
Abstract
Primary aldosteronism is characterized by at least partially autonomous production of the adrenal steroid hormone aldosterone and is the most common cause of secondary hypertension. The most frequent subforms are idiopathic hyperaldosteronism and aldosterone-producing adenoma. Rare causes include unilateral hyperplasia, adrenocortical carcinoma and Mendelian forms (familial hyperaldosteronism). Studies conducted in the last eight years have identified somatic driver mutations in a substantial portion of aldosterone-producing adenomas, including the genes KCNJ5 (encoding inwardly rectifying potassium channel GIRK4), CACNA1D (encoding a subunit of L-type voltage-gated calcium channel CaV1.3), ATP1A1 (encoding a subunit of Na+/K+-ATPase), ATP2B3 (encoding a Ca2+-ATPase), and CTNNB1 (encoding ß-catenin). In addition, aldosterone-producing cells were recently reported to form small clusters (aldosterone-producing cell clusters) beneath the adrenal capsule. Such clusters accumulate with age and appear to be more frequent in individuals with idiopathic hyperaldosteronism. The fact that they are associated with somatic mutations implicated in aldosterone-producing adenomas also suggests a precursor function for adenomas. Rare germline variants of CYP11B2 (encoding aldosterone synthase), CLCN2 (encoding voltage-gated chloride channel ClC-2), KCNJ5, CACNA1H (encoding a subunit of T-type voltage-gated calcium channel CaV3.2), and CACNA1D have been reported in different subtypes of familial hyperaldosteronism. Collectively, these studies suggest that primary aldosteronism is largely due to genetic mutations in single genes, with potential implications for diagnosis and therapy.
Similar content being viewed by others

Introduction
The adrenal salt-retaining steroid hormone aldosterone is produced in the zona glomerulosa (ZG), the outermost zone of the adrenal cortex, from its precursor, cholesterol. In volume depletion, a major stimulus of aldosterone production, angiotensinogen is cleaved by the aspartyl protease renin, leading to the production of angiotensin I, which is then further processed to angiotensin II (ATII) by angiotensin converting enzyme (ACE). The second major stimulus of aldosterone production is hyperkalemia, which directly raises adrenal aldosterone production1. Aldosterone, by binding to mineralocorticoid receptors in the distal convoluted tubule, the connecting tubule and the cortical collecting duct of the kidney, increases the abundance or activity of numerous proteins, including the Na+/K+-ATPase and the epithelial sodium channel2. Consequently, aldosterone leads to increased sodium reabsorption and volume retention as well as potassium secretion, and excessive aldosterone secretion can cause hypertension and hypokalemia3.
In 1955, Conn reported a patient with hypertension, muscular weakness, paralysis, polydipsia, polyuria, and hypokalemic alkalosis who was cured after removal of a 4 cm adrenocortical adenoma (benign tumor of the adrenal cortex)4. He described this as a new syndrome of “primary aldosteronism” (PA). Currently, PA is recognized as the most common cause of secondary hypertension, accounting for up to 6% of hypertensive patients in primary care and up to 11% of patients in specialized referral centers; its prevalence increases with the severity of hypertension5,6. PA is characterized by at least partially autonomous aldosterone production (despite low renin levels), hypertension and low or normal serum potassium levels7. Common causes, include idiopathic hyperaldosteronism (IHA; approximately 60% of the cases) and aldosterone-producing adenoma (APA; approximately 30%)5,6, and rare subforms include unilateral hyperplasia, adrenocortical carcinoma (ACC), or familial hyperaldosteronism (FH). PA is associated with an increased risk of cardiovascular diseases, such as stroke, myocardial infarction, or atrial fibrillation compared with primary hypertension8,9,10. In patients with hypertension, an elevated aldosterone–renin ratio (ARR) is used as a screening parameter, followed by confirmatory testing (salt loading, fludrocortisone or captopril administration, which all fail to sufficiently lower aldosterone levels in PA). Imaging, such as computed tomography, serves to exclude malignancy, whereas so-called adrenal venous sampling, an invasive procedure, is typically required to distinguish unilateral (mostly APA) from bilateral (usually IHA) aldosterone production7. However, adrenal venous sampling remains challenging, and hence, many patients are not diagnosed or treated optimally. While APAs can potentially be cured by surgery, mineralocorticoid receptor antagonists remain the treatment of choice for IHA7,11,12.
Despite the well-established role of aldosterone in renal salt handling and blood pressure regulation, the molecular cause(s) underlying PA, with the exception of a rare familial form13, were unknown until approximately 8 years ago. In recent years, next-generation sequencing approaches have allowed for the identification of somatic (tumor-specific) mutations in APAs as well as germline mutations in FH (Table 1), offering a completely new perspective on the pathophysiology underlying PA and improving our understanding of the regulation of aldosterone production in the adrenal gland14,15,16,17,18,19,20. The purpose of this review is to summarize and discuss these findings.
Somatic mutations in APA
Mutations in potassium channel KCNJ5 in APA
In 2011, Choi et al. conducted a whole-exome sequencing study comparing the blood and tumor DNA of four patients with APAs showing hypertension, high ARR, and unilateral adrenal cortical masses upon CT evaluation. The overall number of somatic (tumor-specific) mutations in APAs was low (2.3 protein-altering and 0.8 silent mutations per tumor) in comparison to malignant tumors. In two APAs, Choi et al.16 discovered heterozygous somatic mutations in the gene KCNJ5 (G151R and L168R) encoding the inwardly rectifying potassium channel Kir3.4. Subsequent targeted Sanger sequencing in 18 APAs revealed six additional somatic KCNJ5 mutations (one G151R and five L168R). These two hotspot mutations were later shown to account for the vast majority of KCNJ5 mutations in APAs; other mutations are very rare (L168R: 23–44%, G151R: 54–79%, others: 0–4.5%21,22,23). Ensuing studies in large cohorts have revealed that KCNJ5 mutations may account for approximately 40% of mutations in APAs21,24,25, with considerable variation among different ethnicities and among women versus men (see below).
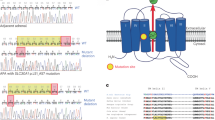
Kir3.4 channels have two transmembrane domains and form tetrameric channels with a central common pore. Potassium selectivity is mostly conferred by the selectivity filter located in the loop between the pore helix and the second transmembrane domain26. The selectivity filter contains a signature sequence (TXGYG) shared among numerous different potassium channels27. The most common KCNJ5 mutations in APAs are located either within (G151R) or close to (L168R) the selectivity filter. Both mutations cause abnormal sodium permeability of the mutant channel, which results in a depolarization of the cell membrane (Fig. 1). Based on these findings, Choi et al. proposed that in APAs with KCNJ5 mutations, tumor formation and autonomous aldosterone production are driven by membrane depolarization of glomerulosa cells, leading to increased calcium influx via voltage-gated calcium channels1 and subsequent changes in the expression of genes implicated in proliferation and aldosterone synthesis. Evidence that KCNJ5 mutations are likely sufficient to cause both aldosterone production and tumor formation is provided by the overall rarity of additional somatic variants in APAs with pathogenic KCNJ5 mutations, specifically the absence of additional mutations that explain proliferation16,28 and the fact that patients with germline KCNJ5 mutations found in APAs typically develop massive adrenal hyperplasia as well as early-onset, therapy-resistant PA (see below)16,29. A proliferative effect of KCNJ5 mutations is also suggested by the peculiar case of a patient with germline mosaicism in whom adrenal hyperplasia was restricted to those areas of the adrenal gland that carried KCNJ5 mutations30. Despite these considerations, a two-hit model of APA formation has been proposed involving the activation of signaling pathways such as shh (sonic hedgehog signaling molecule) or Wnt/β-catenin leading to abnormal proliferation (first hit) and subsequent acquisition of somatic mutations in driver genes such as KCNJ5 leading to increased and autonomous aldosterone production (second hit)31. In very rare cases, this may be due to germline mutations in tumor suppressor genes (published case with APC mutation)32.

a Under resting conditions, voltage-gated calcium channels are closed due to a high potassium conductance and the resulting hyperpolarized membrane potential. b Physiological stimuli of glomerulosa cells include angiotensin II and hyperkalemia, leading to depolarization, followed by the opening of voltage-gated calcium channels, the signal for aldosterone synthase expression and aldosterone production. c KCNJ5 variants (as somatic mutations in APAs and germline mutations in FH-III) change the ion selectivity of the potassium channel, permitting sodium influx and the depolarization of the cell membrane. d Mutations of CACNA1H (as germline mutations in FH-IV) and CACNA1D (as somatic mutations in APAs and germline mutations in PASNA syndrome) directly cause increased calcium permeability. e The higher chloride permeability of CLCN2 variants (as germline mutations in FH-II) depolarizes glomerulosa cells by chloride efflux. f The ACTH-dependent expression of a hybrid variant of CYP11B2 (as germline mutation in FH-I) in zona fasciculata cells directly increases hybrid gene expression. g Variants of ATP1A1 and ATP2B3 (as somatic mutations in APAs) lead to increased permeability for sodium or protons and thereby depolarize glomerulosa cells. h The underlying mechanism of elevated aldosterone production by variants of CTNNB1 (as somatic mutations in APAs) is incompletely understood. AT1R, angiotensin II receptor type I; MC2R, adrenocorticotropic hormone receptor
Mutations in calcium channel CACNA1D in APA
Two groups independently discovered APAs with somatic mutations in CACNA1D, encoding for the α1-subunit of an l-type calcium channel (CaV1.3) via exome sequencing14,17. The α1-subunit consists of four homologous repeats comprising six transmembrane helices (S1–6) each, and calcium selectivity is determined by a pore loop between S5 and S633. Mutations in CACNA1D are more scattered throughout the protein compared with KCNJ5 mutations14,17. By electrophysiology, both groups demonstrated that the somatic mutations discovered caused an increase in calcium permeability, which was inferred to directly lead to increased aldosterone production and proliferation (Fig. 1). In addition to KCNJ5, CACNA1D is the gene with the second highest mutation burden in APAs14,17,25, again with variation among different ethnicities (see below). Studies that relied on Sanger sequencing of only a few exons for somatic mutation detection may have systematically underestimated the prevalence of CACNA1D mutations (21–42% prevalence in studies using panel sequencing of the entire coding sequence34,35 vs. 1.5–10.3% in studies using Sanger sequencing of select regions25,36,37).
Mutations in ATPases ATP1A1 and ATP2B3 in APAs
In 2013, Beuschlein et al. expanded the portfolio of somatic mutations in APAs. They described heterozygous mutations in ATP1A1 (encoding for a Na+–K+–ATPase subunit) and ATP2B3 (encoding for a plasma membrane Ca2+ ATPase)15. Mutations were clustered within the M4 helix involved in ion binding, suggesting a gain-of-function mechanism; however, the authors originally described the loss of pump function in mutant ATPases (which may contribute to depolarization). Subsequent studies demonstrated that mutations in ATP1A114 and ATP2B338 cause abnormal Na+ or H+ permeability (Fig. 1) and enhance aldosterone production in the human adrenocortical H295R cell line, suggesting an underlying pathomechanism similar to that of KCNJ5 mutations. In comparison to KCNJ5 and CACNA1D, ATPases show a lower mutational burden in APAs, accounting for 3–17% and 1.5–4% of APAs, respectively15,25,34,35,37.
Mutations in CTNNB1 in APAs
In addition to ion channels and ATPases, APAs, in rare cases, also carry gain-of-function mutations in CTNNB1, encoding for β-catenin, the effector of the canonical Wnt signaling pathway17,37,39. However, the mechanisms underlying CTNNB1-mediated APA formation have not been clarified. A universal role of CTNNB1 mutations in adrenal tumor formation is supported by the fact that mutations in CTNNB1 are also found in ACC40, cortisol-producing adenomas41,42 and nonproducing adenomas39. Constitutive activation of CTNNB1 in mice has been demonstrated to trigger adrenal hyperplasia, aldosteronism and, at advanced age, malignancy43. In one study, somatic CTNNB1 mutations were reported in two pregnant women. The authors suggested that adenoma formation and excessive aldosterone production may be driven by pregnancy via enhanced activation of LHCGR (luteinizing hormone/choriogonadotropin receptor)44. However, this has been disputed because CTNNB1 mutations are found in APAs of both male and female (typically nonpregnant) patients36,45.
Distribution of mutations in APAs—frequency, sex, and ethnicity
Recent studies suggest that somatic mutations in APAs may be more frequent than previously thought. Using CYP11B2 immunohistochemistry for the identification of aldosterone-producing nodules and next-generation sequencing for somatic mutation discovery, Nanba et al. demonstrated that up to 87% (males) or 90–91% (females) of tumors carry somatic mutations in one of the genes described above34,35. The pathophysiology in the remainder of the cases, however, remains elusive. Further causes may include previously undiscovered, very rare mutations or gene expression changes due to variations in copy number. In fact, a loss of heterozygosity has been reported in a subset of APAs, and such tumors do not carry KCNJ5 mutations16. Mutations in KCNJ5 appear to be more frequent in females (56–63%; compared to 22–31% in males)21,24,25. Higher frequencies in some Asian cohorts (approximately 60–70% of APAs)46,47,48 were in part attributed to different diagnostic criteria; however, a recent study reporting CACNA1D as the gene with the highest mutational burden and lower KCNJ5 mutation frequencies in a cohort of black subjects with APAs suggests that mutation frequencies may indeed be influenced by ethnicity35. The reasons underlying sex- and population-specific variation in APA mutation frequencies are currently unknown.
Implications for diagnosis and therapy
Despite the broad portfolio of somatic mutations and their mechanistic implications discussed above, genetic mutations are currently not exploited for diagnostic or therapeutic purposes. Adrenal venous sampling, the current gold standard for diagnosis, is a complicated and invasive procedure that is available only in specialized centers, impeding high-throughput diagnosis. It has been suggested that selective inhibitors of mutant KCNJ5 channels have the potential to improve this situation: a drop in blood pressure and/or aldosterone upon administration of such inhibitors could allow for the noninvasive identification of patients with adenomas carrying KCNJ5 mutations49. Because KCNJ5-positive tumors are often large, with typical characteristics upon computed tomography37, they could possibly be surgically removed without the need for adrenal venous sampling. Interestingly, in a high-throughput screen, macrolide antibiotics such as roxithromycin and clarithromycin were identified as specific inhibitors of both KCNJ5G151R and KCNJ5L168R, with no significant inhibition of the wild-type channel. Importantly, this effect was also present in macrolide compounds without antibiotic or motilin receptor agonist activity49. In a proof-of-principle clinical trial, the antibiotics roxithromycin and clarithromycin (with established safety) are currently being investigated as diagnostic tools50. Similarly, genotype-specific steroid profiles have been suggested as diagnostic tools. Specifically, tumors carrying KCNJ5 mutations are characterized by higher production of the so-called hybrid steroid 18-oxocortisol51, which could serve to identify APAs for surgery.
DNA methylation and APAs
Genome-wide analysis of methylation and corresponding gene expression comparing APA, normal adrenocortical tissue (including the ZG, fasciculata, and reticularis) and nonproducing adenomas demonstrated, among other differentially methylated and expressed genes, higher expression and decreased methylation of CYP11B2 in APAs52, and these results were confirmed in an independent study53. How this hypomethylation relates to somatic mutations in APAs is less clear54.
APCC and IHA
Studies using an antibody against aldosterone synthase (CYP11B2), one of the rate-limiting factors of aldosterone production, have demonstrated that the human ZG is not always a continuous arrangement of aldosterone-producing cells. Rather, with increasing age, so-called aldosterone-producing cell clusters (APCCs) develop, which express CYP11B2 and protrude into the zona fasciculata55,56,57. A possible role for APCCs in PA is supported by a study investigating CYP11B2 expression in adrenal slices from a cohort of 32 patients with PA58. Six patients did not show signs of APA despite PA, and 4 of these patients had multiple CYP11B2-expressing APCCs. Interestingly, one patient with adenoma showed no expression of CYP11B2 in the tumor but had multiple CYP11B2-expressing APCCs. By subjecting CYP11B2-expressing APCCs of normal adrenal glands to panel sequencing of candidate genes, Nishimoto et al. provided evidence that APCCs are genetically similar to APAs56. They reported 11 somatic mutations (7 in CACNA1D, 3 in ATP1A1 and 1 in ATP2B3) in 31 APCCs, 7 of which were situated at residues affected by known somatic mutations in APAs. This finding also suggests that APCCs may be precursors of APAs. Interestingly, no KCNJ5 variants were detected. Lesions with KCNJ5 mutations may quickly progress to APAs and hence may be underrepresented in this study.
In addition to APA formation, APCCs have also recently been implicated in IHA59. Omata et al.59 identified 15 patients who had undergone unilateral adrenalectomy despite bilateral disease. The resected adrenal glands showed more and larger APCCs than the normotensive controls. Sequencing of 99 APCCs in this cohort revealed CACNA1D mutations in 58% and KCNJ5 mutations in 1% of the lesions. All adrenals had at least one APCC with such a mutation, but the causes of the remaining lesions are currently unknown. Exome sequencing of DNA extracted from APCCs will likely provide additional insight. To confirm the role of APCCs in IHA, it would be interesting to study the adrenal glands of subjects who continue to have elevated aldosterone levels after adrenalectomy (as a potential sign of bilateral disease).
Building on these observations, one could speculate that APCCs may also contribute to subclinical PA in a subset of patients who suffer from hypertension but do not meet the criteria for the diagnosis of PA (see above). If proven to be true, this assumption may help explain findings from the PATHWAY-2 study demonstrating that the mineralocorticoid receptor antagonist spironolactone is the most effective add-on drug in treatment-resistant hypertension60. However, the use of thiazide or thiazide-like diuretics and the resulting upregulation of the renin–angiotensin–aldosterone axis may also lead to increased spironolactone sensitivity.
Unilateral adrenal hyperplasia and PA
Together, bilateral adrenal hyperplasia and APA account for more than 90% of patients with PA. However, a small subset of patients with PA (~2%) may also present with diffuse unilateral adrenal hyperplasia (UAH)61, although such cases should be studied in more detail using CYP11B2 immunohistochemistry. Unlike for APAs, the genetic causes underlying diffuse UAH are largely unknown. One study reported CACNA1D mutations in two patients with PA and UAH37, suggesting a common underlying pathomechanism in UAH and APAs. In contrast, nodular UAH is fairly common. In many cases, one nodule expresses aldosterone synthase and carries a known APA driver mutation, whereas other nodules represent nonproducing adenomas or nodules without such mutations, but two APAs in a single gland with independent driver mutations have also been reported62.
Genome-wide association studies
Spyroglou et al.63 performed a genome-wide association study of probands from the German KORA (Cooperative Health Research in the Augsburg Region) cohort and reported a significant association of the aldosterone:renin ratio (but not PA) with a locus on chromosome 5q32 comprising the SLC26A2 gene. Based on cell and mouse studies, they suggested a functional role of SLC26A2 in aldosterone production. However, to our knowledge, this association has not been independently replicated. Other groups have performed candidate gene association studies64,65.
Adrenocortical carcinoma
ACC is a very rare disease in the general population (incidence of up to 2 cases per million per year) with a dismal prognosis (5-year overall survival 37–47%)66. Clinically, a large proportion of patients present with overproduction of adrenal steroid hormones, most commonly hypercortisolism66. In rare cases, ACC patients may also present with hyperaldosteronism, featuring increased plasma aldosterone despite low plasma renin activity, hypertension and hypokalemia67,68, and one case of aldosterone-producing ACC carrying the KCNJ5L168R mutation has been reported21.
Familial hyperaldosteronism
FH is a rare cause of PA. Thus far, heterozygous variants in the underlying disease genes have been identified in five subtypes, which are all inherited in an autosomal-dominant fashion or occur de novo (see below). The proportion of patients with unexplained FH is difficult to assess, but descriptions of unsolved cases with early-onset PA suggest that additional genes remain to be discovered18.
Familial hyperaldosteronism type I
In 1966, Sutherland et al.69 described a father and his son who presented with hypertension, increased aldosterone levels despite suppressed plasma renin activity and potassium deficiency. Surprisingly, all abnormalities were relieved by dexamethasone administration69; hence, the syndrome was named glucocorticoid-remediable aldosteronism (GRA). After the discovery of additional forms of FH, it was also referred to as FH-I. Many years after the initial description of the syndrome, the molecular basis of the response to glucocorticoids was identified. As a result of unequal crossing over between the highly homologous genes 11ß-hydroxylase (CYP11B1) and aldosterone synthase (CYP11B2), a chimeric gene is formed. This chimeric gene combines the adrenocorticotrophic hormone (ACTH)-responsive regulatory sequences of CYP11B1 with the coding sequence of CYP11B2, leading to ectopic expression of CYP11B2 in the zona fasciculata under the control of ACTH13,70. The chimeric gene was detected in 29 patients from 12 unrelated pedigrees, including the two patients described by Sutherland et al.13,69. Not all subjects with the chimeric gene are hypertensive, but even in those who are normotensive, excessive aldosterone levels lead to increased left ventricular wall thickness and reduced diastolic function compared to normotensive controls71. The diagnosis is based on genetic testing; therapeutic options include mineralocorticoid receptor antagonists and glucocorticoids7.
Familial hyperaldosteronism type II
The term FH-II was initially coined for familial aggregation of PA without response to glucocorticoid administration72. Later, a diagnosis of FH-II was made when at least two first-degree members (offspring, sibling, and parent) of the same family had confirmed PA73; this included both patients with IHA and APA. However, because sporadic PA is not a rare disorder, the majority of such cases are likely chance associations and do not have Mendelian disorders74. Of the published kindreds, one already described in the early 1990s stood out because of the large number of affected subjects72, but linkage analysis failed to identify the underlying disease gene75. Exome sequencing of selected individuals from this kindred demonstrated a mutation in the CLCN2 gene, encoding the chloride channel ClC-2. An investigation of 80 individuals with early-onset unexplained PA revealed seven additional subjects with CLCN2 mutations, and it was suggested to refer to individuals with CLCN2 mutations as FH-II18. In a parallel publication, one further case was identified19. FH-II subjects typically had elevated ARR (although incomplete penetrance was observed) and, where studied, nonlateralizing aldosterone production; massive adrenal hyperplasia was absent18,19.
CLCN2 mutations cause a gain of function. Increased chloride permeability in the presence of high intracellular chloride concentrations leads to depolarization and voltage-gated calcium influx, followed by CYP11B2 upregulation18,19.
Familial hyperaldosteronism type III
In 2008, Geller et al. described a family (a father and his two daughters) with a new form of PA that did not respond to dexamethasone. The affected subjects showed severe early-onset aldosteronism, including the production of hybrid steroids and massive adrenocortical hyperplasia76. Because medical treatments failed, all three subjects underwent bilateral adrenalectomy76. In conjunction with the identification of somatic KCNJ5 mutations in APAs, a germline KCNJ5 mutation (T158A) was found in this family;16 this mutation is also rarely found in APAs77. Since then, other families with mutations in or close to the KCNJ5 selectivity filter have been reported.
With some exceptions, KCNJ5 mutations that are also found in APAs, when present in the germline, appear to be associated with the development of macroscopic adrenal hyperplasia and a severe phenotype. Scholl et al. identified a germline G151R mutation (most common somatic mutation in APAs (see above)) in three individuals of one kindred who all required bilateral adrenalectomy and a de novo variant in a fourth unrelated patient29. Similarly, E145Q (found in an APA21) was reported as a de novo mutation in a female patient diagnosed with PA at 2 years of age due to massively elevated aldosterone levels and blood pressure, treatable only by bilateral adrenalectomy78.
In contrast, mutations that are only found in the germline are typically associated with a milder clinical phenotype without macroscopic adrenal hyperplasia and without requirement for bilateral adrenalectomy. Such mutations include the G151E mutation found in three kindreds who responded to treatment with mineralocorticoid antagonists29,77. Similarly, a Y152C mutation was found in a patient diagnosed at the age of 48 years with hypertension and a milder phenotype79. Functional studies suggest that while all variants affect the ion selectivity of the channel, G151E causes more profound sodium permeability, which may impair cell survival and prevent the development of hyperplasia29. These findings again support a role for APA-associated KCNJ5 mutations not only in aldosterone production but also in cellular proliferation (see above). The term “familial hyperaldosteronism type III” (FH-III) was initially used to describe a phenotype of dexamethasone-refractory hyperaldosteronism with massive adrenal hyperplasia. However, following the description of patients with KCNJ5 mutations with variable phenotypes, FH-III has been used to refer to PA patients with germline KCNJ5 mutations.
Familial hyperaldosteronism type IV
To identify new disease-causing genes in FH, Scholl et al.20 examined 40 unrelated individuals diagnosed with PA until the age of 10 years by exome sequencing. Five unrelated subjects shared the identical novel heterozygous mutation of the voltage-gated T-type calcium channel CACNA1H (M1549V). Family analysis demonstrated the de novo occurrence of the mutation in two kindreds and also suggested incomplete penetrance20. The mutant calcium channel showed dramatically impaired channel inactivation and activation at more hyperpolarized membrane potentials, resulting in increased intracellular Ca2+, the signal for aldosterone production20. The expression of the mutant channel in H295R cells and its subclone HAC15 resulted in increased aldosterone production compared to the wild-type channel, which was abolished by the inhibition of CACNA1H channels with the T-type calcium channel blocker mibefradil80. Further CACNA1H germline variants were identified by whole-exome sequencing in patients with different types of PA: another de novo case carrying M1549I in a patient with early-onset PA; S196L in a brother and a sister; P2083L in brothers diagnosed with FH; and V1951E in a patient with APA81. Whether variants at residues other than M1549 are causative for FH-IV, however, remains to be established.
PA with seizures and neurological abnormalities (PASNA syndrome)
In addition to somatic mutations in APAs, the CACNA1D gene also shows germline variants in PA. Due to the associated very severe phenotype, such variants exclusively occur de novo.
Scholl et al. investigated 100 unrelated individuals with unexplained early-onset PA and identified de novo CACNA1D germline mutations in two subjects: G403D in a patient with hypertension at birth along with cardiac pathophysiology (e.g., ventricular septal defect) and neurological disorders (e.g., seizures and apparent cerebral palsy), and I770M in a subject with cerebral palsy and seizures since birth and elevated blood pressure at 5 years of age17. By electrophysiology, mutant channels showed activation at less depolarized membrane potentials and impaired inactivation in G403D mutants, which were inferred to cause increased Ca2+ influx and CYP11B2 upregulation17. A de novo G403D mutation was also identified in a subject with persistent congenital hyperinsulinaemic hypoglycemia (HH), mild aortic insufficiency, and seizures82. Aldosterone was within the normal range, but renin was not determined. Finally, de novo CACNA1D variants with similar functional consequences as in PA have been implicated in autism and epilepsy83,84,85. These observations likely represent different manifestations of a complicated syndrome.
Genetic mouse models
While genetic mouse models replicating human FH mutations are sparse, PA mouse models with mutations in genes hitherto not implicated in FH have been generated, in part due to species differences in the adrenal expression of ion channels.
Perhaps surprisingly, given the low expression levels of Kcnj5 in rodents, a Kcnj5 knockout model showed reduced aldosterone levels in female but not in male mice86. A model with adrenal expression of wild-type and mutant human KCNJ5 under the control of the Akr1b7 promoter has been reported only in abstract form but did not show adrenal hyperplasia or severe aldosteronism87. Another transgenic mouse model expresses human CYP11B2 under the control of a human CYP11B1 promoter mimicking the condition of FH-I88. Transgenic mice showed increased aldosterone levels and elevated blood pressure induced by a high-salt diet (4% NaCl), which were significantly reduced by treatment with fadrozole (an inhibitor of CYP11B2)88.
Because Kcnj5 is expressed only at low levels in the adrenal gland of rodents, with TWIK-related acid-sensitive potassium (Task) channels as the predominant K+ conductance89, several TASK mouse models have been created and described. Kcnk3 (TASK-1) knockout mice show the expression of Cyp11b2 in the reticulo-fasciculata zone instead of in the ZG and develop severe hyperaldosteronism (including hypokalemia and low renin levels) remediable by glucocorticoids, representing clinical features of FH-I90. The disruption of both Kcnk3 and Kcnk9 (TASK-3) in mice leads to a depolarization of adrenal glomerulosa cells and an overproduction of aldosterone resistant to both salt suppression and treatment with candesartan along with unchanged or lower renin concentrations91. Deletion of only Kcnk9 leads to salt-sensitive hypertension, slightly elevated aldosterone levels and low plasma renin levels92,93. Zona-glomerulosa-specific disruption of Kcnk3 and Kcnk9 causes mild autonomous aldosterone production despite low renin and thereby chronic blood pressure elevation94. In humans, KCNK3 (TASK-1) single-nucleotide polymorphisms have been associated with blood pressure traits and aldosterone levels95.
To investigate the pathological effects of aldosterone on kidney function, a cryptochrome-null mouse (deletion of cryptochrome genes 1 and 2, which are involved in circadian rhythm) was created96. Mice had elevated aldosterone levels and low plasma renin, and kidney injury was present in mice on a normal-salt diet. However, hypertension, as a key feature of PA, was missing96.
Last, by treating a cohort of mice with the mutagen N-ethyl-N-nitrosourea (ENU), a mouse line with elevated aldosterone levels was generated97. These mice were carriers of point mutations in seven candidate genes, of which a variant in SCO-spondin (SSpo) was suggested as the most promising based on a phenotype of increased aldosterone values, associated Cyp11b2 upregulation as well as elevated ARR97.
Discussion
Taken together, the insights gained over the past decade, mainly through next-generation sequencing, suggest that PA is largely a genetic disease caused by somatic and, in rare cases, germline mutations. Sporadic PA is likely a disease continuum that starts with a somatic driver mutation in a single adrenal cell and subsequently progresses to APCC and/or APA. Recent studies suggest that similar pathophysiology applies to IHA. If this is true, cutoff values for the definition of PA7 are, to some degree, arbitrary, and it may be worth retesting patients with borderline results after a few years, in particular if hypertension is difficult to control. Similarly, lesions may initially be too small to be detected by routine imaging. Recent descriptions of APCCs also suggest that the distinction between unilateral and bilateral disease may not be as clear-cut as previously thought, e.g., a patient could have a predominant adenoma on one side and APCCs on the contralateral side.
The partially overlapping mutational spectrum between APAs, APCCs, and FH is also interesting. KCNJ5 variants explain a substantial portion (approximately 40%) of the somatic mutations in APAs but are also the basis of FH-III. CACNA1D variants are involved as somatic mutations in APAs as well as in an inherited form of PA, PASNA syndrome. So far, CACNA1H mutations have been reported only as germline mutations in FH-IV, similar to CLCN2 mutations in FH-II and CYP11B2 mutations in FH-I; whether they account for rare instances of APAs remains to be determined. In addition to ion channels, somatic mutations in ATPases ATP1A1 and ATP2B3 have been implicated in APA formation. Such mutations have not been reported in FH, likely because they would be lethal at the germline level.
Whereas a role of voltage-gated ion channels in adrenal aldosterone production had been well established prior to these studies1, KCNJ5 had not been identified as a key player in aldosterone production, likely due to expression differences between rodents and humans89. Similarly, the study of FH-II first pointed to an anion channel in connection with PA and hypertension.
The underlying pathophysiology of the vast majority of mutations appears to involve increased calcium influx, followed by upregulated CYP11B2 expression and aldosterone production. This increased calcium influx can occur directly, via mutations in voltage-gated calcium channels, or indirectly, via changes in ion permeability or selectivity that cause the depolarization of glomerulosa cells and the subsequent opening of voltage-gated calcium channels. The latter mechanism applies to KCNJ5, CLCN2, and ATPase mutations. These results suggest that calcium signaling could be a useful therapeutic target in PA. However, approved calcium channel inhibitors are not efficient inhibitors of CACNA1D or CACNA1H channels, and widespread extra-adrenal expression could lead to side effects. Distinct from all other mutations is the formation of a hybrid gene by unequal crossing over between CYP11B2 and CYP11B1 along with an aberrant expression pattern of CYP11B2 in the adrenal cortex, leading to ACTH-dependent aldosterone overproduction in FH-I. The exact pathophysiology of aldosteronism due to CTNNB1 mutations is less understood.
Recent studies with improved sensitivity suggest that the vast majority of APAs are explained by somatic mutations in either of the genes discussed above. Mutations in additional genes may account for small percentages of APAs. In contrast, many cases of APCCs and early-onset PA remain unexplained, suggesting that additional mutations remain to be identified.
Beyond the mouse models discussed in this review, future animal models replicating the gain-of-function mechanisms implicated in human FH would be interesting tools to study the physiology and pathophysiology of these genes in the adrenal gland. The translation of the genetic findings from the last decade into the clinic is eagerly awaited.
References
Spat, A. & Hunyady, L. Control of aldosterone secretion: a model for convergence in cellular signaling pathways. Physiol. Rev. 84, 489–539 (2004).
Masilamani, S., Kim, G.-H., Mitchell, C., Wade, J. B. & Knepper, M. A. Aldosterone-mediated regulation of ENaC α, β, and γ subunit proteins in rat kidney. J. Clin. Invest. 104, R19–R23 (1999).
Gekle, M. & Grossmann, C. Actions of aldosterone in the cardiovascular system: the good, the bad, and the ugly? Pflug. Arch. 458, 231–246 (2009).
Conn, J. W. Presidential address. I. Painting background. II. Primary aldosteronism, a new clinical syndrome. J. Lab. Clin. Med. 45, 3–17 (1955).
Monticone, S. et al. Prevalence and clinical manifestations of primary aldosteronism encountered in primary care practice. J. Am. Coll. Cardiol. 69, 1811–1820 (2017).
Rossi, G. P. et al. A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. J. Am. Coll. Cardiol. 48, 2293–2300 (2006).
Funder, J. W. et al. The management of primary aldosteronism: case detection, diagnosis, and treatment: an Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 101, 1889–1916 (2016).
Monticone, S. et al. Cardiovascular events and target organ damage in primary aldosteronism compared with essential hypertension: a systematic review and meta-analysis. Lancet Diabetes Endocrinol. 6, 41–50 (2018).
Ohno, Y. et al. Prevalence of cardiovascular disease and its risk factors in primary aldosteronism: a multicenter study in Japan. Hypertension 71, 530–537 (2018).
Milliez, P. et al. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J. Am. Coll. Cardiol. 45, 1243–1248 (2005).
Williams, T. A. et al. Outcomes after adrenalectomy for unilateral primary aldosteronism: an international consensus on outcome measures and analysis of remission rates in an international cohort. Lancet Diabetes Endocrinol. 5, 689–699 (2017).
Hundemer, G. L., Curhan, G. C., Yozamp, N., Wang, M. & Vaidya, A. Cardiometabolic outcomes and mortality in medically treated primary aldosteronism: a retrospective cohort study. Lancet Diabetes Endocrinol. 6, 51–59 (2018).
Lifton, R. P. et al. A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature 355, 262–265 (1992).
Azizan, E. A. et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat. Genet. 45, 1055–1060 (2013).
Beuschlein, F. et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat. Genet. 45, 440–444 (2013).
Choi, M. et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 331, 768–772 (2011).
Scholl, U. I. et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 45, 1050–1054 (2013).
Scholl, U. I. et al. CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat. Genet. 50, 349–354 (2018).
Fernandes-Rosa, F. L. et al. A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism. Nat. Genet. 50, 355–361 (2018).
Scholl, U. I. et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife 4, e06315 (2015).
Akerstrom, T. et al. Comprehensive re-sequencing of adrenal aldosterone producing lesions reveal three somatic mutations near the KCNJ5 potassium channel selectivity filter. PLoS ONE 7, e41926 (2012).
Williams, T. A. et al. Somatic ATP1A1, ATP2B3, and KCNJ5 mutations in aldosterone-producing adenomas. Hypertension 63, 188–195 (2014).
Boulkroun, S. et al. Prevalence, clinical, and molecular correlates of KCNJ5 mutations in primary aldosteronism. Hypertension 59, 592–598 (2012).
Lenzini, L. et al. A meta-analysis of somatic KCNJ5 K(+) channel mutations In 1636 patients with an aldosterone-producing adenoma. J. Clin. Endocrinol. Metab. 100, E1089–E1095 (2015).
Fernandes-Rosa, F. L. et al. Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension 64, 354–361 (2014).
Bichet, D., Haass, F. A. & Jan, L. Y. Merging functional studies with structures of inward-rectifier K(+) channels. Nat. Rev. Neurosci. 4, 957–967 (2003).
Heginbotham, L., Abramson, T. & Mackinnon, R. A functional connection between the pores of distantly related ion channels as revealed by mutant K+ channels. Science 258, 1152–1155 (1992).
Lerario, A. M. et al. Genetics of aldosterone-producing adenomas with pathogenic KCNJ5 variants. Endocr. Relat. Cancer 26, 463–470 (2019).
Scholl, U. I. et al. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc. Natl Acad. Sci. USA 109, 2533–2538 (2012).
Tamura, A. et al. Somatic KCNJ5 mutation occurring early in adrenal development may cause a novel form of juvenile primary aldosteronism. Mol. Cell. Endocrinol. 441, 134–139 (2017).
Zennaro, M. C., Boulkroun, S. & Fernandes-Rosa, F. Genetic causes of functional adrenocortical adenomas. Endocr. Rev. 38, 516–537 (2017).
Vouillarmet, J. et al. Aldosterone-producing adenoma with a somatic KCNJ5 mutation revealing APC-dependent familial adenomatous polyposis. J. Clin. Endocrinol. Metab. 101, 3874–3878 (2016).
Catterall, W. A., Striessnig, J., Snutch, T. P. & Perez-Reyes, E., International Union of, P. International Union of Pharmacology. XL. Compendium of voltage-gated ion channels: calcium channels. Pharmacol. Rev. 55, 579–581 (2003).
Nanba, K. et al. Targeted molecular characterization of aldosterone-producing adenomas in White Americans. J. Clin. Endocrinol. Metab. 103, 3869–3876 (2018).
Nanba, K. et al. Genetic characteristics of aldosterone-producing adenomas in Blacks. Hypertension 73, 885–892 (2019).
Akerstrom, T. et al. Activating mutations in CTNNB1 in aldosterone producing adenomas. Sci. Rep. 6, 19546 (2016).
Scholl, U. I. et al. Novel somatic mutations in primary hyperaldosteronism are related to the clinical, radiological and pathological phenotype. Clin. Endocrinol. 83, 779–789 (2015).
Tauber, P. et al. Cellular pathophysiology of an adrenal adenoma-associated mutant of the plasma membrane Ca-ATPase ATP2B3. Endocrinology, 157, 2489–2499 (2016).
Tadjine, M., Lampron, A., Ouadi, L. & Bourdeau, I. Frequent mutations of beta-catenin gene in sporadic secreting adrenocortical adenomas. Clin. Endocrinol. 68, 264–270 (2008).
Juhlin, C. C. et al. Whole-exome sequencing characterizes the landscape of somatic mutations and copy number alterations in adrenocortical carcinoma. J. Clin. Endocrinol. Metab. 100, E493–E502 (2015).
Tissier, F. et al. Mutations of β-catenin in adrenocortical tumors: activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res. 65, 7622–7627 (2005).
Goh, G. et al. Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat. Genet. 46, 613 (2014).
Berthon, A. et al. Constitutive beta-catenin activation induces adrenal hyperplasia and promotes adrenal cancer development. Hum. Mol. Genet. 19, 1561–1576 (2010).
Teo, A. E. D. et al. Pregnancy, primary aldosteronism, and adrenal CTNNB1 mutations. N. Engl. J. Med. 373, 1429–1436 (2015).
Murtha, T. D., Carling, T. & Scholl, U. I. Pregnancy, primary aldosteronism, and somatic CTNNB1 mutations. N. Engl. J. Med. 374, 1492–1494 (2016).
Wu, V. C. et al. Prevalence and clinical correlates of somatic mutation in aldosterone producing adenoma-Taiwanese population. Sci. Rep. 5, 11396 (2015).
Taguchi, R. et al. Expression and mutations of KCNJ5 mRNA in Japanese patients with aldosterone-producing adenomas. J. Clin. Endocrinol. Metab. 97, 1311–1319 (2012).
Hong, A. R. et al. Genetics of aldosterone-producing adenoma in Korean patients. PLoS ONE 11, e0147590 (2016).
Scholl, U. I. et al. Macrolides selectively inhibit mutant KCNJ5 potassium channels that cause aldosterone-producing adenoma. J. Clin. Invest. 127, 2739–2750 (2017).
Maiolino, G. et al. Macrolides for KCNJ5-mutated aldosterone-producing adenoma (MAPA): design of a study for personalized diagnosis of primary aldosteronism. Blood Press. 27, 200–205 (2018).
Williams, T. A. et al. Genotype-specific steroid profiles associated with aldosterone-producing adenomas. Hypertension 67, 139–145 (2016).
Howard, B. et al. Integrated analysis of genome-wide methylation and gene expression shows epigenetic regulation of CYP11B2 in aldosteronomas. J. Clin. Endocrinol. Metab. 99, E536–E543 (2014).
Murakami, M. et al. Integration of transcriptome and methylome analysis of aldosterone-producing adenomas. Eur. J. Endocrinol. 173, 185–195 (2015).
Yoshii, Y. et al. Hypomethylation of CYP11B2 in aldosterone-producing adenoma. Hypertension 68, 1432–1437 (2016).
Nishimoto, K. et al. Adrenocortical zonation in humans under normal and pathological conditions. J. Clin. Endocrinol. Metab. 95, 2296–2305 (2010).
Nishimoto, K. et al. Aldosterone-stimulating somatic gene mutations are common in normal adrenal glands. Proc. Natl Acad. Sci. USA 112, E4591–E4599 (2015).
Nanba, K. et al. Molecular heterogeneity in aldosterone-producing adenomas. J. Clin. Endocrinol. Metab. 101, 999–1007 (2016).
Nanba, K. et al. Histopathological diagnosis of primary aldosteronism using CYP11B2 immunohistochemistry. J. Clin. Endocrinol. Metab. 98, 1567–1574 (2013).
Omata, K. et al. Cellular and genetic causes of idiopathic hyperaldosteronism. Hypertension 72, 874–880 (2018).
Williams, B. et al. Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. Lancet 386, 2059–2068 (2015).
Young, W. F. Primary aldosteronism: renaissance of a syndrome. Clin. Endocrinol. 66, 607–618 (2007).
Dekkers, T. et al. Adrenal nodularity and somatic mutations in primary aldosteronism: one node is the culprit? J. Clin. Endocrinol. Metab. 99, E1341–E1351 (2014).
Spyroglou, A. et al. Diastrophic dysplasia sulfate transporter (SLC26A2) is expressed in the adrenal cortex and regulates aldosterone secretion. Hypertension 63, 1102–1109 (2014).
Jung, J. et al. Variations in the potassium channel genes KCNK3 and KCNK9 in relation to blood pressure and aldosterone production: an exploratory study. J. Clin. Endocrinol. Metab. 97, E2160–E2167 (2012).
Imrie, H. et al. Association between aldosterone production and variation in the 11beta-hydroxylase (CYP11B1) gene. J. Clin. Endocrinol. Metab. 91, 5051–5056 (2006).
Fassnacht, M., Libe, R., Kroiss, M. & Allolio, B. Adrenocortical carcinoma: a clinician’s update. Nat. Rev. Endocrinol. 7, 323–335 (2011).
Farge, D. et al. Isolated clinical syndrome of primary aldosteronism in four patients with adrenocortical carcinoma. Am. J. Med. 83, 635–640 (1987).
Seccia, T. M., Fassina, A., Nussdorfer, G. G., Pessina, A. C. & Rossi, G. P. Aldosterone-producing adrenocortical carcinoma: an unusual cause of Conn’s syndrome with an ominous clinical course. Endocr. Relat. Cancer 12, 149–159 (2005).
Sutherland, D. J., Ruse, J. L. & Laidlaw, J. C. Hypertension, increased aldosterone secretion and low plasma renin activity relieved by dexamethasone. Can. Med. Assoc. J. 95, 1109–1119 (1966).
Lifton, R. P. et al. Hereditary hypertension caused by chimaeric gene duplications and ectopic expression of aldosterone synthase. Nat. Genet. 2, 66–74 (1992).
Stowasser, M. et al. Evidence for abnormal left ventricular structure and function in normotensive individuals with familial hyperaldosteronism type I. J. Clin. Endocrinol. Metab. 90, 5070–5076 (2005).
Gordon, R. D. et al. Clinical and pathological diversity of primary aldosteronism, including a new familial variety. Clin. Exp. Pharmacol. Physiol. 18, 283–286 (1991).
Mulatero, P. et al. Prevalence and characteristics of familial hyperaldosteronism. Hypertension 58, 797–803 (2011).
Korah, H. E. & Scholl, U. I. An update on familial hyperaldosteronism. Horm. Metab. Res. 47, 941–946 (2015).
Lafferty, A. R. et al. A novel genetic locus for low renin hypertension: familial hyperaldosteronism type II maps to chromosome 7 (7p22). J. Med. Genet. 37, 831–835 (2000).
Geller, D. S. et al. A novel form of human Mendelian hypertension featuring nonglucocorticoid-remediable aldosteronism. J. Clin. Endocrinol. Metab. 93, 3117–3123 (2008).
Mulatero, P. et al. KCNJ5 mutations in European families with nonglucocorticoid remediable familial hyperaldosteronism. Hypertension 59, 235–240 (2012).
Monticone, S. et al. A case of severe hyperaldosteronism caused by a de novo mutation affecting a critical salt bridge Kir3.4 residue. J. Clin. Endocrinol. Metab. 100, E114–E118 (2015).
Monticone, S. et al. A novel Y152C KCNJ5 mutation responsible for familial hyperaldosteronism type III. J. Clin. Endocrinol. Metab. 98, E1861–E1865 (2013).
Reimer, E. N., Walenda, G., Seidel, E. & Scholl, U. I. CACNA1HM1549VMutant calcium channel causes autonomous aldosterone production in HAC15 cells and is inhibited by mibefradil. Endocrinology 157, 3016–3022 (2016).
Daniil, G. et al. CACNA1H mutations are associated with different forms of primary aldosteronism. EBioMedicine 13, 225–236 (2016).
Flanagan, S. et al. A CACNA1D mutation in a patient with persistent hyperinsulinaemic hypoglycaemia, heart defects, and severe hypotonia. Pediatr. Diabetes 18, 320–323 (2017).
Pinggera, A. et al. New gain-of-function mutation shows CACNA1D as recurrently mutated gene in autism spectrum disorders and epilepsy. Hum. Mol. Genet. 26, 2923–2932 (2017).
O’Roak, B. J. et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250 (2012).
Iossifov, I. et al. De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299 (2012).
Hardege, I., Long, L., Al Maskari, R., Figg, N. & Kevin Targeted disruption of the Kcnj5 gene in the female mouse lowers aldosterone levels. Clin. Sci. 132, 145–156 (2018).
Lichtenauer, U. et al. Establishment of an in vivo model for KCNJ5 dependent hyperaldosteronism. Exp. Clin. Endocrinol. Diabetes 123, P09_25 (2015).
Gu, H. et al. Salt-dependent Blood Pressure in Human Aldosterone Synthase-Transgenic Mice. Sci. Rep. 7, 492 (2017).
Chen, A. X., Nishimoto, K., Nanba, K. & Rainey, W. E. Potassium channels related to primary aldosteronism: expression similarities and differences between human and rat adrenals. Mol. Cell. Endocrinol. 417, 141–148 (2015).
Heitzmann, D. et al. Invalidation of TASK1 potassium channels disrupts adrenal gland zonation and mineralocorticoid homeostasis. EMBO J. 27, 179–187 (2008).
Davies, L. A. et al. TASK channel deletion in mice causes primary hyperaldosteronism. Proc. Natl Acad. Sci. USA 105, 2203–2208 (2008).
Guagliardo, N. A. et al. TASK-3 channel deletion in mice recapitulates low-renin essential hypertension. Hypertension 59, 999–1005 (2012).
Penton, D. et al. Task3 potassium channel gene invalidation causes low renin and salt-sensitive arterial hypertension. Endocrinology 153, 4740–4748 (2012).
Guagliardo, N. A. et al. Adrenal tissue-specific deletion of TASK channels causes aldosterone-driven angiotensin II–independent hypertension. Hypertension 73, 407–414 (2019).
Manichaikul, A. et al. KCNK3 variants are associated with hyperaldosteronism and hypertension. Hypertension 68, 356–364 (2016).
Nugrahaningsih, D. A. A. et al. Chronic hyperaldosteronism in Cryptochrome-null mice induces high-salt- and blood pressure-independent kidney damage in mice. Hypertens. Res. 37, 202–209 (2014).
Perez-Rivas, L. G. et al. Genetic characterization of a mouse line with primary aldosteronism. J. Mol. Endocrinol. 58, 67–78 (2017).
Wu, V. C. et al. The prevalence of CTNNB1 mutations in primary aldosteronism and consequences for clinical outcomes. Sci. Rep. 7, 39121 (2017).
Acknowledgements
This work was supported by grants to UIS from the Stiftung Charité (BIH Johanna Quandt Professorship), the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation, SCHO 1386/2-1) and the Fritz Thyssen Stiftung (10.16.1.027MN). Rockefeller University has filed a patent application (PCT/US2018/033362, Compositions and methods for diagnosing and treating diseases and disorders associated with mutant KCNJ5), listing UIS as an inventor.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Seidel, E., Schewe, J. & Scholl, U.I. Genetic causes of primary aldosteronism. Exp Mol Med 51, 1–12 (2019). https://doi.org/10.1038/s12276-019-0337-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-019-0337-9
This article is cited by
-
Screening and diagnosis of primary aldosteronism. Consensus document of all the Spanish Societies involved in the management of primary aldosteronism
Endocrine (2024)
-
Monogenic forms of low-renin hypertension: clinical and molecular insights
Pediatric Nephrology (2022)
-
Update on the Genetics of Primary Aldosteronism and Aldosterone-Producing Adenomas
Current Cardiology Reports (2022)
-
Rare functional missense variants in CACNA1H: What can we learn from Writer’s cramp?
Molecular Brain (2021)
-
A Chinese pedigree with glucocorticoid remediable aldosteronism
Hypertension Research (2021)
