
Abstract
The metabolic rewiring that occurs during cell transformation is a hallmark of cancer. It is diverse in different cancers as it reflects different combinations of oncogenic drivers, tumor suppressors, and the microenvironment. Metabolic rewiring is essential to cancer as it enables uncontrolled proliferation and adaptation to the fluctuating availability of nutrients and oxygen caused by poor access to the vasculature due to tumor growth and a foreign microenvironment encountered during metastasis. Increasing evidence now indicates that the metabolic state in cancer cells also plays a causal role in tumor growth and metastasis, for example through the action of oncometabolites, which modulate cell signaling and epigenetic pathways to promote malignancy. In addition to altering the metabolic state in cancer cells, some multifunctional enzymes possess non-metabolic functions that also contribute to cell transformation. Some multifunctional enzymes that are highly expressed in cancer, such as pyruvate kinase M2 (PKM2), have non-canonical functions that are co-opted by oncogenic signaling to drive proliferation and inhibit apoptosis. Other multifunctional enzymes that are frequently downregulated in cancer, such as fructose-bisphosphatase 1 (FBP1), are tumor suppressors, directly opposing mitogenic signaling via their non-canonical functions. In some cases, the enzymatic and non-canonical roles of these enzymes are functionally linked, making the modulation of non-metabolic cellular processes dependent on the metabolic state of the cell.
Similar content being viewed by others
Introduction
Metabolism is not just a permissive process that exists exclusively to meet cellular biosynthetic and bioenergetic needs. Instead, metabolism is intricately connected to multiple cellular processes, as certain metabolic intermediates function as cofactors or substrates for the post-translational modification of proteins or the modification of DNA during epigenetic regulation. These metabolic intermediates can become rate-limiting depending on the metabolic state of the cell. Examples of such metabolites include acetyl-CoA (AcCoA), s-adenosylmethionine (SAM), succinate, fumarate, 2-hydroxyglutarate (2HG) and α-ketoglutarate (αKG). The various roles of these molecules, also called oncometabolites, have been studied extensively in the context of cancer1.
However, not only metabolic intermediates have the ability to couple the metabolic state of the cell to other cellular functions. In addition to their canonical enzymatic function within the metabolic network, various multifunctional (moonlighting) metabolic enzymes perform non-canonical functions in a variety of cellular processes. Among the first multifunctional enzymes to be discovered were the glycolytic enzymes that function as crystallins in the lens of the eye2. Since then, it has become clear that the non-canonical functions of metabolic enzymes are quite common. Every single enzyme in the glycolytic cascade and several enzymes from other metabolic pathways have been found to be multifunctional (Table 1). Moreover, some metabolic enzymes, such as PKM2, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and aldolase, carry out multiple non-canonical functions in cellular processes such as transcription, signaling and cytoskeletal dynamics. In some cases, these secondary functions are entirely independent of the canonical enzymatic role and do not involve regulatory processes in the cell, as in the case of the glycolytic enzymes functioning as crystallins. However, the non-canonical functions of metabolic enzymes often regulate processes that are highly relevant for cell transformation and cancer development: they promote uncontrolled cell proliferation, induce resistance to apoptosis or enhance cell migration. Other enzymes have non-canonical functions that oppose mitogenic signaling or promote apoptosis under conditions of stress, thus having a tumor suppressive role. In many of these cases, canonical and non-canonical enzyme functions are often interdependent, thus connecting the activity of cancer-relevant cellular processes to the metabolic state of the cell.
Here, we focus on multifunctional enzymes that have been shown to play a non-canonical role in cancer. These functions represent another layer of complexity within the regulatory network in cancer and provide additional challenges for therapeutic targeting.
Oncogenic non-canonical functions of metabolic enzymes
Of the numerous multifunctional enzymes described, several have pro-proliferative and/or anti-apoptotic roles within various non-metabolic cellular processes and can contribute to cell transformation and tumor development. For some of these enzymes, the switch from canonical to non-canonical function is induced by the action of oncogene-activated signaling cascades via post-translational modifications. For others, the non-canonical function is intrinsic to the enzyme and is promoted in cancer by the elevated expression of the enzyme.
Glycolytic enzymes with non-canonical functions as protein kinases in cancer
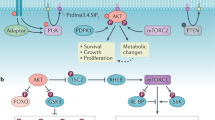
In recent years, several examples of metabolic enzymes acting as phosphate transferases in metabolism but having a secondary role as protein kinases have been uncovered. One of these is ketohexokinase (KHK), the enzyme that converts fructose to fructose-1-phosphate, which subsequently enters glycolysis at the level of aldolase. KHK has a secondary function as a protein kinase3. During the formation of hepatocellular carcinoma (HCC), c-MYC induces an isoform switch from KHKC to KHKA. Interestingly, KHKA, but not KHKC, interacts with and phosphorylates phosphoribosyl pyrophosphate synthetase 1 (PRPS1), a key enzyme in nucleotide synthesis. This phosphorylation prevents the allosteric inhibition of PRPS1 by ADP, resulting in enhanced de novo nucleotide biosynthesis during HCC tumor growth (Fig. 1)3.

Metabolic enzymes can function as protein kinases, transcriptional regulators, scaffolding proteins, and modulators of the actin cytoskeleton to control cancer-relevant phenotypes. Oncogenic functions: Ketohexokinase A (KHKA) phosphorylates phosphoribosyl pyrophosphate synthetase 1 (PRPS1) to prevent its allosteric inhibition by ATP and promote nucleotide synthesis. Phosphoglycerate kinase 1 (PGK1) phosphorylates pyruvate dehydrogenase kinase (PDK1) to promote aerobic glycolysis (the Warburg effect). The low activity of mTORC1 leads to acetylation (ac) of PGK1, phosphorylation of beclin 1 (BCN1) and induction of autophagy. Pyruvate kinase M2 (PKM2) phosphorylates and inactivates AKT1 substrate 1 (AKT1S1), a negative regulator of mTORC1, promoting cell growth. PKM2 also has nuclear functions as a transcriptional co-activator of β-catenin, OCT-4 and HIF-1α (TF) to promote proliferation, transformation, angiogenesis and metabolism. Nuclear translocation of PKM2 is controlled by the Jumonji C domain-containing dioxygenase 5 (JMJD5) and by EGFR signaling. The mitochondrial enzyme 3-hydroxy-3-methylglutaryl-CoA synthase 2 (HMGCS2) is palmitoylated (palm.) and translocated to the nucleus where it functions as a co-activator of PPARα. PGAM1 induces the formation of F-actin and cell migration. Tumor suppressive functions: Fructose-1,6-bisphosphatase 1 (FBP1) translocates to the nucleus and blocks the transcriptional activity of HIF-1α. It also binds to the IQ motif-containing GTPase activating protein 1 (IQGAP1) to prevent activation of the RAS/RAF/MEK/ERK cascade and inhibits proliferation. G6P glucose-6-phosphate, F6P fructose-6-phosphate, F1,6BP fructose-1,6-bisphosphate, G3P glyceraldehyde 3-phosphate, DHAP dihydroxyacetone phosphate, F1P fructose-1-phosphate, 1,3BPG 1,3 bisphosphoglycerate, 3PG 3-phosphoglycerate, 2PG 2-phosphoglycerate, PEP phosphoenolpyruvate, a-KG alpha-ketoglutarate, OAA oxaloacetate
Likewise, the glycolytic enzyme phosphoglycerate kinase 1 (PGK1) is also a protein kinase in glioblastoma cells4,5. Upon phosphorylation by ERK1/2, induced by hypoxia or expression of the oncogenes K-RAS (G12V) or Braf (V600E), PGK1 is transported to the mitochondria where it phosphorylates and activates pyruvate dehydrogenase kinase 1 (PDK1). This, in turn, inhibits the pyruvate dehydrogenase complex (PDC), leading to increased lactate production and glycolytic activity (Fig. 1)4. In addition, glutamine deprivation or hypoxia, but not glucose deprivation, can lead to the acetylation of PGK1, which then phosphorylates beclin 1 (BCN1), a scaffolding protein essential for the formation of autophagosomes during autophagy5. Acetylation of PGK1 is triggered by the low activity of complex 1 of the mammalian target of rapamycin (mTORC1) upon glutamine starvation or treatment with the mTORC1 inhibitor torin 1 (Fig. 1). Analysis of human tumor samples also indicates a strong correlation between PGK-1 acetylation, BCN1 phosphorylation and poor prognosis in glioblastoma5.
Another metabolic enzyme that doubles as a protein kinase is the M2 isoform of the glycolytic enzyme pyruvate kinase (PKM2), which has received much attention in the context of cancer metabolism. This isoform of pyruvate kinase is frequently upregulated in cancer due to a switch in alternative splicing from the PKM1 isoform to PKM2, induced by the transcription factor c-MYC6. In cancer, PKM2 plays a key role in fueling anabolic metabolism by diverting metabolic flux from glycolysis into the pentose phosphate pathway (PPP) and the serine synthesis pathway (SSP)7. This specific role of PKM2 is mediated by its ability to change its conformation between a catalytically low activity dimer and a high activity tetramer. The balance between these two conformations is determined by different allosteric regulators, as well as by intracellular signaling, mostly via post-translational modifications (PTM). The tetrameric form of PKM2 is stabilized by fructose-1,6-bisphosphate (F1,6BP), the product of phosphofructokinase 1 (PFK1)8. This mechanism of allosteric feed-forward regulation couples the last reaction of glycolysis with the first rate-limiting step. Another allosteric activator of PKM2 is serine9, which is mostly taken up by cells but can also be synthesized from 3-phosphoglycerate (3PG), an intermediate of glycolysis. When serine or F1,6BP levels are low, allosteric activation of PKM2 is reduced and glycolytic metabolites can be rerouted into the pentose phosphate and serine synthesis pathways. Once the levels of serine and F1,6BP increase again, PKM2 forms a tetramer and catalyzes the rapid formation of pyruvate, thereby removing glycolytic intermediates. PKM2 can dynamically shift (or oscillate) between its dimeric and tetrameric form in response to alterations in the levels of its allosteric regulators. In fact, this feature of PKM2 was used to generate a FRET biosensor to monitor oscillatory glycolytic activity in pancreatic β-cells10.
Many of the PTMs induced by intracellular signaling promote the dimeric form of PKM2 and do so by interfering with F1,6BP binding. The phosphorylation of fibroblast growth factor receptor type 1 (FGFR1) at Y105 releases F1,6BP from PKM2 and promotes dimerization11. Interestingly, this has been suggested to be a “transprotein” mechanism dependent on the K433 residue in PKM2 that directly interacts with the F1,6BP molecule. Specifically, a modified PKM2 molecule induces the release of F1,6BP from other, unmodified PKM2 molecules by interfering with the interaction of K433 and F1,6BP11. Moreover, this ability to induce the release of F1,6BP from other PKM2 molecules is not unique to tyrosine-phosphorylated PKM2. Different phosphotyrosine peptides with an Src kinase-like motif can induce the release of F1,6BP from PKM212, indicating that various tyrosine-phosphorylated proteins can influence PKM2 oligomerization. In addition to phosphorylation, acetylation at the K433 residue by P300 acetyltransferase and sumoylation by the SUMO-E3 ligase PIAS3, prevent F1,6BP from binding to PKM2, leading to the dimerization and nuclear translocation of PKM213,14. The ability of PKM2 to block glycolysis at the level of pyruvate kinase seems to be essential for cancer development, as cancer cells expressing only the PKM1 isoform have a reduced tumor-forming capacity15. Nevertheless, some non-proliferating populations of cancer cells within a tumor may depend on PKM1 expression, demonstrating the complexity of metabolic regulation in cancer16.
The enzymatically inactive dimeric form of PKM2 is widely reported to have non-canonical functions as either a protein kinase or as a transcriptional co-activator (Fig. 1). Numerous proteins have been identified and studied as targets of PKM2 protein kinase activity, including histone H3, the transcription factor STAT3, the spindle checkpoint protein Bub3 (reviewed in refs.17,18) and the apoptosis regulator Bcl-219.A recent screen for PKM2 substrates identified over 400 proteins as potential targets of PKM2 protein kinase activity. The same study also demonstrated that AKT1 substrate 1 (AKT1S1), a negative regulator of mTORC1, is negatively regulated by PKM2-dependent phosphorylation in renal cancer20. The effects of the PKM2 kinase function are generally pro-mitogenic or anti-apoptotic and are frequently induced by post-translational modifications of PKM2, including ERK1/2 or Aurora B dependent phosphorylation (reviewed in refs.17,18). In contrast to KHKA and PGK1, which use ATP as a phosphate donor for protein phosphorylation, PKM2 has been reported to use its glycolytic substrate, phosphoenolpyruvate (PEP), as a phosphate donor for its non-canonical function as a protein kinase19,20,21,22,23. Nevertheless, the protein kinase function of PKM2 has been called into question by in vitro experiments that failed to find evidence for PEP- or ATP-dependent kinase activity24.
Metabolic enzymes as transcriptional regulators
In addition to its non-canonical role as a protein kinase, PKM2 has also been reported to act as a transcriptional co-activator in the nucleus (Fig. 1). PKM2 interacts with the transcription factor OCT-4 in embryonic carcinoma cells and glioma spheroids and modulates the expression of OCT-4 target genes25,26. In renal cell carcinoma (RCC) and breast cancer cells (MCF7), PKM2 interacts with Hif-1α and promotes its transcriptional activity27,28. Interestingly, in MCF7 cells, Jumonji C domain-containing dioxygenase 5 (JMJD5) is required for PKM2 nuclear translocation and Hif-1α activation; JMJD5 interacts with the dimeric form of PKM2 and prevents its re-tetramerization28. The role of PKM2 as co-activator of Hif-1α is not limited to cancer, as nuclear localization of PKM2 and subsequent activation of Hif-1α transcription has also been observed during macrophage activation29. In this context, the PKM2 nuclear translocation and activation of Hif-1α can be inhibited by agents that promote the tetramerization and activation of PKM229. In glioblastoma cells, activation of the epidermal growth factor receptor (EGFR) leads to phosphorylation of PKM2 and β-catenin by ERK1/2 and c-Src kinases, respectively, and subsequent nuclear localization of both proteins30,31. In the nucleus, PKM2 promotes the transcriptional activity of β-catenin both as a co-activator and as a protein kinase, by phosphorylating histone-H3, which leads to its acetylation22,31.
Other metabolic enzymes also act in transcription within the context of cancer. A recent example of this is the regulation of the peroxisome proliferator-activated receptor α (PPARα) by the enzyme 3-hydroxy-3-methylglutaryl-CoA synthase 2 (HMGCS2), the rate-limiting enzyme in ketogenesis, in hepatic tissue32. HMGCS2, as well as other enzymes of the ketone synthesis pathway, are upregulated in some cancers, including androgen-independent prostate cancer33,34, colorectal carcinoma (CRC) and oral squamous cell carcinoma (OSCC)35. HMGCS2 translocates to the nucleus and, through a process that requires HMGCS2 palmitoylation, acts as a co-transactivator for PPARα to regulate its own expression (Fig. 1)36,37. In CRC and OSCC cells, enhanced invasion and migration induced by the increased expression of HMGCS2 is independent of its catalytic activity but requires interaction with PPARα to promote expression of the tyrosine kinase Src35,36. In addition, the bifunctional peroxisomal enzyme enoyl-CoA-hydratase/3-hydroxyacyl-CoA-dehydrogenase (EHHADH) has also been reported to regulate its own expression via its interaction with PPARα38.
PGAM1 as a modulator of the cytoskeleton
Phosphoglycerate mutase 1 (PGAM1), the glycolytic enzyme responsible for the conversion of 3PG to 2-phosphoglycerate (2PG), is upregulated in a variety of cancers39 and is associated with enhanced cell migration and motility40,41. This effect was attributed to the non-canonical function of PGAM1 as a modulator of the cytoskeleton. A screen in HEK293 cells identified the G-actin isoform of alpha smooth muscle actin (ACTA2), which is associated with metastatic potential in lung adenocarcinoma42, as a novel interaction partner for PGAM1. The interaction between PGAM1 and ACTA2 is essential for cell migration and cytoskeleton remodeling in a breast cancer cell line, and depletion of either PGAM1 or ACTA2 inhibits cell motility by reducing F-actin formation (Fig. 1). Expression of a mutated form of PGAM1 that is unable to bind ACTA2 but retains its enzymatic activity did not restore F-actin assembly and cell migration. Conversely, deletion of the catalytic activity or pharmacological inhibition of PGAM1 did not interfere with its ability to modulate cytoskeletal dynamics39, confirming that its canonical activity is dispensable in this context. In addition, depletion of PGAM1 in an in vivo model of breast cancer significantly reduced the number of lung metastases39 and co-expression of ACTA2 and PGAM1 correlated with poor prognosis in human breast cancer patients.
Non-canonical, tumor suppressive functions of metabolic enzymes
To date, a few enzymes have been found to act as tumor suppressors; well-characterized examples are the TCA cycle enzymes succinate dehydrogenase (SDH) and fumarate hydratase (FH). Loss of function mutations in the genes coding for SDH and FH can lead to cancer, which can be explained by the fact that succinate and fumarate, the substrates for SDH and FH, inhibit the prolyl hydroxylase (PHD) family of enzymes. Inhibition of PHDs leads to the normoxic stabilization of HIF1α, as it is hydroxylated and targeted for degradation by PHDs43. Both SDH and FH act as tumor suppressors via their canonical functions, however, several enzymes have been discovered that possess non-canonical tumor suppressive functions; they either directly oppose mitogenic signaling or promote apoptosis and cell cycle arrest under conditions of stress.
FBPase1 is a tumor suppressor that counteracts the HIF-1α and MAPK/ERK pathways
Fructose-1,6-bisphosphatase 1 (FBP1) is the rate-limiting enzyme in gluconeogenesis and converts F1,6BP to F6P. FBP1 has non-canonical functions that oppose cell transformation. The expression of FBP1 is reduced in several cancer types, including HCC44,45, colon and breast cancers46, renal carcinoma47 and pancreatic cancer48,49. Moreover, processes that promote tumorigenesis also repress FBP1 expression44,45,46,49,50. In ccRCC, FBP1 interacts with HIF-1α on promoters that contain hypoxia response elements (HRE) to repress the transcription of HIF-1α target genes (Fig. 1)47. Ectopic expression of FBP1 in ccRCC cells reduced their glycolytic activity and NADPH production and blocked their ability to form xenograft tumors. Interestingly, the tumor suppressive activity of FBP1 is primarily mediated by its non-canonical nuclear functions; a reduction in tumor growth was still observed after the expression of a catalytically inactive mutant of FBP1, while the addition of a nuclear export signal (NES) completely abolished the effect47. Compared with the wild-type protein, catalytically inactive FBP1 had a reduced ability to suppress ccRCC cell proliferation under low glucose conditions, which suggests that enhancing gluconeogenesis also contributes to tumor suppression by FBP147.
Another non-canonical tumor-suppressing function of FBP1 has been identified in pancreatic cancer. Here, FBP1 interacts with the IQ motif containing GTPase activating protein 1 (IQGAP1), a scaffolding protein that is essential for the RAS/RAF/MEK/ERK signaling cascade that promotes RAS-driven tumorigenesis (Fig. 1)51. Modulation of ERK1/2 phosphorylation by FBP1 overexpression or knock-down is dependent on the presence of IQGAP1 and ERK1/2 and FBP1 compete for binding to its WW domain. Catalytically inactive FBP1, as well as a 45 AA fragment containing the IQGAP1 binding domain of FBP1, were sufficient to block ERK1/2 phosphorylation, inhibit proliferation and the formation of xenograft tumors in PDAC cell lines. However, the catalytic activity of FBP1 also contributes to tumor suppression in this system, as the wild-type protein, which retains its ability to reduce glucose uptake and lactate secretion, had a stronger effect on proliferation and tumor formation compared with the deletion mutants48.
GMPS and MDH1 promote p53 function under conditions of stress
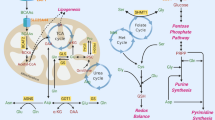
Guanosine 5’-monophosphate synthase (GMPS) catalyzes the glutamine-dependent conversion of xanthosine 5’-monophosphate (XMP) to guanosine 5’-monophosphate (GMP), which is the last reaction in the de novo synthesis of guanine nucleotides. GMPS interacts with the deubiquitylation enzyme USP7 (also called HAUSP)52,53. GMPS is an allosteric activator of USP7 and stabilizes its active state resulting in hyperactivation of USP754. The interaction between GMPS and USP7 promotes the deubiquitylation of H2B and modulates the expression of ecdysteroid target genes55 and homeotic genes52 in Drosophila, as well as expression of the latent origin of replication of the Epstein-Barr virus in HeLa cells53,56.
Interestingly, USP7 can also be found in a complex with the tumor suppressor p53 and its negative regulator MDM2, an E3 ubiquitin ligase that targets p53 for degradation but can also act as transcriptional co-repressor for p5357. In unstressed cells, USP7 deubiquitylates both p53 and MDM2 and contributes to a finely balanced state in which p53 is continuously degraded58. The interaction between MDM2 and p53 is disrupted upon cellular stress, leading to the stabilization of p5357. Knock-down of GMPS prevents p53 stabilization in response to genotoxic stress59, while concomitant overexpression of both USP7 and GMPS leads to p53 stabilization and apoptosis, even in the absence of genotoxic stress59. Mechanistically, this can be explained by a change in complex formation, with GMPS replacing MDM2 and the resulting complex between USP7, GMPS and p53 stabilizes p53 (Fig. 2)59. In unstressed cells, GMPS itself is ubiquitylated by the E3 ligase TRIM21 and sequestered in the cytoplasm. In contrast, genotoxic stress induces GMPS deubiquitylation by USP7, allowing for its nuclear translocation. Interestingly, the enzymatic activity of GMPS is not needed for this non-canonical function; a catalytically inactive form of GMPS stabilizes p53 to the same extent as the wild-type protein when overexpressed with USP759. While it is clear that GMPS is primed to respond to genotoxic stress by activating USP7, it is unclear how genotoxic stress triggers USP7 activation in the first place. Interestingly, inhibition of inosine-5’-monophosphate dehydrogenase (IMPDH), the enzyme directly upstream of GMPS, leads to the nuclear translocation of GMPS and the stabilization of p53, indicating that the trigger for GMPS may be disrupted nucleotide biosynthesis. Remarkably, it has been reported that upon replicative stress and during the G2 phase of the cell cycle, IMPDH itself translocates to the nucleus, where it represses the expression of histones and the master transcriptional regulator of G1/S transition, E2F60. These findings show that GMPS and IMPDH, two enzymes that play a key role in supporting proliferation through their canonical functions in de novo purine synthesis, also play a role in opposing proliferation through their non-canonical functions. It will be interesting to determine, in future studies, whether these functions are interconnected.

In response to DNA damage, guanosine 5’-monophosphate synthase (GMPS) shifts from a complex containing the E3 ligase TRIM21 to interaction with the deubiquitylating enzyme USP7. This leads to deubiquitylation of GMPS and allows its translocation to the nucleus, where it replaces MDM2 and stabilizes p53 to promote cell cycle arrest and apoptosis. Upon glucose starvation, malate dehydrogenase 1 (MDH1) activates p53 by binding to the p53/MDM2 complex and preventing ubiquitylation and cytoplasmic retention of p53. IMP inosine monophosphate; XMP xanthosine monophosphate; GMP guanosine 5’-monophosphate; GDP guanosine 5’-diphosphate; GTP guanosine 5’-triphosphate
Malate dehydrogenase 1 (MDH1), a cytoplasmic enzyme that catalyzes the conversion of malate to oxaloacetate as part of the malate/aspartate shuttle, also regulates p53. MDH1 was identified in a screen of enzymes in central carbohydrate metabolism for p53 interaction partners61. Upon glucose starvation, both p53 and MDH1 are translocated to the nucleus where MDH1 acts as a transcriptional co-activator of p53 target genes by binding to p53-response elements in the promoters of p53 target genes (Fig. 2)61. Interestingly, while MDH1 does not interfere with the p53/MDM2 interaction, overexpression and depletion experiments show that MDH1 inhibits p53 ubiquitylation and prevents MDM2-dependent cytoplasmic retention of p53, thereby promoting apoptosis in response to glucose starvation61. As in the case of GMPS, the catalytic activity of MDH1 is not needed for this non-canonical function61.
Non-canonical enzyme functions mediate cross-talk between metabolism and signaling in cancer
In some cases, the non-canonical functions of metabolic enzymes are intricately linked to their canonical activity; the two functions can be dependent on each other or even be mutually exclusive. Such enzymes can act as sensors that modulate non-metabolic functions in a manner dependent on the metabolic state of a cell. Most of the enzymes that fit this definition belong to the glycolytic pathway. By switching between canonical and non-canonical activities, these enzymes can promote proliferation when glycolytic activity is high but block it when glycolysis is reduced, such as under conditions of nutrient starvation.
Regulation of AMPK and mTORC1 by glycolytic activity
The mammalian target of rapamycin complex 1 (mTORC1) is a serine/threonine kinase and part of a major nutrient-sensing pathway. It integrates information on nutritional availability in the cell (i.e., amino acids, glucose, and oxygen) and growth factor signaling coming from various upstream pathways, including some that are frequently mutated and activated in cancer, such as the PI3K/Akt and the RAS/RAF/MEK/ERK pathways. The net effect of the mTORC1 pathway is to promote growth and anabolic processes (i.e., amino acid, nucleotide, and lipid synthesis) but inhibit catabolic and/or conserving processes such as autophagy62. A major regulator of mTORC1 activity is the AMP-activated protein kinase (AMPK), which inhibits mTORC1 directly and indirectly via activation of the mTORC1 inhibitor tuberous sclerosis complex (TSC). The primary function of AMPK is to respond to altered adenosine nucleotide ratios (AMP/ATP or ADP/ATP) by inhibiting anabolic processes and promoting the generation of energy through catabolic reactions. In addition to mTORC1, AMPK targets various other enzymes including acetyl-CoA carboxylase, the rate-limiting enzyme for fatty acid synthesis63. AMPK is activated by phosphorylation at threonine 172, primarily delivered by the liver kinase B1 (LKB1), and is deactivated by dephosphorylation of threonine 172 by several phosphatases. The binding of AMP and ADP to the gamma subunit of the AMPK complex interferes with dephosphorylation and promotes phosphorylation, resulting in the activation of AMPK64. In addition, AMP also activates AMPK through allosteric mechanisms64.
Apart from this established mechanism of AMPK regulation, aldolase activates AMPK via a non-canonical function even in the absence of an altered adenosine nucleotide ratio65. When cells are starved of glucose, AMPK binds to axin and LKB1, forming a complex on the lysosomal membrane with Ragulator, a protein complex involved in nutrient sensing by mTORC1 and the vacuolar ATPase (v-ATPase)66,67. Interestingly, formation of this complex is mediated by the dissociation of aldolase from the lysosomal membrane, where it can also interact with the v-ATPase. All three mammalian paralogs of aldolase have been found to interact with v-ATPase, and in yeast, this interaction depends on the presence of glucose68,69. The trigger for the removal of aldolase from the lysosomal membrane is a reduction in intracellular levels of F1,6BP, the substrate for aldolase in glycolysis, caused by diminished glycolytic activity. As the lysosomal complex is formed, AMPK is phosphorylated and activated by LKB1, which, in turn, phosphorylates its targets, including raptor and TSC2, leading to inhibition of mTORC1 (Fig. 3). In mouse embryo fibroblasts (MEFs), a reduction in intracellular glucose levels leads to a rapid increase in AMPK activity prior to any significant change in AMP/ATP or ADP/ATP ratios65. Likewise, in the livers of mice starved for 16 h, which causes a large drop in blood glucose levels, AMPK activity, but not nucleotide ratios, is elevated65. The inhibition of mitochondrial activity or the combined starvation of glucose and glutamine in MEFs did increase the nucleotide ratios, as well as AMPK activity. However, this is a delayed response as opposed to the acute increase in AMPK activity observed upon glucose starvation in the absence of altered nucleotide ratios. Hence, this suggests that at least two different mechanisms exist by which AMPK senses energetic stress, an acute response, mediated by the non-canonical function of aldolase, and a delayed response, dependent on altered nucleotide ratios.

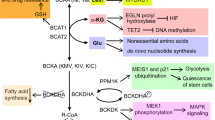
Regulation of AMPK and mTORC1 by glycolytic flux: In the presence of fructose-1,6-bisphosphate (F1,6BP), aldolase interacts with the lysosomal v-ATPase and Ragulator, a component of the mTORC1 nutrient sensing machinery. Upon glucose starvation, F1,6BP levels are reduced and aldolase is removed from this complex, which allows for the association of the v-ATPase-Ragulator complex with AMPK, axin and LKB1, thereby promoting the phosphorylation and activation of AMPK. This mode of regulation contributes to the activation of AMPK by metabolic stress and leads to the inhibition of biosynthesis and the activation of energy-producing processes. Low glucose also promotes the binding of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) to the Ras homolog mTORC1-binding protein (RHEB), resulting in the inactivation of mTORC1. Hexokinase 2 can bind directly to mTORC1 during glucose starvation to block macromolecule biosynthesis during growth and proliferation. Glucose-6-phosphate (G6P), the product of the reaction catalyzed by HK2, prevents this interaction, providing an additional mechanism of nutrient sensing and regulation of mTORC1. Cross-talk between glycolysis, cytoskeletal dynamics and cancer signaling: Aldolase A (ALDOA) has a non-canonical function in regulating the actin cytoskeleton through binding to the Wiskott Aldrich Syndrome protein (WASP) and F-actin. F1,6BP reversibly inhibits this interaction, thereby providing cross-talk between cellular metabolism and cytoskeletal dynamics. The activation of PI3K/Rac signaling in cancer also leads to the release of ALDOA from the actin cytoskeleton to promote glycolytic activity
Until recently, it was believed that the inhibition of mTORC1 signaling observed under glucose starvation is mediated exclusively by AMPK. However, this inhibition can occur not only via AMPK, independent of an altered AMP/ATP ratio (as discussed above) but also entirely independent of AMPK. The two mechanisms identified to date that mediate the glucose starvation-induced inhibition of mTORC1 rely on the non-canonical activities of the glycolytic enzymes HK2 and GAPDH (Fig. 3).
In MEFs deficient for both the α1 and α2 subunits of AMPK, glucose starvation can still inhibit mTORC1 activity70,71, suggesting that an AMPK independent mechanism of glucose sensing must exist. Indeed, it was found that GAPDH binds to the GTPase Rheb and prevents its interaction with mTORC1, similar to the inhibition of mTORC1 by TSC1/272,73. Interestingly, the interaction between GAPDH and Rheb appears to be regulated by glycolytic activity, as it is reduced during culture in high glucose media compared with low glucose media (Fig. 3)72. In addition, cells cultured in the presence of the hexokinase inhibitor 2-deoxyglucose (2-DG) strengthened the interaction between GAPDH and Rheb, while the addition of the GAPDH substrate glyceraldehyde-3-phosphate (G3P) prevented it. Importantly, these effects were also found in AMPKα1-silenced and in TSC1-deficient or TSC2-deficient cells72,73, confirming that this mode of mTORC1 regulation is independent of AMPK and TSC1/2.
The second glycolytic enzyme that regulates mTORC1 activity is hexokinase 2 (HK2). HK2 interacts directly with and inhibits mTORC1 in cardiomyocytes and HEK293 cells during glucose starvation, leading to the activation of autophagy (Fig. 3)74. While the kinase domain of HK2 is dispensable for binding to mTORC1, the interaction between the two proteins is inhibited by glucose-6-phosphate (G6P), the product and allosteric inhibitor of HK74. This mode of regulation is reminiscent of the association of HK2 to with the mitochondria as part of its anti-apoptotic role, which is also regulated by G6P (reviewed in ref.75). However, these two non-canonical roles of HK2 seem to be independent of each other. The HK2-mTOR interaction is dependent on an mTOR signaling (TOS) motif in HK2 (also found in the mTORC1 protein raptor), whereas an HK2 mutant unable to bind the mitochondria, maintains its ability to inhibit mTOR.
The main advantages of this direct modulation of mTORC1 activity by glycolytic flux, as opposed to AMPK signaling, are likely increased speed and specificity of the response. In addition to the energetic state of the cell, AMPK activity is modulated by various cellular signaling pathways76. The induction of biosynthetic processes downstream of mTORC1 requires a sufficient supply of nutrients, including glucose. A direct coupling between mTORC1 activity and glycolytic flux likely provides a safety mechanism that only permits macromolecule assembly when nutrient supplies are adequate. This mode of regulation is also more acute compared with the control by AMPK activity, which is primarily mediated by the cellular energy load. Indeed, GAPDH binding to Rheb occurs within minutes of a reduction in glucose72, resulting in a rapid shut down of biosynthetic processes when nutrients are scarce.
Regulation of cytoskeletal dynamics by aldolase and PI3K/Rac signaling
All three paralogs of aldolase bind F-actin to a different degree, with ALDOA having a much higher affinity for actin than ALDOB or ALDOC77. The binding of ALDOA to actin is regulated by the presence of its substrate F1,6BP, which reversibly inhibits the ALDOA-actin interaction (Fig. 3)77,78,79. The interaction of aldolase with actin may also be functionally important as each aldolase tetramer is able to bind multiple F-actin monomers and crosslink actin filaments into rafts in vitro78,80,81. In addition, aldolase interacts with the Wiskott-Aldrich syndrome protein (WASp), an actin regulator, via its catalytic domain, and this interaction is reversibly inhibited by F1,6BP82,83.
The mobilization of ALDOA from the cytoskeleton is regulated by PI3K signaling84. ALDOA mobilization is essential for the induction of glycolytic activity and is dependent on the activation of Rac, downstream of PI3K, but independent of the Akt/mTORC1 axis. Inhibition of PI3K leads to a rapid and sustained decrease in glycolytic activity, whereas inhibition of Akt or mTOR only leads to a transient and less pronounced reduction in glycolytic activity. Likewise, the mobilization of glycolytic reserves (the rapid increase in glycolytic activity caused by blocking ATP-dependent respiration using oligomycin) is strongly blunted by the inhibition of PI3K, but not by the inhibition of the Akt/mTOR axis. The inhibition of the mobilization of the glycolytic reserve by inhibiting PI3K is abolished in cells expressing mutant forms of ALDOA that are incapable of binding to actin84. Interestingly, in an in vivo mouse model of breast cancer, PI3K inhibition only resulted in a modest decrease in glucose uptake, as determined by PET imaging of FDG uptake. However, the conversion of pyruvate to lactate, determined using MRI and hyperpolarized 13C-pyruvate, was drastically reduced84. In vivo 13C-glucose tracing in tumors confirms that PI3K inhibition blocks the glycolytic pathway at the level of ALDOA84.
These findings indicate that aldolase links glycolytic activity to cytoskeletal dynamics and cell signaling. Under conditions of low glucose availability, ALDOA is bound to the actin cytoskeleton, where it performs its non-canonical role in cytoskeletal organization. When glucose becomes available and the F1,6BP level increases, the interaction between ALDOA and the cytoskeleton is disrupted and the enzyme performs its canonical function in glycolytic metabolism. However, in response to elevated F1,6BP levels, the activation of PI3K/Rac signaling is likely needed to enable the mobilization of ALDOA from the cytoskeleton, via an as-yet-unknown mechanism (possibly involving post-translational modification). Hence, the non-canonical function of ALDOA may have a dual role, linking glycolytic activity with cytoskeletal dynamics and allowing for the rapid elevation of glycolytic activity in response to PI3K signaling. This could be particularly important in cancer cells where PI3K signaling is continuously active.
The YAP/TAZ complex senses glycolytic activity via PFK1
YAP /TAZ are transcriptional coactivators that interact primarily with the TEA domain (TEAD) family of transcription factors to regulate the transcription of genes involved in proliferation and survival85. The HIPPO pathway is the best-characterized upstream regulator of YAP/TAZ but they are also influenced by the TGFβ, BMP, and Wnt signaling pathways85. In addition, YAP/TAZ play an important role in mechanotransduction and are directly regulated by the extracellular matrix (ECM) and the cytoskeleton85. Upregulation of YAP/TAZ activity is frequently found in cancer and is believed to be important for resistance to anoikis, the epithelial to mesenchymal transition (EMT) and cell migration85,86.
A recently discovered additional player in YAP/TAZ regulation is AMPK, which phosphorylates components of the HIPPO pathway, as well as YAP itself in response to glucose starvation, leading to inhibition of YAP/TAZ-dependent transcription87,88,89. Similar to the regulation of mTORC1 by glycolytic activity via GAPDH and HK2 (discussed above), the activity of YAP/TAZ is also modulated by glycolytic flux via phosphofructokinase 1 (PFK1) (Fig. 4). PFK1 catalyzes the irreversible conversion of F6P to F1,6BP in glycolysis, thereby controlling a key regulatory node that determines the entry of metabolites into glycolysis and the associated PPP and the hexosamine pathways90. In breast cancer and mammary epithelial cell lines, YAP/TAZ activity has been found to correlate with glycolytic activity independent of AMPK91. This regulation of YAP/TAZ activity is mediated by PFK1, which binds to TEAD1 and 4 in the nucleus. This interaction is required for the stability of the YAP/TAZ/TEAD complex, as depletion of PFK1 leads to its dissociation. Likewise, glucose starvation or inhibition of glycolysis using 2DG inhibits the interaction of PFK1 with TEAD and the interaction of TEAD with YAP/TAZ91. Furthermore, overexpression of 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3(PFKFB3), which produces F2,6BP, a potent allosteric activator of PFK1, promotes YAP/TAZ-dependent transcription, whereas a mutant PFK1 that is unable to bind F2,6BP has a low affinity for TEAD91. These data strongly suggest that YAP/TAZ-dependent transcription is regulated by glycolytic activity via a non-canonical function of PFK1. The regulation of YAP/TAZ by glycolysis may be of particular significance in breast cancer, as analysis of the expression data from breast tumors revealed a strong correlation between a glycolysis-dependent signature (i.e., genes downregulated by 2DG treatment in breast cancer cell lines) and genes upregulated by YAP/TAZ91. In addition, genes that are common to both the glycolysis-dependent and the YAP/TAZ signature are highly expressed in high-grade tumors, while expression of the remaining genes from both signatures has no prognostic value91. As the YAP/TAZ coactivators have also been associated with the induction of stem cell-related properties92, the modulation of their activity by glycolytic flux in cancer can affect treatment resistance and disease recurrence.

Regulation of transcription: Phosphofructokinase 1 (PFK1) regulates the transcriptional activity of YAP/TAZ through a mechanism that is dependent on the presence of its allosteric regulator fructose-2,6-bisphosphate (F2,6BP), which is generated by the bifunctional enzyme 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3). In the presence of F2,6BP, PFK1 translocates to the nucleus and acts as a transcriptional co-activator for YAP/TAZ and TEAD to drive the expression of genes involved in proliferation, the epithelial to mesenchymal transition (EMT) and stem cell maintenance. Inhibition of glycolysis promotes the nuclear translocation of enolase 1 (ENO1) and its alternative gene product c-Myc promoter-binding protein 1 (MBP1). Both proteins function as transcriptional repressors to block transcription of the genes coding for Forkhead Box P1 and c-Myc. Regulation of translation: Inhibition of glycolysis also promotes the binding of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) to the mRNA of interferon α (IFNα) to inhibit translation. This mechanism of regulation mediates the regulation of IFNα production by glycolytic flux in activated T cells. In the absence of substrate, thymidylate synthase (TS) and dihydrofolate reductase (DHFR) block translation of their own mRNAs. This mechanism of feedback regulation ensures the availability of the enzyme when substrate levels accumulate. DHF dihydrofolate, THF tetrahydrofolate, 5,10-me-THF 5,10-methylene tetrahydrofolate, dUMP deoxyuridine monophosphate, dTMP deoxythymidine monophosphate
Enolase1/MPB1 is a transcriptional repressor of Myc
Enolase 1 (ENO1, also known as α-enolase) catalyzes the conversion of 3PG to PEP in glycolysis. However, an alternate translation product of the ENO1 mRNA has been identified as Myc promoter binding protein 1 (MBP1)93. MPB1 binds to the c-MYC promoter and represses transcription (Fig. 4)93. Both full-length ENO1 and MBP1 localize to the nucleus in leukemia cells, where they interact with the Notch1 intracellular domain (NICD) and suppress transcription of c-MYC and other Notch1 target genes94. In addition, overexpression of MBP1 or ENO1 suppresses NICD-induced soft agar colony formation in leukemia cells94. The regulation of this non-canonical function of ENO1/MBP1 by glycolytic activity was not assessed in these studies. However, an investigation into the role of metabolism in the differentiation of conventional T cells (Tconv cells) into regulatory T cells (Treg cells) in response to T cell antigen receptor (TCR) stimulation, revealed that elevated glycolysis promotes Treg development by modulating the activity of ENO1/MPB1 as a transcriptional repressor (Fig. 4)95. The development of Treg cells is accompanied by an increase in glycolytic activity and increased expression of the transcription factor forkhead box P3 (FOXP3) and its splicing variant FOXP3-E2. Inhibition of glycolysis by 2DG during the development of Treg cells attenuates FOXP3/FOXP3-E2 expression and impairs Treg cell function95. Interestingly, compared with untreated cells, 2DG-treated Treg cells display elevated nuclear localization of both MBP1 and ENO1, as well as increased recruitment of ENO1 to the FOXP3 promoter. In addition, the effect of 2DG on FOXP3/FOXP3-E2 expression is rescued by silencing ENO195. These data suggest that, at least in T cells, the non-canonical function of ENO1 as a transcriptional repressor and its canonical function as a glycolytic enzyme may be mutually exclusive. It will be interesting to see whether this also applies to the ENO1/MBP1-mediated repression of c-MYC expression in cancer cells.
PKM2 may integrate mitogenic signaling with the metabolic state of the cell
As discussed in the “Oncogenic non-canonical functions of metabolic enzymes” section of this review, the non-canonical functions of PKM2 include protein kinase and transcription factor (in its dimeric form). The oligomeric state of PKM2 is allosterically regulated by F1,6BP and altering the amount of glucose available to the cell impacts both the intracellular F1,6BP levels and the oligomeric state of PKM28,10. Some of the non-canonical functions performed by dimeric PKM2 are preceded by posttranslational modifications that lock PKM2 into the dimeric state13,96, effectively abolishing the metabolic regulation of the PKM2 oligomeric state and the non-canonical functions carried out by the PKM2 dimeric state. However, there are indications that some of the non-canonical functions of PKM2 in the nucleus can be regulated by the metabolic state of the cell. In a study investigating the nuclear interaction of PKM2 and Oct4 in glioma spheroids enriched in cancer stem cells (CSC), treatment with the pyruvate dehydrogenase kinase (PDK) inhibitor dichloroacetate (DCA) induced the nuclear translocation of PKM2 and promoted the interaction between PKM2 and Oct426. Likewise, the finding that PKM2 uses PEP as a phosphate donor for its protein kinase activity raises the possibility that the kinase function of PKM2 is dependent on glycolytic flux via the availability of PEP; PKM2 may, therefore, integrate mitogenic signaling with metabolic activity. In this scenario, PKM2 protein kinase activity would not only represent a driver for proliferation but would also act as an indicator for a nutrient- and energy-rich metabolic state that can support macromolecule biosynthesis and cell proliferation. PKM2 would thus act as a safety valve that only propagates mitogenic signals when nutrient levels are sufficient to support proliferation.
Metabolic enzymes as RNA binding proteins
Through recent mRNA interactome studies in HeLa, HEK, and mESC cells, it is evident that multiple enzymes from intermediary metabolism also function as mRNA-binding proteins (reviewed in ref.97). The functional importance of most of these interactions is not fully understood, as the identity of many of the interacting mRNAs remains largely unknown. However, the non-canonical functions as mRNA-binding proteins of some metabolic enzymes have been studied in greater detail (reviewed in refs.98,99). In some enzymes, the catalytic activity and mRNA-binding activity are interdependent, providing a potential mechanism for the functional linkage of metabolic activity and posttranscriptional regulation.
Of the metabolic enzymes that can also bind mRNA, the best characterized is GAPDH, which has been shown to interact with numerous, different mRNAs and regulates their stability or efficiency of translation (reviewed in ref.100). For some of these interactions, the effect of the catalytic activity of GAPDH on its non-canonical function as an mRNA-binding protein has not been addressed. However, it has been shown that in monocytes and activated T cells, elevated glycolytic activity promotes cytokine production in a manner that is dependent on GAPDH mRNA binding101,102. In T cells, the switch from oxidative phosphorylation to aerobic glycolysis during T cell activation is required in order to produce the cytokines IFN-γ and IL-2. When activated T cells are cultured on galactose instead of glucose, which results in reduced glycolytic activity, GAPDH binds to the IFN-γ and IL-2 mRNAs and suppresses their translation. However, when the cells are returned to glucose-containing media and can switch to aerobic glycolysis, GAPDH binding to IFN-γ and IL-2 mRNA is reduced and translation occurs (Fig. 4)102. Furthermore, GAPDH-mediated suppression of cytokine production in T cells cultured in galactose was reversed by supplementing the medium with G3P, the substrate for GAPDH in glycolysis, showing that cytokine production is controlled by the competition between glycolytic flux and GAPDH mRNA binding102.
Thymidylate synthase (TS) and dihydrofolate reductase (DHFR) are enzymes involved in the de novo synthesis of dTMP and one-carbon metabolism, respectively. Both enzymes have been found to bind their own mRNAs and repress translation (Fig. 4)103. In both cases, mRNA binding is inhibited by the respective substrate of the enzyme: dUMP and 5,10-methylentetrahydrofolate for TS and dihydrofolate for DHFR. This type of auto-regulation couples the translation of TS and DHFR with the availability of their substrates, thereby facilitating enzyme production when the need for its enzymatic activity is highest, such as during DNA synthesis103. Interestingly, TS has also been reported to bind other mRNAs including the p53 mRNA and overexpression of TS leads to reduced p53 translation104.
Another example of mRNA-binding enzymes is mevalonate kinase (MVK), which catalyzes the conversion of mevalonate to phospho-mevalonate as part of the cholesterol biosynthesis pathway. MVK, also called LH receptor mRNA binding protein (LRBP) binds to the luteinizing hormone receptor (LHR) mRNA, resulting in its degradation105,106. LHR mRNA stability is regulated by both estrogen and ERK1/2 signaling, which increase and decrease MVK levels, respectively106,107. The binding of MVK to LHR mRNA is inhibited by mevalonate, the substrate for MVK, while treatment with mevastatin, an inhibitor of mevalonate synthesis, results in increased LHR expression. Furthermore, the same amino acids in MVK that are essential for its catalytic function are also required for its mRNA-binding activity 108. These data indicate that mRNA-binding activity and the catalytic activity of MVK are mutually exclusive98,105. Considering the frequent deregulation of enzymes of the mevalonate pathway in various human cancers109, it will be interesting to see whether MVK might also interact with other mRNAs to modulate the expression of cancer-relevant genes.
Concluding remarks
It is now evident that the non-canonical functions of metabolic enzymes play important roles in cellular processes that are closely linked to oncogenic signaling and cell transformation. These additional activities need to be considered when studying metabolic alterations in cancer cells. One example is the large-scale screening studies that aim to identify metabolic enzymes that are essential for the survival of cancer cells. Such screens generally use gene silencing or knockout strategies and it is likely that, in some cases, the essentiality of the identified enzymes is caused by their non-canonical activities. In such a case, drug screens are faced with the additional challenge that non-canonical functions often depend on protein/protein interactions, which are difficult to target with small molecules. One possible solution for this problem could be the use of drugs that induce the selective ubiquitylation and degradation of a target protein through molecules known as proteolysis targeting chimeras (PROTACs)110. Indeed, PROTAC strategies should be particularly successful for the targeting of metabolic enzymes, as these proteins contain catalytic centers that can selectively bind small molecule ligands. Another strategy for the targeting of non-canonical functions of metabolic enzymes could be to exploit the known mechanisms of allosteric regulation. For example, compounds that lock PKM2 into its highly active tetrameric conformation have already been shown to block tumor growth in a xenograft model of non-small cell lung cancer111. In addition to inducing constant activation of its catalytic activity, these compounds should also block the non-canonical functions of PKM2 that may depend on the formation of the dimer. Non-canonical enzyme functions that are regulated by substrate availability (as in the case of mRNA binding by TS or DHFR) or that require the binding of a specific ligand (as in the case of transcriptional coactivation by PFK1) can be targeted by small molecules that either block or mimic these interactions. Finally, non-canonical enzyme functions that are regulated by metabolic flux or respond to cellular signaling pathways can be modulated by anti-metabolites or targeted therapies. However, in most cases, it will be challenging to confirm that the therapeutic effect is indeed linked to the non-canonical function of the enzyme. Additional research is needed to reveal the full extent of the contribution of non-canonical functions of metabolic enzymes in cancer and to identify suitable strategies for their therapeutic targeting.
References
Nowicki, S. & Gottlieb, E. Oncometabolites: tailoring our genes. FEBS J. 282, 2796–2805 (2015).
Piatigorsky, J. Crystallin genes: specialization by changes in gene regulation may precede gene duplication. J. Struct. Funct. Genom. 3, 131–137 (2003).
Li, X. et al. A splicing switch from ketohexokinase-C to ketohexokinase-A drives hepatocellular carcinoma formation. Nat. Cell Biol. 18, 561–571 (2016).
Li, X. et al. Mitochondria-translocated PGK1 functions as a protein kinase to coordinate glycolysis and the TCA cycle in tumorigenesis. Mol. Cell 61, 705–719 (2016).
Qian, X. et al. Phosphoglycerate kinase 1 phosphorylates Beclin1 to induce autophagy. Mol. Cell 65, 917–931.e916 (2017).
David, C. J., Chen, M., Assanah, M., Canoll, P. & Manley, J. L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 463, 364–368 (2010).
Israelsen, W. J. & Vander Heiden, M. G. Pyruvate kinase: function, regulation and role in cancer. Semin Cell Dev. Biol. 43, 43–51 (2015).
Ashizawa, K., Willingham, M. C., Liang, C. M. & Cheng, S. Y. In vivo regulation of monomer-tetramer conversion of pyruvate kinase subtype M2 by glucose is mediated via fructose 1,6-bisphosphate. J. Biol. Chem. 266, 16842–16846 (1991).
Chaneton, B. et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 491, 458–462 (2012).
Merrins, M. J., Van Dyke, A. R., Mapp, A. K., Rizzo, M. A. & Satin, L. S. Direct measurements of oscillatory glycolysis in pancreatic islet beta-cells using novel fluorescence resonance energy transfer (FRET) biosensors for pyruvate kinase M2 activity. J. Biol. Chem. 288, 33312–33322 (2013).
Hitosugi, T. et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci. Signal 2, ra73 (2009).
Christofk, H. R., Vander Heiden, M. G., Wu, N., Asara, J. M. & Cantley, L. C. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 452, 181–186 (2008).
Lv, L. et al. Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Mol. Cell 52, 340–352 (2013).
Spoden, G. A. et al. The SUMO-E3 ligase PIAS3 targets pyruvate kinase M2. J. Cell Biochem 107, 293–302 (2009).
Christofk, H. R. et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452, 230–233 (2008).
Israelsen, W. J. et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 155, 397–409 (2013).
Dong, G. et al. PKM2 and cancer: the function of PKM2 beyond glycolysis. Oncol. Lett. 11, 1980–1986 (2016).
Yang, W. & Lu, Z. Pyruvate kinase M2 at a glance. J. Cell Sci. 128, 1655–1660 (2015).
Liang, J. et al. Mitochondrial PKM2 regulates oxidative stress-induced apoptosis by stabilizing Bcl2. Cell Res 27, 329–351 (2017).
He, C. L. et al. Pyruvate kinase M2 activates mTORC1 by phosphorylating AKT1S1. Sci. Rep. 6, 21524 (2016).
Gao, X., Wang, H., Yang, J. J., Liu, X. & Liu, Z. R. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol. Cell 45, 598–609 (2012).
Yang, W. et al. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 150, 685–696 (2012).
Jiang, Y. et al. PKM2 phosphorylates MLC2 and regulates cytokinesis of tumour cells. Nat. Commun. 5, 5566 (2014).
Hosios, A. M., Fiske, B. P., Gui, D. Y. & Vander Heiden, M. G. Lack of evidence for PKM2 protein kinase activity. Mol. Cell 59, 850–857 (2015).
Lee, J., Kim, H. K., Han, Y. M. & Kim, J. Pyruvate kinase isozyme type M2 (PKM2) interacts and cooperates with Oct-4 in regulating transcription. Int. J. Biochem. Cell Biol. 40, 1043–1054 (2008).
Morfouace, M. et al. Control of glioma cell death and differentiation by PKM2-Oct4 interaction. Cell Death Dis. 5, e1036 (2014).
Luo, W. et al. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 145, 732–744 (2011).
Wang, H. J. et al. JMJD5 regulates PKM2 nuclear translocation and reprograms HIF-1alpha-mediated glucose metabolism. Proc. Natl Acad. Sci. USA 111, 279–284 (2014).
Palsson-McDermott, E. M. et al. Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 21, 65–80 (2015).
Yang, W. et al. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat. Cell Biol. 14, 1295–1304 (2012).
Yang, W. et al. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature 480, 118–122 (2011).
Grabacka, M., Pierzchalska, M., Dean, M. & Reiss, K. Regulation of ketone body metabolism and the role of PPARalpha. Int. J. Mol. Sci. 17, E2093 (2016).
Saraon, P. et al. Quantitative proteomics reveals that enzymes of the ketogenic pathway are associated with prostate cancer progression. Mol. Cell Proteom. 12, 1589–1601 (2013).
Saraon, P. et al. Evaluation and prognostic significance of ACAT1 as a marker of prostate cancer progression. Prostate 74, 372–380 (2014).
Chen, S. W. et al. HMGCS2 enhances invasion and metastasis via direct interaction with PPARalpha to activate Src signaling in colorectal cancer and oral cancer. Oncotarget 8, 22460–22476 (2017).
Meertens, L. M., Miyata, K. S., Cechetto, J. D., Rachubinski, R. A. & Capone, J. P. A mitochondrial ketogenic enzyme regulates its gene expression by association with the nuclear hormone receptor PPARalpha. EMBO J. 17, 6972–6978 (1998).
Kostiuk, M. A., Keller, B. O. & Berthiaume, L. G. Palmitoylation of ketogenic enzyme HMGCS2 enhances its interaction with PPARalpha and transcription at the Hmgcs2 PPRE. FASEB J. 24, 1914–1924 (2010).
Juge-Aubry, C. E., Kuenzli, S., Sanchez, J. C., Hochstrasser, D. & Meier, C. A. Peroxisomal bifunctional enzyme binds and activates the activation function-1 region of the peroxisome proliferator-activated receptor alpha. Biochem. J. 353, 253–258 (2001).
Zhang, D. et al. Phosphoglycerate mutase 1 promotes cancer cell migration independent of its metabolic activity. Oncogene 36, 2900–2909 (2017).
Zhang, D. et al. Phosphoglycerate mutase 1 predicts the poor prognosis of oral squamous cell carcinoma and is associated with cell migration. J. Cancer 8, 1943–1951 (2017).
Xu, Z. et al. The diagnostic value and functional roles of phosphoglycerate mutase 1 in glioma. Oncol. Rep. 36, 2236–2244 (2016).
Lee, H. W. et al. Alpha-smooth muscle actin (ACTA2) is required for metastatic potential of human lung adenocarcinoma. Clin. Cancer Res 19, 5879–5889 (2013).
King, A., Selak, M. A. & Gottlieb, E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene 25, 4675–4682 (2006).
Chen, M. et al. Promoter hypermethylation mediated downregulation of FBP1 in human hepatocellular carcinoma and colon cancer. PLoS ONE 6, e25564 (2011).
Yang, J. et al. Inhibiting histone deacetylases suppresses glucose metabolism and hepatocellular carcinoma growth by restoring FBP1 expression. Sci. Rep. 7, 43864 (2017).
Dong, C. et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 23, 316–331 (2013).
Li, B. et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 513, 251–255 (2014).
Jin, X. et al. Fructose-1,6-bisphosphatase Inhibits ERK activation and bypasses gemcitabine resistance in pancreatic cancer by blocking IQGAP1-MAPK interaction. Cancer Res 77, 4328–4341 (2017).
Zhu, Y. et al. NPM1 activates metabolic changes by inhibiting FBP1 while promoting the tumorigenicity of pancreatic cancer cells. Oncotarget 6, 21443–21451 (2015).
Zhang, J. et al. Down-regulation of FBP1 by ZEB1-mediated repression confers to growth and invasion in lung cancer cells. Mol. Cell Biochem. 411, 331–340 (2016).
Jameson, K. L. et al. IQGAP1 scaffold-kinase interaction blockade selectively targets RAS-MAP kinase-driven tumors. Nat. Med. 19, 626–630 (2013).
van der Knaap, J. A. et al. GMP synthetase stimulates histone H2B deubiquitylation by the epigenetic silencer USP7. Mol. Cell 17, 695–707 (2005).
Sarkari, F. et al. EBNA1-mediated recruitment of a histone H2B deubiquitylating complex to the Epstein-Barr virus latent origin of DNA replication. PLoS Pathog. 5, e1000624 (2009).
Faesen, A. C. et al. Mechanism of USP7/HAUSP activation by its C-terminal ubiquitin-like domain and allosteric regulation by GMP-synthetase. Mol. Cell 44, 147–159 (2011).
van der Knaap, J. A., Kozhevnikova, E., Langenberg, K., Moshkin, Y. M. & Verrijzer, C. P. Biosynthetic enzyme GMP synthetase cooperates with ubiquitin-specific protease 7 in transcriptional regulation of ecdysteroid target genes. Mol. Cell Biol. 30, 736–744 (2010).
Frappier, L. & Verrijzer, C. P. Gene expression control by protein deubiquitinases. Curr. Opin. Genet Dev. 21, 207–213 (2011).
Shi, D. & Gu, W. Dual Roles of MDM2 in the regulation ofp53: ubiquitination dependent and ubiquitination independent mechanisms of MDM2 repression of p53 activity. Genes Cancer 3, 240–248 (2012).
Li, M., Brooks, C. L., Kon, N. & Gu, W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol. Cell 13, 879–886 (2004).
Reddy, B. A. et al. Nucleotide biosynthetic enzyme GMP synthase is a TRIM21-controlled relay of p53 stabilization. Mol. Cell 53, 458–470 (2014).
Kozhevnikova, E. N. et al. Metabolic enzyme IMPDH is also a transcription factor regulated by cellular state. Mol. Cell 47, 133–139 (2012).
Lee, S. M., Kim, J. H., Cho, E. J. & Youn, H. D. A nucleocytoplasmic malate dehydrogenase regulates p53 transcriptional activity in response to metabolic stress. Cell Death Differ. 16, 738–748 (2009).
Saxton, R. A. & Sabatini, D. M. mTOR Signaling in growth, metabolism, and disease. Cell 168, 960–976 (2017).
Hardie, D. G., Schaffer, B. E. & Brunet, A. AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 26, 190–201 (2016).
Jeon, S. M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med 48, e245 (2016).
Zhang, C. S. et al. Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 548, 112–116 (2017).
Zhang, Y. L. et al. AMP as a low-energy charge signal autonomously initiates assembly of AXIN-AMPK-LKB1 complex for AMPK activation. Cell Metab. 18, 546–555 (2013).
Zhang, C. S. et al. The lysosomal v-ATPase-Ragulator complex is a common activator for AMPK and mTORC1, acting as a switch between catabolism and anabolism. Cell Metab. 20, 526–540 (2014).
Lu, M., Holliday, L. S., Zhang, L., Dunn, W. A. Jr. & Gluck, S. L. Interaction between aldolase and vacuolar H+-ATPase: evidence for direct coupling of glycolysis to the ATP-hydrolyzing proton pump. J. Biol. Chem. 276, 30407–30413 (2001).
Lu, M., Sautin, Y. Y., Holliday, L. S. & Gluck, S. L. The glycolytic enzyme aldolase mediates assembly, expression, and activity of vacuolar H+-ATPase. J. Biol. Chem. 279, 8732–8739 (2004).
Kalender, A. et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 11, 390–401 (2010).
Efeyan, A. et al. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature 493, 679–683 (2013).
Lee, M. N. et al. Glycolytic flux signals to mTOR through glyceraldehyde-3-phosphate dehydrogenase-mediated regulation of Rheb. Mol. Cell Biol. 29, 3991–4001 (2009).
Buller, C. L., Heilig, C. W. & Brosius, F. C. 3rd GLUT1 enhances mTOR activity independently of TSC2 and AMPK. Am. J. Physiol. Ren. Physiol. 301, F588–F596 (2011).
Roberts, D. J., Tan-Sah, V. P., Ding, E. Y., Smith, J. M. & Miyamoto, S. Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Mol. Cell 53, 521–533 (2014).
Pastorino, J. G. & Hoek, J. B. Regulation of hexokinase binding to VDAC. J. Bioenerg. Biomembr. 40, 171–182 (2008).
Mihaylova, M. M. & Shaw, R. J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 13, 1016–1023 (2011).
Kusakabe, T., Motoki, K. & Hori, K. Mode of interactions of human aldolase isozymes with cytoskeletons. Arch. Biochem Biophys. 344, 184–193 (1997).
Wang, J., Morris, A. J., Tolan, D. R. & Pagliaro, L. The molecular nature of the F-actin binding activity of aldolase revealed with site-directed mutants. J. Biol. Chem. 271, 6861–6865 (1996).
Kao, A. W., Noda, Y., Johnson, J. H., Pessin, J. E. & Saltiel, A. R. Aldolase mediates the association of F-actin with the insulin-responsive glucose transporter GLUT4. J. Biol. Chem. 274, 17742–17747 (1999).
Taylor, K. A. & Taylor, D. W. Formation of two-dimensional complexes of F-actin and crosslinking proteins on lipid monolayers: demonstration of unipolar alpha-actinin-F-actin crosslinking. Biophys. J. 67, 1976–1983 (1994).
Pagliaro, L. & Taylor, D. L. Aldolase exists in both the fluid and solid phases of cytoplasm. J. Cell Biol. 107, 981–991 (1988).
Buscaglia, C. A., Penesetti, D., Tao, M. & Nussenzweig, V. Characterization of an aldolase-binding site in the Wiskott-Aldrich syndrome protein. J. Biol. Chem. 281, 1324–1331 (2006).
Ritterson Lew, C. & Tolan, D. R. Aldolase sequesters WASP and affects WASP/Arp2/3-stimulated actin dynamics. J. Cell Biochem. 114, 1928–1939 (2013).
Hu, H. et al. Phosphoinositide 3-Kinase regulates glycolysis through mobilization of aldolase from the actin cytoskeleton. Cell 164, 433–446 (2016).
Moroishi, T., Hansen, C. G. & Guan, K. L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 15, 73–79 (2015).
Zanconato, F., Cordenonsi, M. & Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 29, 783–803 (2016).
Mo, J. S. et al. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat. Cell Biol. 17, 500–510 (2015).
Wang, W. et al. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 17, 490–499 (2015).
DeRan, M. et al. Energy stress regulates hippo-YAP signaling involving AMPK-mediated regulation of angiomotin-like 1 protein. Cell Rep. 9, 495–503 (2014).
Mor, I., Cheung, E. C. & Vousden, K. H. Control of glycolysis through regulation of PFK1: old friends and recent additions. Cold Spring Harb. Symp. Quant. Biol. 76, 211–216 (2011).
Enzo, E. et al. Aerobic glycolysis tunes YAP/TAZ transcriptional activity. Embo J. 34, 1349–1370 (2015).
Cordenonsi, M. et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 147, 759–772 (2011).
Feo, S., Arcuri, D., Piddini, E., Passantino, R. & Giallongo, A. ENO1 gene product binds to the c-myc promoter and acts as a transcriptional repressor: relationship with Myc promoter-binding protein 1 (MBP-1). FEBS Lett. 473, 47–52 (2000).
Hsu, K. W. et al. The activated Notch1 receptor cooperates with alpha-enolase and MBP-1 in modulating c-myc activity. Mol. Cell Biol. 28, 4829–4842 (2008).
De Rosa, V. et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat. Immunol. 16, 1174–1184 (2015).
Yu, Z. et al. PKM2 Thr454 phosphorylation increases its nuclear translocation and promotes xenograft tumor growth in A549 human lung cancer cells. Biochem Biophys. Res Commun. 473, 953–958 (2016).
Castello, A., Hentze, M. W. & Preiss, T. Metabolic enzymes enjoying new partnerships as rna-binding proteins. Trends Endocrinol. Metab. 26, 746–757 (2015).
Ciesla, J. Metabolic enzymes that bind RNA: yet another level of cellular regulatory network? Acta Biochim Pol. 53, 11–32 (2006).
Hentze, M. W. & Preiss, T. The REM phase of gene regulation. Trends Biochem Sci. 35, 423–426 (2010).
White, M. R. & Garcin, E. D. The sweet side of RNA regulation: glyceraldehyde-3-phosphate dehydrogenase as a noncanonical RNA-binding protein. Wiley Interdiscip. Rev. RNA 7, 53–70 (2016).
Millet, P., Vachharajani, V., McPhail, L., Yoza, B. & McCall, C. E. GAPDH binding to TNF-alpha mRNA contributes to posttranscriptional repression in monocytes: a novel mechanism of communication between inflammation and metabolism. J. Immunol. 196, 2541–2551 (2016).
Chang, C. H. et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251 (2013).
Tai, N. et al. Translational autoregulation of thymidylate synthase and dihydrofolate reductase. Front Biosci. 9, 2521–2526 (2004).
Liu, J. et al. Thymidylate synthase as a translational regulator of cellular gene expression. Biochim. Biophys. Acta 1587, 174–182 (2002).
Nair, A. K. & Menon, K. M. Isolation and characterization of a novel trans-factor for luteinizing hormone receptor mRNA from ovary. J. Biol. Chem. 279, 14937–14944 (2004).
Ikeda, S. et al. Effect of estrogen on the expression of luteinizing hormone-human chorionic gonadotropin receptor messenger ribonucleic acid in cultured rat granulosa cells. Endocrinology 149, 1524–1533 (2008).
Menon, B., Franzo-Romain, M., Damanpour, S. & Menon, K. M. Luteinizing hormone receptor mRNA down-regulation is mediated through ERK-dependent induction of RNA binding protein. Mol. Endocrinol. 25, 282–290 (2011).
Nair, A. K., Young, M. A. & Menon, K. M. Regulation of luteinizing hormone receptor mRNA expression by mevalonate kinase--role of the catalytic center in mRNA recognition. FEBS J. 275, 3397–3407 (2008).
Bathaie, S. Z., Ashrafi, M., Azizian, M. & Tamanoi, F. Mevalonate Pathway and Human Cancers. Curr. Mol. Pharmacol. 10, 77–85 (2017).
Collins, I., Wang, H., Caldwell, J. J. & Chopra, R. Chemical approaches to targeted protein degradation through modulation of the ubiquitin-proteasome pathway. Biochem J. 474, 1127–1147 (2017).
Anastasiou, D. et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat. Chem. Biol. 8, 839–847 (2012).
Courteau, L. et al. Hexokinase 2 controls cellular stress response through localization of an RNA-binding protein. Cell Death Dis. 6, e1837 (2015).
Fairbank, M., St-Pierre, P. & Nabi, I. R. The complex biology of autocrine motility factor/phosphoglucose isomerase (AMF/PGI) and its receptor, the gp78/AMFR E3 ubiquitin ligase. Mol. Biosyst. 5, 793–801 (2009).
Rangarajan, E. S., Park, H., Fortin, E., Sygusch, J. & Izard, T. Mechanism of aldolase control of sorting nexin 9 function in endocytosis. J. Biol. Chem. 285, 11983–11990 (2010).
Caspi, M. et al. Aldolase positively regulates of the canonical Wnt signaling pathway. Mol. Cancer 13, 164 (2014).
Ma, D. et al. Upregulation of the ALDOA/DNA-PK/p53 pathway by dietary restriction suppresses tumor growth. Oncogene 37, 1041–1048 (2017).
Zheng, L., Roeder, R. G. & Luo, Y. S phase activation of the histone H2B promoter by OCA-S, a coactivator complex that contains GAPDH as a key component. Cell 114, 255–266 (2003).
Tristan, C., Shahani, N., Sedlak, T. W. & Sawa, A. The diverse functions of GAPDH: views from different subcellular compartments. Cell Signal 23, 317–323 (2011).
Lay, A. J. et al. Phosphoglycerate kinase acts in tumour angiogenesis as a disulphide reductase. Nature 408, 869–873 (2000).
Li, L., Zhang, Y., Qiao, J., Yang, J. J. & Liu, Z. R. Pyruvate kinase M2 in blood circulation facilitates tumor growth by promoting angiogenesis. J. Biol. Chem. 289, 25812–25821 (2014).
Castonguay, Z., Auger, C., Thomas, S. C., Chahma, M. & Appanna, V. D. Nuclear lactate dehydrogenase modulates histone modification in human hepatocytes. Biochem. Biophys. Res. Commun. 454, 172–177 (2014).
Chueh, F. Y. et al. Nuclear localization of pyruvate dehydrogenase complex-E2 (PDC-E2), a mitochondrial enzyme, and its role in signal transducer and activator of transcription 5 (STAT5)-dependent gene transcription. Cell. Signal 23, 1170–1178 (2011).
Hentze, M. W., Muckenthaler, M. U., Galy, B. & Camaschella, C. Two to tango: regulation of Mammalian iron metabolism. Cell 142, 24–38 (2010).
Li, X., Qian, X. & Lu, Z. Fructokinase A acts as a protein kinase to promote nucleotide synthesis. Cell Cycle 15, 2689–2690 (2016).
Gustafsson Sheppard, N. et al. The folate-coupled enzyme MTHFD2 is a nuclear protein and promotes cell proliferation. Sci. Rep. 5, 15029 (2015).
Acknowledgements
This study was funded by the German Research Foundation (FOR2314) and the German Cancer Aid (111917).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Snaebjornsson, M.T., Schulze, A. Non-canonical functions of enzymes facilitate cross-talk between cell metabolic and regulatory pathways. Exp Mol Med 50, 1–16 (2018). https://doi.org/10.1038/s12276-018-0065-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s12276-018-0065-6
This article is cited by
-
Glycolytic enzymes in non-glycolytic web: functional analysis of the key players
Cell Biochemistry and Biophysics (2024)
-
Lactate-dependent transcriptional regulation controls mammalian eye morphogenesis
Nature Communications (2023)
-
A functional loop between YTH domain family protein YTHDF3 mediated m6A modification and phosphofructokinase PFKL in glycolysis of hepatocellular carcinoma
Journal of Experimental & Clinical Cancer Research (2022)
-
Pyruvate kinase, a metabolic sensor powering glycolysis, drives the metabolic control of DNA replication
BMC Biology (2022)
-
A cross-species spatiotemporal proteomic analysis identifies UBE3A-dependent signaling pathways and targets
Molecular Psychiatry (2022)
