspase1, and NLRP3, remained unchanged11. Treatment of TFAM-deficient BMDMs with synthetic mtDNA fragments containing the oxidized nucleotide 8-OH-dGTP restored NLRP3 inflammasome activation, supporting the notion that mtDNA synthesis during the priming step is essential for inflammasome activation11. Indeed, LPS promotes mtDNA synthesis by activating mitochondrial deoxyribonucleotide kinase UMP-CMPK (CMPK2), which provides dCTP for mtDNA replication and increases mtROS levels11. The IL-1β and IFN pathways are interlinked as indicated by IL-1β induction of IFN production and suppression of viral replication96,97. IL-1β also triggers mtDNA release and activates the cGAS-STING pathway by reducing mitochondrial membrane potential and increasing mitochondrial mass97. Thus, mtDNA release and IL-1β secretion promote chronic inflammation by driving a feed-forward loop.
Disruption of mitochondrial integrity
Oxidatively damaged mitochondria contain small mtDNA fragments that are released. When the permeability of mitochondrial membranes was increased98,99, TFAM protected mtDNA from damage, but surprisingly, Tfam ± cells released more mtDNA fragments into the cytosol and exhibited higher cGAS-STING activity, as evidenced by the upregulation of ISG expression and antiviral response17. On the other hand, mitochondrial fission, which clears damaged mitochondria via mitophagy, suppressed the upregulation of ISG expression, but depletion of the mtDNA quality-control enzyme endonuclease G-like 1 (EXOG) increased ISG expression in these cells17.
YME1L deficiency also induces mtDNA release into the cytosol and triggers the innate immune response through the cGAS–STING pathway100. YME1L is an ATP-dependent metalloprotease located on the inner mitochondrial membrane and coordinates mitochondrial biogenesis and dynamics by balancing the fusion and fission of mitochondria100. YME1L deficiency causes the accumulation of numerous YME1L substrate proteins associated with mtDNA metabolism, including solute carrier family 25 member 33 (SLC25A33), CMPK2, and nucleoside diphosphate kinase (NME4)100. More direct evidence for the role of oxidative damage in mtDNA release has been demonstrated by studies showing the role of thioredoxin-2 (Trx2), which scavenges mtROS, in regulating mtDNA release101. Trx2 deficiency increased the level of mitochondrial superoxide, hydrogen peroxide and cytosolic mtDNA in brown adipose tissue101. Increased levels of NLRP3, cleaved caspase-1, and mature IL-1β in these cells suggested NLRP3 inflammasome activation, which, as we described above, required the release of oxidized mtDNA101.
Channels on the mitochondrial membrane involved in innate immunity
Early understanding of mtDNA release came from studying the mitochondrial apoptosis pathway, which requires mitochondrial outer membrane permeabilization (MOMP) to release cytochrome C, which initiates the apoptotic cascade102. The Bcl-2 family of proteins, BCL2 antagonist/killer (BAK) and BCL2-associated X (BAX), which accumulate in the mitochondrial outer membrane (MOM) in response to apoptotic stimuli, were the first mediators of MOMP to be identified103,104. Subsequent studies demonstrated that voltage-dependent anion channels (VDACs) were required for BAX/BAK-mediated MOMP and apoptosis104. At the time of its discovery, it was thought that VDAC was a component of the mitochondrial permeability transition pore (mPTP) in the mitochondrial inner membrane, the opening of which leads to mitochondrial swelling and rupture. However, VDAC has since been shown to not be a component of the mPTP99, raising the question, how is VDAC involved in MOMP?
mtDNA efflux through BAK/BAX macropores
During apoptosis, cytochrome c is released from mitochondria through the BAK/BAX pore and binds to apoptosis protease activator-1 (Apaf-1) and pro-caspase-9 to induce the apoptosome complex105. The assembly of apoptosomes activates caspase-9, an apoptosis-initiating caspase, which in turn activates the effector caspase-3 or caspase-7, leading to the biochemical and morphological features of apoptosis105. However, immunogenic mitochondrial components (e.g., mtDNA) are released during apoptosis, raising the possibility that apoptosis can trigger systemic inflammation102,105. Such harmful outcomes are averted by a several built-in safety mechanism. For example, infection by thrombocytopenia syndrome virus (SFTSV) activates BAK/BAX and the NLRP3 inflammasome106. Activation of BAK/BAX increases +intracellular [AMP]/[ATP] ratio, which activates AMPK and induces autophagy in an ATG5/7-dependent manner107, thereby removing damaged mitochondria, which are likely to be sources of released mtDNA. Moreover, caspase-3 is activated during apoptosis and cleaves cGAS at D319 and IRF-3 at D121/125102,108,109, and caspase-1 cleaves cGAS at D140/D157, further inactivating the type I IFN pathway9 (Fig. 5).

Mitochondrial stress induces the release of mtDNA and subsequently activates the cGAS-STING pathway. This signaling pathway can be regulated by various posttranslational modifications and can also be suppressed by caspases during apoptosis.
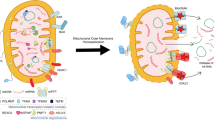
Microscopy studies of MOMP after treatment with ABT-737, a BAK/BAX activator, showed that the elongated mitochondrial network becomes hyperfragmented during apoptosis and then, BAK/BAX is oligomerized on the MOM, forming macropores109,110. The mitochondrial inner membrane (MIM) then herniates through the BAK/BAX macropores, releasing mitochondrial matrix components, including mtDNA, into the cytosol109,110 (Fig. 6).

(Left) During BAK/BAX-mediated apoptosis, mitochondria undergo hyperfragmentation, and mitochondrial matrix components, including mtDNA, are released into the cytosol through BAK/BAX macropores. mPTP opening is not required for MIM disruption. The released mtDNA does not activate the type-I IFN pathway because of caspase activation. (Right) Mitochondrial stress triggers the formation of VDAC-oligomer pores and activates the type-I IFN pathway. mPTP opening is required for MIM disruption.
mtDNA release through VDAC oligomer pores
VDAC is the most abundant MOM protein and is the primary channel through which Ca2+, anions, cations, ATP, and metabolites are transported between mitochondria and other compartments of a cell111. VDAC is a β-barrel protein expressed as three isoforms (VDAC1, VDAC2 and VDAC3), of which VDAC1 is most highly and ubiquitously expressed16,99. Although VDACs are monomers involved in channel functions, they exist in dynamic equilibrium with the oligomeric forms112. Equilibrium shifts towards the oligomeric form during apoptosis, which, as mentioned above, is accompanied by MOMP and cytochrome C release113. Although the structure of VDAC oligomers in apoptotic cells is not known, it is reasonable to propose that they form macropores similar to BAK/BAX. All VDACs contain a flexible amphipathic N-terminal domain that lies inside the channel but is thought to translocate out of the pore when voltage is applied across the VDAC channel 98 or during apoptosis16. Evidence suggests that the translocated N-terminal domain may generate a hydrophilic surface around the circumference of the VDAC macropore, stabilizing the VDAC–water interface98. The crucial difference between VDAC and BAK/BAX pores is that VDAC oligomers can also form in living (not apoptotic) cells and require mitochondrial permeability transition pore (mPTP) opening for permeabilization of the MIM98. Thus, even in normally growing cells, VDAC oligomers form on stressed mitochondria, which in combination with the opening of the mPTP releases mtDNA98. Because these oligomers form in BAK/BAX-deficient cells, the BAK/BAX macropore is not required for MOMP in living cells98. Because mtDNA is released in the absence of apoptosis or caspase activity, the cGAS-STING signal remains intact and activates the type I IFN pathway.
Consistent with the notion that VDAC oligomers release mtDNA, VBIT-4, a small- molecule inhibitor of VDAC, inhibits mtDNA release and type-I IFN signaling, ameliorating inflammatory diseases such as SLE in a mouse model 98. The requisite release of mtDNA for the NLRP3 inflammasome activation has been shown to be mediated by VDAC oligomerization and blocked by VBIT-495. More recently, it was discovered that COVID-19 caused type-I IFN pathology in endothelial cells by inducing VDAC oligomerization, which led to released mtDNA and activation of the cGAS-STING pathway114 (Fig. 6).
The pathological role of VDAC oligomerization-induced release of mtDNA extends beyond conditions classically considered to be immunity-released diseases. Amyotrophic lateral sclerosis (ALS) is a motor neuron disease that leads to paralysis and nearly 100% mortality115. One of the etiologies of ALS is mislocalization of TAR DNA-binding protein 43 (TDP-43) to mitochondria, causing neurotoxicity115. TDP-43–mediated inflammation depends on cGAS/STING, which is activated by released mtDNA115. TDP-43 does not induce apoptosis, and BAK/BAX are not required for TDP-43-dependent mtDNA release or inflammatory cytokine gene expression115. VBIT-4 or genetic deletion of VDAC1 decreases cytoplasmic mtDNA levels and inflammation in motor neurons derived from iPSCs of ALS patients and MEFs, and blockade of the cGAS/STING pathway ameliorates the related neurodegeneration115.
Conclusions and perspectives
The discovery of the cGAS-STING pathway has led to a paradigm shift in which ectopically localized self-DNA, similar to microbial DNA, causes disease. In this review, we highlight the biological properties of mtDNA released into the cytosol and the mechanism by which mtDNA crosses the MOM. In addition, we presented comprehensive updates on the regulation of the cGAS-STING pathway. The mechanism by which MIM permeabilization (MIMP) occurs is not understood as well as that of MOMP. In the context of MOMP induced by the BAK/BAX activator ABT-737, MIM and the matrix herniate and rupture without opening the mPTP. On the other hand, VDAC oligomer-induced MOMP requires mPTP opening, suggesting that an influx of small molecules and water through the mPTP into the matrix may mediate MIM disruption. The requirement for mPTP opening for VDAC oligomer-mediated mtDNA release may be an indication that the VDAC macropore is much smaller than the BAK/BAX macropore and may require an outward force applied to the MIM to drive mtDNA release. Thus, inhibitors of VDAC oligomerization, along with inhibitors of the cGAS-STING pathway, may become valuable tools in treating inflammatory diseases. One potential benefit of VDAC oligomerization inhibitors in the treatment of inflammation and autoimmunity is that this strategy of targeting mtDNA release may not diminish antimicrobial immunity to the same extent as the use of anti-cGAS-STING inhibitors. Understanding how the VDAC oligomer forms and the nature of its interactions with other MOM proteins, such as Bcl-2 family members (e.g., BAK and BAX), may provide further insights for targeting MOMP as a therapeutic strategy.
References
Ishikawa, H. & Barber, G. N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 (2008).
Andreeva, L. et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature 549, 394–398 (2017).
Civril, F. et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498, 332–337 (2013).
West, A. P., Shadel, G. S. & Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 11, 389–402 (2011).
Takeuchi, O. & Akira, S. Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010).
Ning, X. et al. Apoptotic caspases suppress type I interferon production via the cleavage of cGAS, MAVS, and IRF3. Mol. Cell 74, 19–31.e17 (2019).
Liu, S. et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630 (2015).
Zhou, R., Yazdi, A. S., Menu, P. & Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011).
Wang, Y. et al. Inflammasome activation triggers caspase-1-mediated cleavage of cGAS to regulate responses to DNA virus infection. Immunity 46, 393–404 (2017).
Shimada, K. et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414 (2012).
Zhong, Z. et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560, 198–203 (2018).
Gao, P. et al. Cyclic [G(2’,5’)pA(3’,5’)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153, 1094–1107 (2013).
Herzner, A. M. et al. Sequence-specific activation of the DNA sensor cGAS by Y-form DNA structures as found in primary HIV-1 cDNA. Nat. Immunol. 16, 1025–1033 (2015).
Zhang, C. et al. Structural basis of STING binding with and phosphorylation by TBK1. Nature 567, 394–398 (2019).
Sun, L., Wu, J., Du, F., Chen, X. & Chen, Z. J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013).
Shoshan-Barmatz, V., Keinan, N., Abu-Hamad, S., Tyomkin, D. & Aram, L. Apoptosis is regulated by the VDAC1 N-terminal region and by VDAC oligomerization: release of cytochrome c, AIF and Smac/Diablo. Biochim. Biophys. Acta 1797, 1281–1291 (2010).
West, A. P. et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557 (2015).
Sun, W. et al. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc. Natl Acad. Sci. USA 106, 8653–8658 (2009).
Zhong, B. et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29, 538–550 (2008).
Woodward, J. J., Iavarone, A. T. & Portnoy, D. A. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328, 1703–1705 (2010).
Li, X. D. et al. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341, 1390–1394 (2013).
Li, X. et al. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced ligomerization. Immunity 39, 1019–1031 (2013).
Ablasser, A. et al. cGAS produces a 2’-5’-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384 (2013).
Kranzusch, P. J., Lee, A. S., Berger, J. M. & Doudna, J. A. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep. 3, 1362–1368 (2013).
Lahaye, X. et al. NONO detects the nuclear HIV capsid to promote cGAS-mediated innate immune activation. Cell 175, 488–501.e422 (2018).
Barnett, K. C. et al. Phosphoinositide interactions position cGAS at the plasma membrane to ensure efficient distinction between self- and viral DNA. Cell 176, 1432–1446.e1411 (2019).
Mankan, A. K. et al. Cytosolic RNA:DNA hybrids activate the cGAS-STING axis. EMBO J. 33, 2937–2946 (2014).
Luecke, S. et al. cGAS is activated by DNA in a length-dependent manner. EMBO Rep. 18, 1707–1715 (2017).
Xie, W. et al. Human cGAS catalytic domain has an additional DNA-binding interface that enhances enzymatic activity and liquid-phase condensation. Proc. Natl. Acad. Sci. USA 116, 11946–11955 (2019).
Du, M. & Chen, Z. J. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361, 704–709 (2018).
Seo, G. J. et al. Akt kinase-mediated checkpoint of cGAS DNA sensing pathway. Cell Rep. 13, 440–449 (2015).
Sun, X. et al. DNA-PK deficiency potentiates cGAS-mediated antiviral innate immunity. Nat. Commun. 11, 6182 (2020).
Li, T. et al. Phosphorylation and chromatin tethering prevent cGAS activation during mitosis. Science 371, eabc5386 (2021).
Li, M. & Shu, H. B. Dephosphorylation of cGAS by PPP6C impairs its substrate binding activity and innate antiviral response. Protein Cell 11, 584–599 (2020).
Xia, P. et al. Glutamylation of the DNA sensor cGAS regulates its binding and synthase activity in antiviral immunity. Nat. Immunol. 17, 369–378 (2016).
Hu, M. M. et al. Sumoylation promotes the stability of the DNA sensor cGAS and the adaptor STING to regulate the kinetics of response to DNA virus. Immunity 45, 555–569 (2016).
Wang, Q. et al. The E3 ubiquitin ligase RNF185 facilitates the cGAS-mediated innate immune response. PLoS Pathog. 13, e1006264 (2017).
Chen, M. et al. TRIM14 inhibits cGAS degradation mediated by selective autophagy receptor p62 to promote innate immune responses. Mol. Cell 64, 105–119 (2016).
Ohkuri, T. et al. Intratumoral administration of cGAMP transiently accumulates potent macrophages for anti-tumor immunity at a mouse tumor site. Cancer Immunol. Immunother. 66, 705–716 (2017).
Li, T. et al. Antitumor activity of cGAMP via stimulation of cGAS-cGAMP-STING-IRF3 mediated innate immune response. Sci. Rep. 6, 19049 (2016).
Dorostkar, F. et al. Co-administration of 2'3’-cGAMP STING activator and CpG-C adjuvants with a mutated form of HPV 16 E7 protein leads to tumor growth inhibition in the mouse model. Infect. Agent. Cancer 16, 7 (2021).
Carozza, J. A. et al. Extracellular cGAMP is a cancer cell-produced immunotransmitter involved in radiation-induced anti-cancer immunity. Nat. Cancer 1, 184–196 (2020).
Kato, K. et al. Structural insights into cGAMP degradation by Ecto-nucleotide pyrophosphatase phosphodiesterase 1. Nat. Commun. 9, 4424 (2018).
Carozza, J. A. et al. ENPP1’s regulation of extracellular cGAMP is a ubiquitous mechanism of attenuating STING signaling. Proc. Natl Acad. Sci. USA 119, e2119189119 (2022).
Ritchie, C., Cordova, A. F., Hess, G. T., Bassik, M. C. & Li, L. SLC19A1 is an importer of the immunotransmitter cGAMP. Mol. Cell 75, 372–381.e375 (2019).
Luteijn, R. D. et al. SLC19A1 transports immunoreactive cyclic dinucleotides. Nature 573, 434–438 (2019).
Zhou, Y. et al. Blockade of the phagocytic receptor MerTK on tumor-associated macrophages enhances P2X7R-dependent STING activation by tumor-derived cGAMP. Immunity 52, 357–373.e359 (2020).
Drill, M. et al. Inhibition of purinergic P2X receptor 7 (P2X7R) decreases granulocyte-macrophage colony- stimulating factor (GM-CSF) expression in U251 glioblastoma cells. Sci. Rep. 10, 14844 (2020).
Zhou, C. et al. Transfer of cGAMP into bystander cells via LRRC8 volume-regulated anion channels augments STING-mediated interferon responses and anti-viral immunity. Immunity 52, 767–781.e766 (2020).
Xie, Y. et al. Gut epithelial TSC1/mTOR controls RIPK3-dependent necroptosis in intestinal inflammation and cancer. J. Clin. Invest 130, 2111–2128 (2020).
Li, X. et al. An alternatively spliced STING isoform localizes in the cytoplasmic membrane and directly senses extracellular cGAMP. J. Clin. Invest 132, e144339 (2022).
Ablasser, A. et al. Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 503, 530–534 (2013).
Kalamvoki, M., Du, T. & Roizman, B. Cells infected with herpes simplex virus 1 export to uninfected cells exosomes containing STING, viral mRNAs, and microRNAs. Proc. Natl. Acad. Sci. USA 111, E4991–E4996 (2014).
Bridgeman, A. et al. Viruses transfer the antiviral second messenger cGAMP between cells. Science 349, 1228–1232 (2015).
Liu, H. et al. cGAS facilitates sensing of extracellular cyclic dinucleotides to activate innate immunity. EMBO Rep. 20, e46293 (2019).
Shang, G., Zhang, C., Chen, Z. J., Bai, X. C. & Zhang, X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature 567, 389–393 (2019).
Yin, Q. et al. Cyclic di-GMP sensing via the innate immune signaling protein STING. Mol. Cell 46, 735–745 (2012).
Ouyang, S. et al. Structural analysis of the STING adaptor protein reveals a hydrophobic dimer interface and mode of cyclic di-GMP binding. Immunity 36, 1073–1086 (2012).
Huang, Y. H., Liu, X. Y., Du, X. X., Jiang, Z. F. & Su, X. D. The structural basis for the sensing and binding of cyclic di-GMP by STING. Nat. Struct. Mol. Biol. 19, 728–730 (2012).
Shang, G. et al. Crystal structures of STING protein reveal basis for recognition of cyclic di-GMP. Nat. Struct. Mol. Biol. 19, 725–727 (2012).
Ergun, S. L., Fernandez, D., Weiss, T. M. & Li, L. STING polymer structure reveals mechanisms for activation, hyperactivation, and inhibition. Cell 178, 290–301.e210 (2019).
Dobbs, N. et al. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe 18, 157–168 (2015).
Zhao, B. et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature 569, 718–722 (2019).
Zhao, B. et al. Structural basis for concerted recruitment and activation of IRF-3 by innate immune adaptor proteins. Proc. Natl. Acad. Sci. USA 113, E3403–E3412 (2016).
Uzé, G., Lutfalla, G. & Gresser, I. Genetic transfer of a functional human interferon alpha receptor into mouse cells: cloning and expression of its cDNA. Cell 60, 225–234 (1990).
Novick, D., Cohen, B. & Rubinstein, M. The human interferon alpha/beta receptor: characterization and molecular cloning. Cell 77, 391–400 (1994).
Cohen, B., Novick, D., Barak, S. & Rubinstein, M. Ligand-induced association of the type I interferon receptor components. Mol. Cell. Biol. 15, 4208–4214 (1995).
Ivashkiv, L. B. & Donlin, L. T. Regulation of type I interferon responses. Nat. Rev. Immunol. 14, 36–49 (2014).
Schoggins, J. W. et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485 (2011).
Hou, Y. et al. Non-canonical NF-κB antagonizes STING sensor-mediated DNA sensing in radiotherapy. Immunity 49, 490–503.e494 (2018).
Fang, R. et al. NEMO-IKKβ are essential for IRF3 and NF-κB activation in the cGAS-STING pathway. J. Immunol. 199, 3222–3233 (2017).
Abe, T. & Barber, G. N. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J. Virol. 88, 5328–5341 (2014).
Balka, K. R. et al. TBK1 and IKKε act redundantly to mediate STING-induced NF-κB responses in myeloid cells. Cell Rep. 31, 107492 (2020).
de Oliveira Mann, C. C. et al. Modular architecture of the STING C-terminal tail allows interferon and NF-κB signaling adaptation. Cell Rep. 27, 1165–1175.e1165 (2019).
Cerboni, S. et al. Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J. Exp. Med. 214, 1769–1785 (2017).
Bakhoum, S. F. et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472 (2018).
Tsuchida, T. et al. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double- stranded DNA. Immunity 33, 765–776 (2010).
Yang, L. et al. UBXN3B positively regulates STING-mediated antiviral immune responses. Nat. Commun. 9, 2329 (2018).
Yang, B. et al. RNF90 negatively regulates cellular antiviral responses by targeting MITA for degradation. PLoS Pathog. 16, e1008387 (2020).
Zhong, B. et al. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity 30, 397–407 (2009).
Zhang, J., Hu, M. M., Wang, Y. Y. & Shu, H. B. TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J. Biol. Chem. 287, 28646–28655 (2012).
Wang, Q. et al. The E3 ubiquitin ligase AMFR and INSIG1 bridge the activation of TBK1 kinase by modifying the adaptor STING. Immunity 41, 919–933 (2014).
Zhang, Z. D. et al. RNF115 plays dual roles in innate antiviral responses by catalyzing distinct ubiquitination of MAVS and MITA. Nat. Commun. 11, 5536 (2020).
Qin, Y. et al. RNF26 temporally regulates virus-triggered type I interferon induction by two distinct mechanisms. PLoS Pathog. 10, e1004358 (2014).
Ni, G., Konno, H. & Barber, G. N. Ubiquitination of STING at lysine 224 controls IRF3 activation. Sci. Immunol. 2, eaah7119 (2017).
Wang, Y. et al. TRIM30α is a negative-feedback regulator of the intracellular DNA and DNA virus- triggered response by targeting STING. PLoS Pathog. 11, e1005012 (2015).
Li, Q. et al. TRIM29 negatively controls antiviral immune response through targeting STING for degradation. Cell Disco. 4, 13 (2018).
Li, X. et al. The transmembrane endoplasmic reticulum-associated E3 ubiquitin ligase TRIM13 restrains the pathogenic-DNA-triggered inflammatory response. Sci. Adv. 8, eabh0496 (2022).
Kraus, F., Roy, K., Pucadyil, T. J. & Ryan, M. T. Function and regulation of the divisome for mitochondrial fission. Nature 590, 57–66 (2021).
Gehrke, N. et al. Oxidative damage of DNA confers resistance to cytosolic nuclease TREX1 degradation and potentiates STING-dependent immune sensing. Immunity 39, 482–495 (2013).
Stetson, D. B., Ko, J. S., Heidmann, T. & Medzhitov, R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598 (2008).
Peschke, K. et al. Loss of Trex1 in dendritic cells is sufficient to trigger systemic autoimmunity. J. Immunol. 197, 2157–2166 (2016).
Caielli, S. et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J. Exp. Med. 213, 697–713 (2016).
Lood, C. et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 22, 146–153 (2016).
Xian, H. et al. Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity 55, 1370–1385.e8 (2022).
Orzalli, M. H. et al. An antiviral branch of the IL-1 signaling pathway restricts immune-evasive virus replication. Mol. Cell 71, 825–840.e826 (2018).
Aarreberg, L. D. et al. Interleukin-1β Induces mtDNA release to activate innate immune signaling via cGAS-STING. Mol. Cell 74, 801–815.e806 (2019).
Kim, J. et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus- like disease. Science 366, 1531–1536 (2019).
Baines, C. P., Kaiser, R. A., Sheiko, T., Craigen, W. J. & Molkentin, J. D. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 9, 550–555 (2007).
Sprenger, H. G. et al. Cellular pyrimidine imbalance triggers mitochondrial DNA-dependent innate immunity. Nat. Metab. 3, 636–650 (2021).
Huang, Y. et al. Brown adipose TRX2 deficiency activates mtDNA-NLRP3 to impair thermogenesis and protect against diet-induced insulin resistance. J. Clin. Invest 132, e148852 (2022).
Rongvaux, A. et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159, 1563–1577 (2014).
Merry, D. E. & Korsmeyer, S. J. Bcl-2 gene family in the nervous system. Annu. Rev. Neurosci. 20, 245–267 (1997).
Shimizu, S., Narita, M. & Tsujimoto, Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399, 483–487 (1999).
Schafer, Z. T. & Kornbluth, S. The apoptosome: physiological, developmental, and pathological modes of regulation. Dev. Cell 10, 549–561 (2006).
Li, S. et al. SFTSV infection induces BAK/BAX-dependent mitochondrial DNA release to trigger NLRP3 inflammasome activation. Cell Rep. 30, 4370–4385.e4377 (2020).
Lindqvist, L. M. et al. Autophagy induced during apoptosis degrades mitochondria and inhibits type I interferon secretion. Cell Death Differ. 25, 784–796 (2018).
White, M. J. et al. Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell 159, 1549–1562 (2014).
McArthur, K. et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359, eaao6047 (2018).
Riley, J. S. et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. 37, e99238 (2018).
Shoshan-Barmatz, V., Krelin, Y. & Shteinfer-Kuzmine, A. VDAC1 functions in Ca(2+) homeostasis and cell life and death in health and disease. Cell Calcium 69, 81–100 (2018).
Krause, J., Hay, R., Kowollik, C. & Brdiczka, D. Cross-linking analysis of yeast mitochondrial outer membrane. Biochim. Biophys. Acta 860, 690–698 (1986).
Zalk, R., Israelson, A., Garty, E. S., Azoulay-Zohar, H. & Shoshan-Barmatz, V. Oligomeric states of the voltage-dependent anion channel and cytochrome c release from mitochondria. Biochem. J. 386, 73–83 (2005).
Domizio, J. D. et al. The cGAS-STING pathway drives type I IFN immunopathology in COVID-19. Nature 603, 145–151 (2022).
Yu, C. H. et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 183, 636–649 e618 (2020).
Acknowledgements
This work was supported by the intramural research program of National Heart Lung and Blood Institute (NHLBI) in NIH, and by National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. RS-2023-00210934, No. 2022R1F1A1072984 and No. 2022R1A2C1010531), by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (grant number HI14C1176), and by the Catholic Medical Center Research Foundation made in the program year of 2022 and 2023. The cartoons were generated using BioRender.com.
Funding
Open Access funding provided by the National Institutes of Health (NIH).
Author information
Authors and Affiliations
Contributions
J.K. curated the information and wrote and supervised the manuscript. H-S.K. provided conceptual feedback. J.H.C. revised and supervised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, J., Kim, HS. & Chung, J.H. Molecular mechanisms of mitochondrial DNA release and activation of the cGAS-STING pathway. Exp Mol Med 55, 510–519 (2023). https://doi.org/10.1038/s12276-023-00965-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-023-00965-7
This article is cited by
-
The cGAS-STING pathway in cardiovascular diseases: from basic research to clinical perspectives
Cell & Bioscience (2024)
-
A break in mitochondrial endosymbiosis as a basis for inflammatory diseases
Nature (2024)
-
Advances and challenges in modeling inherited peripheral neuropathies using iPSCs
Experimental & Molecular Medicine (2024)
-
Cytosolic Escape of Mitochondrial DNA Triggers cGAS-STING Pathway-Dependent Neuronal PANoptosis in Response to Intermittent Hypoxia
Neurochemical Research (2024)
-
SS-31 inhibits mtDNA–cGAS–STING signaling to improve POCD by activating mitophagy in aged mice
Inflammation Research (2024)
