
Abstract
The NLRC4 inflammasome, a member of the nucleotide-binding and oligomerization domain-like receptor (NLR) family, amplifies inflammation by facilitating the processing of caspase-1, interleukin (IL)–1β, and IL-18. We explored whether NLRC4 knockdown alleviated inflammatory injury following intracerebral hemorrhage (ICH). Furthermore, we investigated whether NLRC4 inflammasome activation can be adjusted by the regulator of G protein signaling 2/leucine-rich repeat kinase-2 pathway. Fifty microliters of arterial blood was drawn and injected into the basal ganglion to simulate the ICH model. NLRC4 small interfering RNAs (siRNAs) were utilized to knockdown NLRC4. An LRRK2 inhibitor (GNE7915) was injected into the abdominal cavity. Short hairpin (sh) RNA lentiviruses and lentiviruses containing RGS2 were designed and applied to knockdown and promote RGS2 expression. Neurological functions, brain edema, Western blot, enzyme-linked immunosorbent, hematoxylin and eosin staining, Nissl staining, immunoprecipitation, immunofluorescence assay and Evans blue dye extravasation and autofluorescence assay were evaluated. It was shown that the NLRC4 inflammasome was activated following ICH injury. NLRC4 knockdown extenuated neuronal death, damage to the blood-brain barrier, brain edema and neurological deficiency 3 days after ICH. NLRC4 knockdown reduced myeloperoxidase (MPO) cells as well as tumor necrosis factor (TNF)-α, interleukin (IL)-6, IL-1β and IL-18 following ICH. GNE7915 reduced pNLRC4 and NLRC4 inflammasome activation. RGS2 suppressed the interaction of LRRK2 and NLRC4 and NLRC4 inflammasome activation by regulating pLRRK2. Our study demonstrated that the NLRC4 inflammasome may aggravate the inflammatory injury induced by ICH and that RGS2/LRRK2 may relieve inflammatory injury by restraining NLRC4 inflammasome activation.
Similar content being viewed by others
Introduction
Intracerebral hemorrhage (ICH) is characterized by high mortality and high disability1, although it accounts for 10–15% of all stroke types2. A considerable amount of evidence has shown that intracerebral hemorrhage leads to a series of pathophysiological changes, including inflammation, edema, apoptosis, and necrosis3. Inflammation plays a key role in ICH-induced injury by releasing proinflammatory cytokines, especially interleukin (IL)-1β4. Increased expression of IL‐1β and IL‐18 is often observed upon brain injury5. The inflammasome, a part of the innate immune system, can cut pro-caspase-1 into cleaved caspase-1, which makes pro-interleukin-1β and pro-interleukin-18 mature and causes inflammatory responses6.
The nucleotide-binding and oligomerization domain-like receptor (NLR) family responds to innate immunity by forming inflammasomes7. NLRC4 (CARD12, IPAF) recruits pro-caspase‐1 directly through its CARDs without ASC (Pycard, PYD and CARD domain containing), although ASC contribute to the maturation of IL‐1β, cleaved caspase‐1 and IL‐188. Moreover, phosphorylation at NLRC4-Ser533 is critical in the activation of the NLRC4 inflammasome9. The NLRC4 inflammasome is activated in bacterial inflammation10. However, neuroinflammation in ICH is sterile inflammation. The phosphorylation at NLRC4-Ser533 and the activation of the NLRC4 inflammasome after ICH remain to be elucidated.
Regulator of G protein signaling 2 (RGS2) consists of a single RGS domain with minimal flanking amino and carboxy-terminal regions11. The protective role of RGS2 in anxiety, panic disorder, and suicide has been reported12. Our previous study showed that RGS2 expression was upregulated and relieved inflammatory injury in a collagenase-induced intracerebral hemorrhage model13. However, the mechanism of the protective role of RGS2 in ICH remains unclear. Leucine-rich repeat kinase-2 (LRRK2) consists of GTPase domains, functional kinase domains, and multiple domains for protein–protein interactions14. LRRK2 is a well-known kinase closely related to Parkinson’s disease (PD)15,16. Intriguingly, Cui H and his colleagues have shown that LRRK2 kinase can activate the NLRC4 inflammasome in acute Salmonella typhimurium infection by promoting the phosphorylation of NLRC4 at Ser533 with interaction with NLRC417. A report indicated that the level of phosphorylation of Ser935 can be used to assess the activity of LRRK218. RGS2 modulates LRRK2 function by restricting the phosphorylation of LRRK2 in Parkinson’s disease19.
Therefore, our study aimed to investigate whether the NLRC4 inflammasome is activated by phosphorylation at NLRC4-Ser533 after ICH. We also explored whether LRRK2 aggravates the NLRC4 inflammasome and whether RGS2 regulates activation of the NLRC4 inflammasome after ICH via LRRK2.
Materials and methods
Rats
Sprague–Dawley rats (300–350 g, healthy, male and adult) were purchased from Chongqing Medical University with the license of the institutional animal care and use committee. All animal procedures were reviewed by the Institutional Animal Ethics Committee (IACUC) of Chongqing Medical University, Chongqing, China (NIH Publication No. 85–23, revised 1996). All animals used in this experiment were cared for in strict accordance with the Guide for the Care and Use of Laboratory Animals. All rats were housed under constant temperature (25–26 °C) with sufficient food and water. All efforts were made to relieve pain and unintentional death20.
Experimental design
A schematic of the experimental design, including the numbers of animals, groups and experimental methods, is shown in Fig. 1b.

a A schematic of the ICH model including indication of the stereotactic injection coordinates, hematoma volume, and the regions of the perihematomal area. b A schematic of the experimental design and animal groups.
Experiment 1
To investigate the expression of NLRC4 and pNLRC4 and the interaction of NLRC4 with pro-caspase-1 after ICH, 36 rats were randomly divided into six groups: sham, 12, 24, 48, 72 h, and 7 d after ICH (n = 6). The samples of each group were used for Western blot (WB). In addition, the sham and ICH 72 h samples were shared for coimmunoprecipitation (Co-IP).
Experiment 2
To explore the effects of NLRC4 on neuroinflammation, the number of nerve cells, and brain injury, 96 rats were randomly assigned to four groups: sham, ICH, ICH + NC siRNA and ICH + NLRC4 siRNA. Nine rats in each group were evaluated with a modified neurological severity score (mNSS). All rats were sacrificed at 72 h after ICH for brain water content (n = 6), immunohistochemistry (IHC)/immunofluorescence (IF) (n = 6), Evans Blue Dye Extravasation and Autofluorescence (EB) (n = 6), and Western blot (WB)/enzyme-linked immunosorbent assay (ELISA) (n = 6).
Experiment 3
To explore the mechanisms of NLRC4 inflammasome activation, 66 rats were randomly divided into eleven groups: sham, ICH, ICH + GNE7915, ICH + vehicle, ICH + shRGS2, ICH + NC shRNA, ICH + shRGS2+si-NLRC4, ICH + shRGS2+NC siRNA, ICH + Lenti-RGS2, ICH + shRGS2 + GNE7915, and ICH + shRGS2 + vehicle. The samples of the sham, ICH, ICH + shRGS2, ICH + NC shRNA and ICH + Lenti-RGS2 groups were shared in Experiment 3. All rats were sacrificed at 72 h after ICH for Western blot (WB)/enzyme-linked immunosorbent assay (ELISA)/coimmunoprecipitation (Co-IP) (n = 6). An additional 30 rats were divided into five groups (sham, ICH, ICH + shRGS2, ICH + NC shRNA, ICH + Lenti-RGS2) for double immunofluorescence staining at 72 h after ICH (n = 6).
ICH model
The rats first underwent intraperitoneal anesthesia with sodium pentobarbital. The surgical instruments were autoclaved, and Hamilton syringes and needles were sterilized with 84 disinfectants. Hamilton syringes and needles were rinsed in sterile water three times before use. The hair of the rats was removed, and the surface of the rats was wiped with 75% ethanol and allowed to dry. Then, 50 µL of autologous blood was taken from the femoral artery by a microinjection pump (Shenzhen Ruiwode Life Technology Co., Ltd., China) on the operation side with a microsyringe washed with heparin. Afterward, these rats were fixed on a stereotaxic apparatus (Shenzhen Ruiwode Life Technology Co., Ltd., China). The blood was automatically infused into the right basal ganglia within 5 min at 3.00 mm from the midline, 0.2 mm behind bregma, and 5.80 mm beneath the cortex. The needle was left for 10 min to prevent blood reflux before suturing the scalp. Throughout the experiment, the rats were placed in thermostatic blankets to keep the body temperature at approximately 37 °C. The sham group was only given a needle insertion. A schematic of the ICH model, including indication of the stereotactic injection coordinates, hematoma volume, and the regions of the perihematomal area, is shown in Fig. 1a.
Brain water content
The brain-water content was assessed using the wet-dry method21. The brain was divided into the ipsilateral (hemorrhagic) side and contralateral side, weighed on an analytical microbalance and then dried at 100 °C for 48 h to determine the dry weight. The brain-water content (%) was measured accordingly (wet weight–dry weight)/wet weight × 100%.
The modified neurological severity score
Motor, sensory (visual, tactile, and proprioceptive), balance and reflex tests were included in the modified Neurological Severity Score22. The neurological severity of the rats on day 3 after ICH was assessed by a score of 0 to 18, with 0 indicating a normal score and 18 indicating a maximum neurological deficit23.
Hematoxylin and eosin (H&E) staining
After perfusion of normal saline from the heart, 4% paraformaldehyde was slowly infused into the whole body for internal fixation at 72 h after ICH. The brains were removed, fixed with 4% paraformaldehyde for 48 h, and dehydrated using 75, 80, 95 and 100% alcohol. Then, the brains were transparentized in dimethylbenzene, dipped in wax and cut into brain Section (5 μm thick) by a slicer. After dewaxing, debenzene, hematoxylin staining and eosin staining, the brain sections were dehydrated, cleared, and observed under a microscope.
Nissl staining
The brain sections were made using the above method. After being debenzened and deparaffinized, brain sections underwent staining in a tar purple solution for 15 min before being washed in ddH2O. The brain sections underwent a color separation reaction and were then dehydrated in 70, 80, 95, and 100% ethanol. Finally, the sections were transparentized, mounted and observed under a microscope (×400 magnification). Neurons in the area around the hematoma were counted per microscopic field.
siRNA transfections
NLRC4-siRNA was infused at a concentration of 2 μg/µL and retained for 10 min into the right lateral ventricle at 1 mm anterior–posteriorly, 2 mm mediolaterally, and 3.5 mm dorsoventrally24,25. siRNA-NLRC4 and scramble siRNA (si-NC or si-negative control) were transfected 24 h before and 36 h after ICH model establishment. Both were obtained from GenePharma (China): NLRC4-siRNA(sense: GCUGAGGCCCACGUAUAAATT; antisense: UUUAUACGUGGGCCUCAGCTT); Negative control siRNA(sense: UUCUCCGAACGUGUCACGUTT; antisense: ACGUGACACGUUCGGAGAATT).
Inhibitor GNE7915 injection
DMSO (5%), 30% PEG 300, 5% Tween 80 and ddH2O were added to the inhibitor GNE-7915 at a concentration of 3.1 mg/mL GNE7915 and intraperitoneally injected (50 mg/kg) into the rats 60 h after ICH26.
Lentivirus transfection
Rats were placed in a stereotactic frame (Shenzhen Ruiwode Life Technology Co., Ltd., China) after being anesthetized using 4% C2H3Cl3O2. The lentivirus vectors (Lenti-RGS2) for RGS2 overexpression and the short hairpin (sh) RNA (sh-RGS2) for RGS2 knockdown were produced (GenePharma Technology Corporation, PRC): sh-NC (sense: TTCTCCGAACGTGTCACGT); sh-RGS2 (sense: GCTCTGGGCAGAAGCATTTGA). Holes were made in the rat pericranium 1.9 mm behind the coronal suture and 0.9 mm from the sagittal suture. Then, a 10 μL microinjection pump was placed stereotactically 3.5 mm deeper under the cortex. Five microliters of lentivirus with 1 × 109 genomic copies of Lenti-RGS2 and short hairpin (sh) RNA-RGS2 (sh-RGS2) were injected into the right lateral cerebral ventricle ipsilaterally at 0.5 μL/min27. Four weeks later, these rats underwent ICH surgery28.
Co-Immunoprecipitation assay
To detect whether LRRK2 interacted with NLRC4, an LRRK2 antibody (1:80; Millipore Biologicals, USA) was incubated with magnetic beads to form a complex in solution. Then, the magnetic beads were separated, and the antibody was recycled. Next, the tissue sample was incubated with beads that would bind to the antibody to form an antibody/antigen complex, and the tissue sample was dissociated. To pull down the complex from the beads, loading buffer was diluted with PBS and added to the complex with the beads, which was subsequently abandoned. The LRRK2 proteins were separated by SDS–PAGE for Western blot analysis using anti-NLRC4 (1:80, Novus Biologicals, USA) to determine NLRC4. The same method was used to detect whether NLRC4 was combined with LRRK2 and whether NLRC4 interacted with pro-caspase-1.
Immunofluorescence staining
Consecutive coronal sections of the brain (8 μm thick) were then blocked in 5% bovine serum albumin (BSA) at 37 °C for 1 h. They were incubated with rabbit anti-mouse MPO antibody (1:50, Abcam, USA), rabbit anti-mouse NLRC4 antibody (1:100; Novus Biologicals, USA), mouse anti-mouse LRRK2 antibody (1:100; Millipore Biologicals, USA), rabbit anti-mouse IBA-1 antibody (1:1000; Abcam, USA) and rabbit anti-GFAP antibody (1:200; Abconal, China) overnight at 4 °C. The samples were then incubated after three cycles of PBS washing with the corresponding fluorescence-conjugated secondary antibodies (1:100; Proteintech, USA) for 1 h at 37 °C. Three cycles of PBS washing were again performed. Microphotographs were analyzed with ImageJ software, and Pearson’s coefficient was used to evaluate the combination of NLRC4 and LRRK2.
Western blotting
Proteins were loaded (50 μg), separated by 6, 8, and 15% SDS–PAGE and then transferred to polyvinylidene fluoride membranes (PVDF, Millipore, USA). The primary antibodies: rabbit anti-NLRC4 (1:800, Novus Biologicals, USA), mouse anti-pNLRC4 (1:400, ECM Biologicals, USA), mouse anti-RGS2 (1:500, Santa Cruz, USA), rabbit anti-Pro-caspase-1 (1:1000, Abconal, China), rabbit anti-Pro-IL-1β (1:1000, Abconal), rabbit anti-IL-18 (1:1000, Abconal, China), rabbit anti-cleaved caspase-1 (1:500, Cell Signaling, USA), rabbit anti-cleaved IL-1β (1:500, Affinity, USA), rabbit anti-cleaved IL-18 (1:200, R&D, USA), rabbit anti-TNF-α (1:500, Boster, China), rabbit anti-IL-6 (1:500, Boster, China), rabbit anti-LRRK2 (1:1000, Abcam, USA), rabbit anti-pLRRK2 s935 (1:500, Abcam, USA) and rabbit anti-β-actin (1:1000, Proteintech, USA). Secondary antibody (1:2000, Proteintech, USA)-linked horseradish peroxidase (HRP) was added to the bands for 2 h. ECL detection reagents (Thermo, USA) were used to visualize the bands. ImageJ software was used to determine the relative density of these proteins.
Evans blue dye extravasation and autofluorescence
Evans blue dye (4%, 5 mL/kg, Sigma–Aldrich) was injected into the femoral vein under anesthesia. After 1 h, the rats were perfused with normal saline, and brain tissues were collected. Then, 50% trichloroacetic acid was put onto the brain tissues around the hematoma before the sample was homogenized and centrifuged at 12,000 × g for 30 min. The absorbance of the resulting supernatant was measured by a spectrophotometer at 620 nm. Meanwhile, the brains were cut into 8 µm slices by slicer (Leica, CM1860) and observed under a fluorescence microscope29.
Enzyme-linked immunosorbent assay (ELISA)
Brain samples were harvested at day 3 after ICH, and the levels of IL-18 and IL-1β were examined with ELISA kits (Jiangsu Meibiao Biotechnology Co., Jiangsu, China).
Statistical analysis
Data were described as the mean ± SD. GraphPad Prism software (version 7.0) was employed to conduct the statistical analyses. One-way ANOVA and Tukey’s multiple comparisons test were used to analyze parametric data. P < 0.05 indicated significant difference.
Results
The NLRC4 inflammasome is activated following ICH
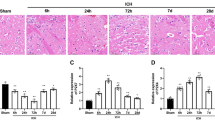
To investigate whether the NLRC4 inflammasome was activated after ICH, Western blotting was used to detect the levels of pNLRC4 and NLRC4, and coimmunoprecipitation was used to detect the interaction of NLRC4 with pro-caspase-1. The results indicated increased pNLRC4 levels after ICH, which reached a peak at approximately 72 h after intracerebral hemorrhage (Fig. 2a, c). There was no significant difference in NLRC4 levels between the six groups (Fig. 2a, b). The results of coimmunoprecipitation showed that NLRC4 interacted with and pro-caspase-1 in brain tissue at 72 h after ICH (Fig. 2d). These results revealed that ICH induced the phosphorylation of NLRC4 at Ser533 and activation of NLRC4 inflammasomes after ICH.

a–c NLRC4 and pNLRC4 expressions in the peri-hematoma area of sham and ICH rats were detected by Western blot at 12, 24, 48, 72 h, and 7 days after surgery, respectively (six rats for each group after ICH). *P < 0.05, compared with Sham group. d The results of Co-Immunoprecipitation showed the interaction of NLRC4 and pro-caspase-1 after ICH.
NLRC4 promotes the accumulation of glial cells, neurological injury, brain edema, neuronal death, and damage to the blood-brain barrier following ICH
To explore the role of NLRC4 in ICH, a specific and efficient siRNA targeting NLRC4 was designed. We subsequently used modified neurological severity scores (mNSSs) and brain water content to detect the role of NLRC4 in ICH-induced neurological injury. The results showed that, compared with the sham group, the ICH group at 72 h demonstrated severe neurological deficits and higher brain edema (Fig. 3a, b). Significant neurological improvement was seen in the NLRC4 siRNA+ICH group compared with the ICH group (Fig. 3a). After NLRC4 siRNA injection, brain edema in the ipsilateral brain and contralateral brain decreased significantly compared to that in the ICH group (Fig. 3b). Correspondingly, compared with the sham group, HE staining evidenced disorderly cytoplasmic loosening, karyopyknosis, and edema of the neurons in the ICH group. This alteration could be significantly reversed after treatment with NLRC4 siRNA (Fig. 3e). Nissl staining showed decreased Nissl bodies in the ICH group compared to the Sham samples. The number of Nissl bodies was significantly increased after treatment with NLRC4 siRNA (Fig. 3c, f). NLRC4 siRNA injection decreased EB leakage from blood vessels into the brain tissue at 72 h after ICH (Fig. 3d). The autofluorescence intensity (×400) of Evans blue declined with NLRC4 siRNA treatment (Fig. 3g). We tested the changes in microglial cells using the astrocyte marker GFAP and the microglial marker Iba-1 under this circumstance. We observed numerous accumulations of microglia and astrocytes after ICH (×400). There was a significant reduction in microglial accumulation in the ICH + NLRC4 siRNA group compared with the ICH group (Fig. 3h, j). Similarly, there was also a reduction in astrocytes in the ICH + NLRC4 siRNA group compared with the ICH group (Fig. 3i, k). These results indicated that NLRC4 exacerbates the accumulation of microglia and astrocytes after ICH. Altogether, these results showed that NLRC4 may aggravate ICH-induced brain injury in rats.

a Modified Neurological Severity Scores (mNSS) for neurological function, b Brain edema in ipsilateral brain and contralateral brain for brain water content, e HE staining for the morphology (×400), f Nissl staining for the morphology (×400) and c Nissl staining for the number of nissl bodies, d Evans blue Dye extravasation and g autofluorescence (×400) for the integrity of blood brain barrier, h IF of Iba-1 staining for microglia. i IF of GFAP staining for astrocytes. Quantification of microglial accumulation (j) and astrogliosis (k) in the Sham, ICH, ICH + NC, and ICH + NLRC4 siRNA groups at 72 h after ICH. The data represent the means ± SEM. (six rats for each group). *P < 0.05, compared with ICH.
NLRC4 facilitates neutrophil infiltration and the expression of TNF-α, IL-6, IL-1β, and IL-18 after intracerebral hemorrhage
To explore the effect of NLRC4 on ICH-reduced inflammation, siRNAs were injected to knock down NLRC4. Western blot analysis revealed that the expression of NLRC4 and pNLRC4 was effectively reduced (Fig. 4a–c). Cleaved caspase-1, IL-1β and IL-18 levels were reduced by NLRC4 siRNA injection (Fig. 4a, d, e, j). Accordingly, ELISA showed that, compared with the ICH group, the levels of IL-18 and IL-1β were decreased (Fig. 4k, l) in the ICH + NLRC4 siRNA group. However, the expression of pro-caspase-1, pro-IL-1β and pro-IL-18 was not significantly different among the four groups. Additionally, TNF-α and IL-6 were also reduced by NLRC4 siRNA injection (Fig. 4a, f, g). We used immunostaining to detect neutrophil infiltration. Immunostaining (× 200 and × 400) results showed that NLRC4 siRNA treatment reduced MPO-positive cells around the hematoma compared to the ICH group (Fig. 4h, i). These data suggested that NLRC4 knockdown may mitigate neuroinflammation after ICH.

a–g, j Western blot assay to detect NLRC4, pNLRC4, Pro-IL-1β, IL-1β, Pro-caspase-1, caspase-1, Pro-IL-18, IL-18, TNF-α, and IL-6, (k, l) ELISA for IL–18 and IL–1β (h, i) Immunostaining for MPO-positive cells (×400 and ×200) at peri-hematoma area in sham, ICH, negative control siRNA, and NLRC4 siRNA groups at 72 h after ICH (six rats for each group). *P < 0.05, compared with ICH.
RGS2 regulates the phosphorylation of NLRC4 and affects the NLRC4-dependent inflammasome activation
To explore the effect of RGS2 on NLRC4 inflammasome activation, short hairpin (sh) RNA was used to knock down the expression level of RGS2, and siRNA was injected to knock down NLRC4. Since phosphorylation at NLRC4-Ser533 is critical in the activation of the NLRC4 inflammasome9, our results showed that shRNA-RGS2 increased the phosphorylation of NLRC4 at Ser533 but failed to reduce the expression of NLRC4 (Fig. 5a–c). In addition, shRNA-RGS2 increased the expression of IL-1β, caspase-1 and IL-18 (Fig. 5a, d–f). However, it seemed that shRNA-RGS2 failed to affect the expression of pro-caspase-1, pro-IL-1β, and pro-IL-18 (Fig. 5a). Moreover, siRNA-NLRC4 reversed the effect of shRNA-RGS2 on increasing the levels of IL-1β, caspase-1 and IL-18 (Fig. 5d–f). These results demonstrated that shRNA-RGS2 treatment significantly enhanced the phosphorylation of NLRC4 and the maturation of IL‐1β and IL-18 induced by NLRC4. Thus, RGS may regulate NLRC4 inflammasome activation.

a–f Western blot assay was used to detect the expression of RGS2, pNLRC4, NLRC4, Pro-caspase-1, caspase-1, Pro-IL-1β, IL-1β, Pro-IL-18, and IL-18, in perihematomal area in ICH, Sham, ICH + shRGS2, ICH + negative control shRNA (ICH + NC shRNA), ICH + shRGS2 + si-NLRC4 and ICH + shRGS2 + NC siRNA at 72 h after ICH (6 rats for each group).
LRRK2 is critical for the activation of NLRC4 inflammasomes following ICH
LRRK2 is a certain and specific kinase that promotes the phosphorylation of NLRC4 and activates the NLRP4 inflammasome but not the NLRP3 inflammasome, accompanied by the formation of a complex with NLRC4 after S. Typhimurium infection17. Interestingly, it seems that the expression of LRRK2 did not change significantly after ICH. However, pLRRK2 at Ser935 was increased after ICH (Fig. 6a, b). This result indicated that LRRK2 may play a role dependent on its phosphorylation at Ser935 after ICH. We used GNE7915 (a specific inhibitor of pLRRK2) to verify whether pLRRK2 is needed for NLRC4 inflammasome activation following ICH.

a–g Western blot assay to detect LRRK2, pLRRK2, NLRC4, pNLRC4, pro- IL-1β, IL-1β, Pro- caspase-1, caspase-1, Pro-IL-18, and IL-18 (h and i) ELISA for IL-18 and IL-1β (j) Co-Immunoprecipitation assay for NLRC4 and LRRK2, in peri-hematoma area in sham, ICH, ICH + GNE7915, and ICH + Vehicle groups at 72 h after ICH (six rats for each group). *P < 0.05, compared with ICH.
GNE7915 effectively reduced the expression of LRRK2 Ser935 but failed to reduce the expression of NLRC4 (Fig. 6a–c). Of note, Western blot and ELISA results showed that GNE7915 decreased the phosphorylation of NLRC4 at Ser533 and attenuated the expression of caspase-1, IL-1β, and IL-18 following ICH (Fig. 6a, d–i). However, the expression of pro-caspase-1, pro-IL-1β, and pro-IL-18 was not significantly different among the four groups. These data indicated that LRRK2 promoted the phosphorylation of NLRC4 and the maturation of IL-1β and IL-18.
Since the NLRC4 and LRRK2 complex is required for NLRC4 inflammasome activation17. We next used coimmunoprecipitation to detect the interaction of NLRC4 and LRRK2. Our results revealed that LRRK2 interacted with NLRC4 in the ICH group. GNE7915 reduced the interaction of NLRC4 and LRRK2 (Fig. 6j). These results verified that LRRK2 regulated the phosphorylation of NLRC4 and the activation of the NLRC4 inflammasome following ICH.
RGS2 may restrain NLRC4 inflammasome activation via LRRK2
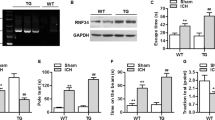
To determine how RGS2 affects NLRC4 inflammasome activation, short hairpin (sh) RNA was designed to knock down the expression level of RGS2, and lentivirus containing RGS2 was designed to overexpress RGS2. The Western blot results showed that Lenti-RGS2 reduced the phosphorylation of NLRC4 and the levels of IL-1β, caspase-1 and IL-18 (Fig. 7a, b, d–i). In contrast, shRNA-RGS2 enhanced the phosphorylation of NLRC4 and the expression of caspase-1, IL-1β, and IL-18 (Fig. 7a, b, d–i). The expression of pro-caspase-1, pro-IL-1β and pro-IL-18 was not significantly different in these seven groups. These results demonstrated that lenti-RGS2 treatment significantly decreased the phosphorylation of NLRC4 and the activation of the NLRC4 inflammasome, while treatment with shRNA-RGS2 had the opposite effect.

a–g Western blot assay was used to detect the expression of RGS2, LRRK2, pLRRK2, NLRC4, pNLRC4, pro- IL-1β, IL-1β, Pro-caspase-1, caspase-1, Pro-IL-18, and IL-18, h, i ELISA for IL-1β and IL-18 in perihematomal area in ICH, Sham, ICH + shRGS2, ICH + Lentil-RGS2, negative control lentiviral (ICH + NC), ICH + shRGS2 + GNE7915 and ICH + shRGS2 + Vehicle groups at 72 h after ICH (six rats for each group).
Next, we explored whether RGS2 affects NLRC4 inflammasome activation via LRRK2; notably, RGS2 failed to regulate the expression of LRRK2 (Fig. 7a). We found that Lenti-RGS2 reduced the phosphorylation of LRRK2; conversely, shRNA-RGS2 enhanced the phosphorylation of LRRK2 (Fig. 7a–c). Then, we used GNE7915 to reduce pLRRK2. Our results found that GNE7915 reversed the effect of shRNA-RGS2 on increasing the phosphorylation of NLRC4 and the expression of IL-1β, caspase-1 and IL-18 (Fig. 7a, b, d–i). The results demonstrated that RGS2 may regulate the phosphorylation of LRRK2 and that RGS2 may restrain NLRC4 inflammasome activation via pLRRK2.
RGS2 regulates the interaction of NLRC4 and LRRK2
Since the NLRC4 and LRRK2 complex is required for NLRC4 inflammasome activation, our aforementioned results showed that LRRK2 interacted with NLRC4 after ICH (Fig. 6j). Next, we detected the effect of RGS2 on the interaction of NLRC4 and LRRK2. Coimmunoprecipitation results revealed that Lenti-RGS2 prevented the interaction of LRRK2 and NLRC4; conversely, shRNA-RGS2 promoted the interaction of LRRK2 and NLRC4 (Fig. 8a). The immunofluorescence colocalization results (Fig. 8d, e) were consistent with the coimmunoprecipitation results. The Pearson’s coefficient and overlap coefficient were lower in the ICH + Lenti-RGS2 group than in the ICH group (Fig. 8b, c). Conversely, the Pearson’s coefficient and overlap coefficient were higher in the ICH + shRGS2 group than in the ICH group (Fig. 8b, c). These results suggested that RGS2 may regulate the interaction of LRRK2 and NLRC4 during intracerebral hemorrhage.

a Co-Immunoprecipitation assay for NLRC4 and LRRK2, d The location of the image around the perihematomal area e Representative images of Immunofluorescence (IF) colocalization for NLRC4 and LRRK2. b, c Pearson’s coefficient and overlap coefficient for NLRC4 and LRRK2 in peri-hematoma area in ICH, Sham, ICH + shRGS2, ICH + Lentil-RGS2 and negative control lentiviral (ICH + NC) groups at 72 h after ICH (six rats for each group). *P < 0.05, compared with ICH.
Discussion
The pathogenesis of ICH-induced brain injury includes primary brain injury and secondary brain injury (SBI). Increasing evidence has shown that SBI is a key factor in the deterioration of neurological function after ICH30. SBI consists of inflammation, oxidation, autophagy, and apoptosis31, leading to destruction of the blood-brain barrier and massive neuronal cell death32. Inflammation induced by the innate immune system plays an important role in SBI after ICH33,34. The NLR family, intracellular innate immune sensors, is responsible for processing pro-IL-1β and pro-IL-18 into a maturation state to promote inflammation35,36. The role of the NLR family in neuroinflammation is well known, especially NLRP1 and NLRP3. However, there are currently few studies on the role of NLRC4 in neuroinflammation. Our data revealed that NLRC4 could be activated by recognizing a damage-associated molecular pattern (DAMP) sensor after ICH. In this study, we found that NLRC4 reached the peak of phosphorylation at Ser533 on the 3rd day and that the NLRC4 inflammasome was activated after ICH, which corresponded to the peak of the proinflammatory cytokine IL-1β at 2–3 days after ICH37. A study on NLRC4−/− mice after ischemic stroke demonstrated that the NLRC4 inflammasome contributes to acute brain injury38. Moreover, the NLRC4 inflammasome is reported to promote alcohol-induced liver injury39 and breast cancer progression40. Similar to these studies, our results showed that NLRC4 knockdown reduced brain damage. Furthermore, NLRC4 knockdown also reduced the expression of TNF-a and IL-6. Meanwhile, neutrophil infiltration, neuronal cell death and blood-brain barrier injury were alleviated in NLRC4 knockdown rats. These results might be related to NLRC4 playing a role in inflammation independent of inflammasome activation41. Overall, our results indicated that NLRC4 may result in the aggravation of intracerebral hemorrhage-related inflammation.
It is relatively clear that increased release of Il-1β and IL-18 from microglia has been identified as an important cytokine in ICH pathology42,43. Inhibitors and antagonists of IL-1β and IL-18 could extenuate brain edema, neuroinflammation, and neurodegeneration in experimental rats44,45. Furthermore, our results indicated that NLRC4 promoted the release of the cytokines IL-1β and IL-18, accumulation of microglia, neuronal death, Evans blue extravasation blood–brain barrier permeability, and brain water content for brain edema (Figs. 3 and 4). Taken together, we speculated that NLRC4 might promote all these outcomes via NLRC4 inflammasome-derived IL-1β and IL-18 processing. As mentioned before, both IL-1β and IL-18 play a crucial role in brain edema, neuroinflammation, and neurodegeneration44,45. Our experiment showed that IL-1β and IL-18 were decreased, and both may be involved in ICH injury. However, a previous study indicated that both IL-1β and IL-18 are required for NLRC4-derived inflammation, but IL-1β is more effective than IL-18 in treating bone erosion in Nlrc4-H443P-Tg Mice46. As far as the current evidence, we could not conclude whether IL-1β or IL-18 is more effective in NLRC4-induced inflammation after ICH.
We concluded that NLRC4 inflammasomes are involved in intracerebral hemorrhage-induced inflammation. Nevertheless, the molecular mechanisms of NLRC4 inflammasome activation in ICH are poorly known. GNE7915, a highly selective and BBB-penetrable LRRK2 inhibito47, was used to decrease the phosphorylation of LRRK2. We found that GNE7915 reduced pLRRK2 at Ser935 and the phosphorylation of NLRC4. Accordingly, IL-1β, caspase-1, and IL-18 levels were also decreased by GNE7915, as expected. Many studies are consistent with our results. George T and his colleagues reported that LRRK2 may result in neuronal apoptosis after cerebral ischemia by modulating the phosphorylation of Tau48. Similarly, LRRK2 contributes to traumatic brain injury (TBI)-induced neuronal apoptosis, BBB permeability, brain edema and neurological impairment49. The phosphorylation of LRRK2 may be related to NLRC4 inflammasome activation after ICH.
LRRK2, due to its complex structure, is also a Roco protein. It is an atypical G-protein that is a member of the Ras/GTPase superfamily called the Ras of complex proteins (Roc)50. It has been shown that Rab GTPase activity is regulated by GEF, GAP and GDI proteins51. RGS2 has been reported as a physiological GTPase activation protein (GAP) of LRRK219. RGS2 has been regarded as a controller of GPCR and linked G protein signaling. RGS terminates the signal by speeding the intrinsic activity of GTPase in the G protein, which returns the G protein to the receptor in its GDP-bound form (inactive)52. RGS2 was considered as a key regulator of AHR53. This resulted from the specific interaction between RGS2 and G proteins, which was dependent on the linked GPCR’s selective recognition54. RGS2 was also reported to play a protective role in airway inflammation by reducing the number of granulocytes (neutrophils and eosinophils) and the release of inflammatory cytokines and chemokines55. Similarly, we discovered that RGS2 exerted a protective role in the neuroinflammation induced by the NLRC4 inflammasome following ICH by regulating LRRK2, which may not be related to the phosphorylation of LRRK2. These findings indicated that blocking NLRC4 may be a novel potential therapeutic target after ICH.
In summary, our study indicated that the NLRC4 inflammasome may play a role in ICH-induced inflammatory activation by increasing neutrophil infiltration and the expression of TNF-α, IL-6, IL-1β, and IL-18. Furthermore, our research provided novel insights into the NLRC4 inflammasome after intracerebral hemorrhage and suggested the role of RGS2 in NLRC4 inflammasome activation by LRRK2 after ICH (Fig. 9). It also identified RGS2 and LRRK2 as targets for the NLRC4 inflammasome in ICH.

A pattern diagram showed that the NLRC4 inflammasome plays a role in ICH-induced neuroinflammation and that RGS2 regulates NLRC4 inflammasome activation by LRRK2 after ICH.
References
Feigin, V. L., Lawes, C. M., Bennett, D. A. & Anderson, C. S. Stroke epidemiology: a review of population-based studies of incidence, prevalence, and case-fatality in the late 20th century. Lancet Neurol. 2, 43–53 (2003).
Feigin, V. L., Lawes, C. M., Bennett, D. A., Barker-Collo, S. L. & Parag, V. Worldwide stroke incidence and early case fatality reported in 56 population-based studies: a systematic review. Lancet Neurol. 8, 355–369 (2009).
Qureshi, A. I. et al. Apoptosis as a form of cell death in intracerebral hemorrhage. Neurosurgery 52, 1041–1047 (2003).
Chen, S., Yang, Q., Chen, G. & Zhang, J. H. An update on inflammation in the acute phase of intracerebral hemorrhage. Transl. Stroke Res. 6, 4–8 (2015).
Heneka, M. T., McManus, R. M. & Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 19, 610–621 (2018).
Broz, P. & Dixit, V. M. Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16, 407–420 (2016).
Kanneganti, T. D., Lamkanfi, M. & Núñez, G. Intracellular NOD-like receptors in host defense and disease. Immunity 27, 549–559 (2007).
Broz, P., von Moltke, J., Jones, J. W., Vance, R. E. & Monack, D. M. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe 8, 471–483 (2010).
Qu, Y. et al. Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 490, 539–542 (2012).
Mariathasan, S. et al. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430, 213–218 (2004).
Gerber, K. J., Squires, K. E. & Hepler, J. R. Roles for regulator of G protein signaling proteins in synaptic signaling and plasticity. Mol. Pharmacol. 89, 273–286 (2016).
Hohoff, C. et al. RGS2 ggenetic variation: association analysis with panic disorder and dimensional as well as intermediate phenotypes of anxiety. Am. J. Med. Genet. B. 168b, 211–222 (2015).
Gan, H. et al. Experimental study of RGS2 inhibiting inflammatory response after cerebral hemorrhage in rats. J. Chongqing Med. Univ. 44, 588–591 (2019).
Liao, J. & Hoang, Q. Q. Roco proteins and the Parkinson’s disease-associated LRRK2. Int. J. Mol. Sci. 19, 4074 (2018).
Li, J. Q., Tan, L. & Yu, J. T. The role of the LRRK2 gene in Parkinsonism. Mol. Neurodegener. 9, 47 (2014).
Paisán-Ruíz, C. et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44, 595–600 (2004).
Liu, W. et al. LRRK2 promotes the activation of NLRC4 inflammasome during Salmonella Typhimurium infection. J. Exp. Med. 214, 3051–3066 (2017).
Dzamko, N. et al. Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14-3-3 binding and altered cytoplasmic localization. Biochem. J. 430, 405–413 (2010).
Dusonchet, J. et al. A Parkinson’s disease gene regulatory network identifies the signaling protein RGS2 as a modulator of LRRK2 activity and neuronal toxicity. Hum. Mol. Genet. 23, 4887–4905 (2014).
Lively, S. & Schlichter, L. C. Age-related comparisons of evolution of the inflammatory response after intracerebral hemorrhage in rats. Transl. Stroke Res. 3, 132–146 (2012).
Wang, B. F. et al. Curcumin attenuates brain edema in mice with intracerebral hemorrhage through inhibition of AQP4 and AQP9 expression. Acta Pharmacol. Sin. 36, 939–948 (2015).
Liu, Y. et al. Characterization of axon damage, neurological deficits, and histopathology in two experimental models of intracerebral hemorrhage. Front. Neurosci. 12, 928 (2018).
Song, Y. et al. Lipoxin A4 methyl ester reduces early brain injury by inhibition of the nuclear factor Kappa B (NF-κB)-dependent matrix metallopeptidase 9 (MMP-9) pathway in a rat model of intracerebral hemorrhage. Med. Sci. Monit. 25, 1838–1847 (2019).
Meng, C. et al. Effects of NLRP6 in cerebral Ischemia/Reperfusion (I/R) injury in rats. J. Mol. Neurosci. 69, 411–418 (2019).
He, Q. et al. Resveratrol alleviates cerebral ischemia/reperfusion injury in rats by inhibiting NLRP3 inflammasome activation through Sirt1-dependent autophagy induction. Int. Immunopharmacol. 50, 208–215 (2017).
Cao, J. et al. Leucine-rich repeat kinase 2 aggravates secondary brain injury induced by intracerebral hemorrhage in rats by regulating the P38 MAPK/Drosha pathway. Neurobiol. Dis. 119, 53–64 (2018).
Wang, S. et al. C1q/Tumor necrosis factor-related protein-3 attenuates brain injury after intracerebral hemorrhage via AMPK-dependent pathway in rat. Front. Cell. Neurosci. 10, 237 (2016).
He, Q. et al. Parkin-dependent mitophagy is required for the inhibition of ATF4 on NLRP3 inflammasome activation in cerebral Ischemia-Reperfusion injury in rats. Cells 8, 897 (2019).
Chen, Y. et al. Norrin protected blood-brain barrier via frizzled-4/β-catenin pathway after subarachnoid hemorrhage in rats. Stroke 46, 529–536 (2015).
Aronowski, J. & Zhao, X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke 42, 1781–1786 (2011).
Felberg, R. A. et al. Cell death in experimental intracerebral hemorrhage: the “black hole” model of hemorrhagic damage. Ann. Neurol. 51, 517–524 (2002).
Babu, R., Bagley, J. H., Di, C., Friedman, A. H. & Adamson, C. Thrombin and hemin as central factors in the mechanisms of intracerebral hemorrhage-induced secondary brain injury and as potential targets for intervention. Neurosurg. Focus 32, E8 (2012).
Wang, J. & Doré, S. Inflammation after intracerebral hemorrhage. J. Cereb. Blood Flow. Metab. 27, 894–908 (2007).
Zhou, Y., Wang, Y., Wang, J., Anne Stetler, R. & Yang, Q. W. Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Prog. Neurobiol. 115, 25–44 (2014).
Lamkanfi, M. & Dixit, V. M. Mechanisms and functions of inflammasomes. Cell 157, 1013–1022 (2014).
Lamkanfi, M. & Dixit, V. M. Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 28, 137–161 (2012).
Holmin, S. & Mathiesen, T. Intracerebral administration of interleukin-1beta and induction of inflammation, apoptosis, and vasogenic edema. J. Neurosurg. 92, 108–120 (2000).
Denes, A. et al. AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proc. Natl Acad. Sci. USA 112, 4050–4055 (2015).
DeSantis, D. A. et al. Alcohol-induced liver injury is modulated by Nlrp3 and Nlrc4 inflammasomes in mice. Mediat. Inflamm. 2013, 751374 (2013).
Kolb, R. et al. Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nat. Commun. 7, 13007 (2016).
Janowski, A. M. et al. NLRC4 suppresses melanoma tumor progression independently of inflammasome activation. J. Clin. Invest. 126, 3917–3928 (2016).
Gao, Z. et al. Microglial activation and intracerebral hemorrhage. Acta Neurochir. Suppl. 105, 51–53 (2008).
Voet, S., Srinivasan, S., Lamkanfi, M. & van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 11, e10248 (2019).
Yatsiv, I. et al. Elevated intracranial IL-18 in humans and mice after traumatic brain injury and evidence of neuroprotective effects of IL-18-binding protein after experimental closed head injury. J. Cereb. Blood Flow. Metab. 22, 971–978 (2002).
Clausen, F. et al. Neutralization of interleukin-1β reduces cerebral edema and tissue loss and improves late cognitive outcome following traumatic brain injury in mice. Eur. J. Neurosci. 34, 110–123 (2011).
Sasaki, Y., Otsuka, K., Arimochi, H., Tsukumo, S. I. & Yasutomo, K. Distinct roles of IL-1β and IL-18 in NLRC4-induced autoinflammation. Front. Immunol. 11, 591713 (2020).
Estrada, A. A. et al. Discovery of highly potent, selective, and brain-penetrable leucine-rich repeat kinase 2 (LRRK2) small molecule inhibitors. J. Med. Chem. 55, 9416–9433 (2012).
Kim, T. & Vemuganti, R. Mechanisms of Parkinson’s disease-related proteins in mediating secondary brain damage after cerebral ischemia. J. Cereb. Blood Flow. Metab. 37, 1910–1926 (2017).
Rui, Q. et al. LRRK2 contributes to secondary brain injury through a p38/Drosha signaling pathway after traumatic brain injury in rats. Front. Cell. Neurosci. 12, 51 (2018).
Bosgraaf, L. & Van Haastert, P. J. Roc, a Ras/GTPase domain in complex proteins. Biochim. Biophys. Acta 1643, 5–10 (2003).
Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 10, 513–525 (2009).
Hollinger, S. & Hepler, J. R. Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol. Rev. 54, 527–559 (2002).
Jiang, H. et al. Regulator of G-protein signaling 2 repression exacerbates airway hyper-responsiveness and remodeling in asthma. Am. J. Respir. Cell Mol. Biol. 53, 42–49 (2015).
Heximer, S. P., Watson, N., Linder, M. E., Blumer, K. J. & Hepler, J. R. RGS2/G0S8 is a selective inhibitor of Gqalpha function. Proc. Natl Acad. Sci. USA 94, 14389–14393 (1997).
George, T. et al. Protective roles for RGS2 in a mouse model of house dust mite-induced airway inflammation. PLoS ONE 12, e0170269 (2017).
Funding
This work was supported by Chongqing Science and Technology Committee, China (No. cstc2016shmszx0432), Natural Science Foundation of Chongqing, China (cstc2015jcyjBX0144) and the National Natural Science Foundation of China (Nos. 81671158 and 81771261).
Author information
Authors and Affiliations
Contributions
Hui Gan designed the study, performed the surgical operation, Western blot, ELISA, and data analysis and drafted the article. Li Zhang, Shuyue Zheng, and Ning Jiang participated in HE staining, Nissal staining and immunofluorescence. Hui Chen, Han Xiao, and Lu Wang participated in the surgical operation. Xuan Zhai participated in article modification. Ping Liang and Jing Zhao provided funding and edited the manuscript for this study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gan, H., Zhang, L., Chen, H. et al. The pivotal role of the NLRC4 inflammasome in neuroinflammation after intracerebral hemorrhage in rats. Exp Mol Med 53, 1807–1818 (2021). https://doi.org/10.1038/s12276-021-00702-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-021-00702-y
This article is cited by
-
Treatment of Intracerebral Hemorrhage with Traditional Chinese Medicine Monomer Wogonin by Modifying NLRP3 with METTL14 to Inhibit Neuronal Cell Pyroptosis
Applied Biochemistry and Biotechnology (2024)
-
Cystatin C Attenuates Perihematomal Secondary Brain Injury by Inhibiting the Cathepsin B/NLRP3 Signaling Pathway in a Rat Model of Intracerebral Hemorrhage
Molecular Neurobiology (2024)
