
Abstract
Hypertrophic cardiomyopathy is a heterogeneous disease caused by gene mutations. Most of the disease-causing mutations were found in the genes for sarcomeric proteins, but there are several cases carrying mutations in genes for extra-sarcomeric cytoskeletons. Desmin is a member of extra-sarcomeric cytoskeletons and plays an important role in muscle contraction. Mutations in the desmin gene cause various type of general myopathy and/or cardiomyopathy, known as desmin-related myopathies. We identified a novel desmin missense mutation, Thr219Pro, in the homozygous state in a patient, who first manifested with hypertrophic cardiomyopathy and later progressed to general myopathy. His parents were heterozygous for the mutation, but showed no clinical abnormality, suggesting the recessive inheritance of the mutation. We here report a severe phenotype of hypertrophic cardiomyopathy preceded the onset of general myopathy caused by a novel homozygous missense mutation in the 1B α-helix domain of desmin.
Similar content being viewed by others

Introduction
Hypertrophic cardiomyopathy (HCM) is a cardiac disease characterized by left ventricular thickening in the absence of pressure- or volume-overload such as systemic hypertension and valvular heart disease. More than half of patients had family history of the disease, consistent with the autosomal dominant inheritance trait, and genetic linkage studies as well as candidate gene analyses during the last three decades have revealed that mutations in several genes (disease-causing genes) cause HCM [1,2,3]. Most of the disease-causing genes encode sarcomere proteins such as β-cardiac myosin heavy chain (MYH7), cardiac myosin-binding protein C (MYBPC3) and cardiac troponin T (TNNT2), which consist of the contractile element sarcomere, while there are other disease-causing genes encoding extra-sarcomeric cytoskeletons, such as components of intercalated desk and desmin intermediate filaments [1, 3, 4].
Desmin is an essential component of extra-sarcomeric cytoskeleton. Human desmin gene (DES) locates on chromosome 2q35 and encodes a desmin monomer of 470 amino acids, which is composed of several domains including a head, four α-helical segments (coil 1A, 1B, 2A, and 2B), three short non-helical rod segments (L1, L12, and L2) and a tail domain [4, 5]. Two desmin monomers form a dimer associated with the same direction and two dimer forms a tetramer in anti-parallel and half-staggered fashion with overlapping of two coiled coils via coil domains 1A and 1B. A unit length filament composed of eight tetramers polymerizes longitudinally and assembles into an extended desmin intermediate filament. The desmin intermediate filaments connect the contractile apparatus through Z-discs, nuclei, intercalated discs, costameres of the plasma membrane, mitochondria, and other membranous organelles to form a network, that exerts a mechano-chemical signaling, in which proper propagation and sensing of mechanical forces require a coordination of multiple cytoplasmic and nuclear components in myocytes [4]. DES mutations result in systemic myopathy or cardiomyopathy called as desminopathy, in which mutant desmin proteins aggregate in cytoplasm. Many of the disease-causing DES mutations were found in the α-helical domains, especially in 2B α-helical domain. It was reported that cardiac manifestation was associated with more than 70% of the DES mutations [5], in which various cardiac phenotypes, including HCM, dilated cardiomyopathy, restrictive cardiomyopathy, and arrhythmogenic right ventricular (RV) cardiomyopathy, were inherited as autosomal dominant or recessive traits [4, 5].
In this study, we found a novel homozygous missense mutation in DES, Thr219Pro, in a patient among 46 juvenile cases of HCM, who were diagnosed before 16-years-old. The patients carrying the DES mutation manifested with HCM phenotype and later progressed to general myopathy, of which inheritance was consistent with the autosomal recessive trait.
Materials and methods
Clinical examinations
Chest radiography, electrocardiogram (ECG) and echocardiogram were performed to evaluate cardiac function. The patient was followed cardiac catheter examination at his ages of 13 and 19. RV biopsy was performed at the cardiac catheter examinations. Although the second examination of RV biopsy at 19-years-old was failed, a skeletal muscle biopsy sample was obtained from biceps brachii muscle.
Analysis of skeletal muscle specimen
In light microscopic immunochemical analysis of skeletal muscle specimen, desmin was stained by a standard ABC method [6] using anti-human desmin antibody purchased from Dako, Denmark as a primary antibody. Electron microscopic data was obtained as described previously [7].
Analysis of mutation in candidate genes
We analyzed a total of 46 juvenile HCM patients, who were diagnosed before 16-years-old, for mutations in cardiomyopathy-associated genes including genes for components of sarcomere, Z-disc and intercalated disc [3], which were screened for mutations in exons and exon–intron boundaries by using next-generation sequencing. In brief, 67 genes were examined (Supplementary Table S1). Detected variations were filtered out that the minor allele frequency was less than 0.002 in public nucleotide databases including dbSNP common [8], 1000 genomes project [9], Exome aggregation consortium [10] and Human Genetic Variation Database [11]. Identified mutations were confirmed by Sanger sequencing with di-deoxy chain termination method and automated nucleotide sequencer. Primer pairs used in the confirmation of DES and obscurin gene (OBSCN) mutations were DES-219-F, AGCGGTGAGTGCCCTTCTTT with DES-219-R, GCACTTTCTTAAGGAACGCG and OBSCN-512-F, AAGAGAGCTGGGGATCCTGT with OBSCN-512-R, TGGGGACCAGCTGAGAATCA, respectively. Functional alteration caused by gene mutation was assessed by in silico prediction programs [12,13,14,15]: Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/), Mutation Taster (http://www.mutationtaster.org/), PROVEAN (http://provean.jcvi.org/index.php), and fathmm (http://fathmm.biocompute.org.uk). Informed consent for genetic testing was given from each individual including the proband patient and his parents. The study protocol was approved by the Ethics Committee of Medical Research Institute, Tokyo Medical and Dental University.
Statistical analysis
Data for chest X-ray and echocardiography from the patient were obtained once or several times per year during the follow-up period. Changes over the years of cardio-thoracic ratio, left ventricular ejection fraction (LVEF), and interventricular septal thickness were statistically evaluated with Kruskal–Wallis analysis of non-parametrical method.
Results
Clinical course of a juvenile HCM case with the DES mutation
The case was a male patient of 20-years-old at the genetic examination. His parents were clinically normal and were consanguineous marriage of cousins, and no obvious family history of cardiomyopathy, sudden cardiac death or skeletal myopathy was found in the family relatives of both parents. The first ECG abnormality in the case had been pointed out at his age of 6 in check-up at school, although no detailed information on ECG was available because clinical records had been scrapped. However, it was inquired from his parents that no obvious structural abnormality was detected by echocardiogram at that time. He actively exercised, such as playing baseball, in his school life. At his age of 13, the 12-lead ECG showed sinus rhythm, normal axial deviation, and narrow QRS complex (0.08 s), whereas increased voltages and ST depression were found in I, II, III, aVF, and V4 to V6 leads and T-wave inversion were noted in V1 toV3 leads. Severe dual atrial overload was also detected in ECG (Supplementary Fig. S1). Left ventricular wall thickness was 12~13 mm, and bi-ventricular hypertrophy, right atrial hypertrophy and tricuspid valve regurgitation were found in the echocardiogram. Cardio-thoracic ratio was slightly increased to 55%. However, no histopathological change was found in RV biopsy. Thus, he was diagnosed as an atypical HCM with hypertrophy of both bi-ventricle and right atrium.
He then had repeatedly hospitalized due to congestive heart failure with paroxysmal atrial fibrillation (Af) since age of 16. His ECG showed Af with wide QRS complex (120 ms) and RV bundle branch block along with ST depression at I, II, III, aVF, V5, and V6 accompanied by ST elevation in aVR and negative T in V5 and V6 (Fig. 1). LVEF retained at the range of normal contraction in his clinical progress, however, LVEF decreased rapidly from 82% to less than 63% for a year after emergence of paroxysmal atrial fibrillation at his age of 17 (p = 0.0558, Fig. 2a). Obvious hypertrophy of interventricular septal thickness, as same as posterior wall thickness, had gradually become thin reminiscent of dilated-phase of HCM since past his age of 17 (p = 0.0009, Fig. 2a).

Electrocardiogram (ECG) of HCM patient CM588. a ECG at age of 16. b ECG at age of 20 (Color figure online)

Clinical examinations of chest X-ray and echocardiogram of HCM patient CM588. a Changes of cardio-thoracic ratio (CTR), left ventricular ejection fraction (LVEF), and interventricular septal thickness (IVST) in the clinical course. b A four chamber view of echocardiogram at his age of 20. AF atrial fibrillation, NSVT non-sustained ventricular tachycardia, PAF paroxysmal atrial fibrillation. The values are median and 1.5 interquartile range (IQR) of the obtained data for each year. Changes over the years were statistically evaluated by Kruskal–Wallis one-way analysis of variance (Color figure online)
At age of 18, he experienced syncope probably due to paroxysmal rapid Af or non-sustained ventricular tachycardia. Thereafter, Af was fixed and QRS complex became wide (up to 160 ms). From that time, atrophy of appendicular skeletal muscle was emerged and gradually progressed. Skeletal muscle biopsy from biceps at age 19 showed reticular pattern of loss of activity of succinate dehydrogenase and cytochrome c oxidase. In desmin staining, aggregated desmin was clearly stained in the skeletal muscle cell (Fig. 3). The structure accumulation of which showed the same intensity as Z-band was found coincidence with desmin staining portion. In the electron microscopic (EM) analysis, it was demonstrated that there were findings of sarcomere degeneration and granuro-filamentous materials in subsarcolemma, where a lot of electron dense granular deposits and a few mitochondria were observed (Fig. 4). These are typical EM findings of skeletal muscle associated with desminopathy.

Desmin staining of skeletal muscle specimen of HCM patient CM588. a Sample from CM588 showed desmin aggregations indicated by arrowheads. b Control sample (Color figure online)

Electron microscopic findings of skeletal muscle specimen of HCM patient CM588. Typical deposition of desmin is shown in a. b Is a high-magnitude view. a The granuro-filamentous material in subsarcolemma (center) and sarcomere degeneration (right). b Inside of the granuro-filamentous material, there are a lot of electron dense granular deposits and a few mitochondria
In the disease progression process, LVEF retained in normal range, while the cardio-thoracic ratio gradually increased from 55.6% at age 13 to 68.3% at age 20, and the left ventricular wall thickness became thin. Since his age of 19, congestive heart failure had deteriorated rapidly (Fig. 1b) and he died suddenly after a restroom at home at his age of 20.
Targeted gene analysis for cardiomyopathy-associated genes
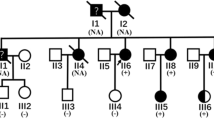
To explore a causative mutation, 67 cardiomyopathy-associated genes were searched for mutations in genomic DNAs of peripheral blood obtained from 46 juvenile HCM patients (details will be reported elsewhere). A novel missense mutation of DES, Thr219Pro (ACT to CCT at codon 219), was found in a case (proband CM588) (Fig. 5). Because he was homozygous for the mutation, we also analyzed his healthy parents (CM367 and CM368) and found that both were heterozygous for the mutation. In order to investigate whether the mutation would cause functional alteration, we performed in silico prediction analyses by using five different programs, Polyphen-2, SIFT, Mutation Taster, PROVEAN, and fathmm. We obtained predictions of “probably damaging” (score 1.00), “tolerated” (score 0.12), “disease causing”, “deleterious” (score −3.96), and “damaging” (score −1.75), respectively. Because cardiomyopathy and general myopathy were observed only in CM588 carrying the homozygous mutation, but not in other members including patients and grand-parents, who were obligatory carriers of the mutation, it was speculated that the mutation would not manifest with any clinical phenotypes under the heterozygous state. Although we did not perform genetic testing, when we had interviewed his family relatives, his two elder sisters and eight cousins had been healthy without any clinical signs of cardiomyopathy or skeletal myopathy (Fig. 5a).

Family pedigree and nucleotide sequence of DES. a Pedigree of CM588 family. Open squares and open circles indicate subjects without any signs of cardiomyopathy or myopathy. Closed square with slash indicates the proband patient CM588 homozygous for the desmin mutation. Parents of CM588 are first cousins and carriers of the mutation. b Sequencing analysis demonstrated the DES mutation. (a) Control subject showing ACT at codon 219, (b) CM588 showing CCT at codon 219, and (c) father of CM588 showing superimpose of CCT and ACT. Mother of CM588 showed the same heterozygous pattern as (c) (Color figure online)
In addition to the DES mutation, a missense mutation of OBSCN, Pro512Leu (CCC to CTC at codon 512), was found in the proband (CM588) and his unaffected father (CM367), but not in his mother (CM368). In silico analysis by Mutation Taster and PROVEAN predicted “disease causing” and “deleterious” (score −4.69), however, three other programs, Polyphen-2, SIFT, and fathmm, showed “benign” (score 0.142), “tolerated” (score 0.63) and “tolerated” (score −1.29), respectively. These data suggested that the OBSCN mutation may not cause large functional changes leading to clinical phenotype, although we cannot exclude a possibility that the OBSCN mutation might modify clinical phenotypes in the proband patient. There was no additional rare variant found in the patient.
Discussion
In this study, we identified a novel homozygous DES mutation, Thr219Pro, in a patient with HCM. The mutation was considered to be causative for earlier-onset cardiomyopathy and later-onset myopathy, because (i) this mutation was not found in the general population, (ii) his unaffected parents were both heterozygous for the mutation, while the proband patient was homozygous for the mutation, (iii) no other mutations co-segregated with HCM and myopathy were found in the tested 67 candidate genes, and (iv) in silico bioinformatics program predicted a functional alteration by the mutation.
The patient showed ventricular thickness of 12–13 mm at age 13. In adult HCM, especially for HCM subtypes, Maron type 1 and type II with asymmetric hypertrophy, criteria for ventricular thickness was usually over 15 mm and ratio of interventricular septal thickness (IVST) to ventricular posterior wall thickness (VPWT) over 1.5 are used [2]. However, such criteria are not defined for Maron type III and type IV and apical hypertrophy. On the other hand, both IVST and VPWT for subjects of twenties in Japanese general population were reported to be 8 ± 1 mm [16]. In addition, IVST and VPWT adjusted by body surface area for children were reported [17]. The body surface area of the patient at age 13 was about 1.25 m2 for which adjusted IVST and VPWT were calculated to be 6.6 ± 1.8 and 6.9 ± 2.6 mm. From these observations, we considered ventricular thickness of 12–13 mm at age 13 was consistent with the diagnosis of HCM.
Previous reports indicated that DES mutations showed various inheritance patterns and cardiac phenotypes [4, 5]. The Thr219Pro mutation in our case was found in the 1B helix domain1B helix domain, which was suggested to be important for nebulin Z-disc assembly and stability of actin thin filaments [5], and its inheritance was considered as the autosomal recessive trait, albeit that we could not formally exclude a possibility of autosomal dominant inheritance with low penetrance, because the majority of inheritance pattern of desmin-related myopathy is autosomal dominant and mutation of autosomal recessive trait was quite rare for the mutations in the 1B helix domain of desmin [4, 5]. Although the reported number of mutation in the 1B helix domain was less than that in the 2B helix domain, a few mutations manifesting with cardiac hypertrophy were found in the 1B helix domain [4, 5].
The case with a novel homozygous DES mutation manifested with unique clinical phenotypes and severe disease course, in which first manifestation was cardiac hypertrophy diagnosed as atypical HCM at age of 13. Subsequently, the cardiac hypertrophy did not progress as found for the ordinary HCM cases, and the left ventricular wall thickness became gradually thin with transition to dilated-phase of HCM. On the other hand, atrophy of appendicular skeletal muscle emerged later than the cardiac hypertrophy at age of 18 and general myopathy developed gradually. Although there are a few DES mutations manifesting with cardiomyopathy alone [4], little was reported for the natural history or development of desmin-related cardiomyopathy.
Does the autosomal recessive trait of DES mutation would develop malignant phenotype? Although there was an attenuated phenotype of autosomal recessive trait [18], there were reports of a homozygous mutation Arg16Cys located in the head domain [19] and a compound heterozygous mutation of Ala360Pro and Asn393Ile located in the 2B helix domain, which showed severe clinical phenotypes including conduction defects with pacemaker implantation, cardiac transplantation due to severe heart failure, syncopal episode and sudden death [20,21,22]. The patient with the homozygous Arg16Cys mutation was first diagnosed as restrictive cardiomyopathy with complete atrioventricular block (cAVB) at age 30, but not suffered from obvious general myopathy. The case with the compound heterozygous mutation of Ala360Pro and Asn393Ile was first diagnosed as cAVB at her age 2 and general myopathy emerged later at age of 22. Her two brothers also manifested with cAVB at ages of 9 and 10, and later onset of general myopathy at ages of 20 and 24, respectively. Novel points of Thr219Pro mutation in our case were the manifestation of HCM prior to general myopathy at age 13, which progressed to dilated-phase.
In conclusion, to the best of our knowledge, this is the first report of HCM patient carrying the homozygous missense mutation in DES, who showed severe prognosis, such as early onset of HCM, severe repetitive heart failure with LV wall thinning and conduction disturbance, arrhythmias with syncope, later onset of general myopathy and sudden death. Because his parents did not manifest with any clinical symptoms even though they carried the DES mutation in heterozygous state, it was suggested that the DES mutation was inherited as an autosomal recessive trait.
References
Kimura A. Molecular basis of hereditary cardiomyopathy: abnormalities in calcium sensitivity, stretch response, stress response and beyond. J Hum Genet. 2010;55:81–90.
Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years. J Am Coll Cardiol. 2012;60:705–15.
Kimura A. Molecular genetics and pathogenesis of cardiomyopathy. J Hum Genet. 2016;61:40–51.
Clemen CS, Herrmann H, Strelkov SV, Schroder R. Desminopathies: pathology and mechanisms. Acta Neuropathol. 2013;125:47–75.
Capetanaki Y, Papathanasiou S, Diokmetzidou A, Vatsellas G, Tsikitis M. Desmin related disease: a matter of cell survival failure. Curr Opin Cell Biol. 2015;32:113–20.
Hsu SM, Raine L, Fanger H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J Histochem Cytochem. 1981;29:577–80.
Ikezoe K, Nakagawa M, Osoegawa M, Kira J, Nonaka I. Ultrastructural detection of DNA fragmentation in myonuclei of fatal reducing body myopathy. Acta Neuropathol. 2004;107:439–42.
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucl Acids Res. 2001;29:308–11.
Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, et al. A global reference for human genetic variation. Nature. 2015;526:68–74.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Higasa K, Miyake N, Yoshimura J, Okamura K, Niihori T, Saitsu H, et al. Human genetic variation database, a reference database of genetic variations in the Japanese population. J Hum Genet. 2016;61:547–53.
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012;7:e46688.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2.
Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GL, Edwards KJ, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34:57–65.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–49.
Daimon M, Watanabe H, Abe Y, Hirata K, Hozumi T, Ishii K, et al. Normal values of echocardiographic parameters in relation to age in a healthy Japanese population: the JAMP study. Circ J. 2008;72:1859–66.
Kampmann C, Wiethoff CM, Wenzel A, Stolz G, Betancor M, Wippermann CF, et al. Normal values of M mode echocardiographic measurements of more than 2000 healthy infants and children in central Europe. Heart. 2000;83:667–72.
Olivé M, Goldfarb L, Moreno D, Laforet E, Dagvadorj A, Sambuughin N, et al. Desmin-related myopathy: clinical, electrophysiological, radiological, neuropathological and genetic studies. J Neurol Sci. 2004;219:125–37.
Arbustini E, Pasotti M, Pilotto A, Pellegrini C, Grasso M, Previtali S, et al. Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Eur J Heart Fail. 2006;8:477–83.
Goldfarb LG, Park K-Y, Cervenáková L, Gorokhova S, Lee H-S, Vasconcelos O, et al. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet. 1998;19:402–3.
Dalakas MC, Park K-Y, Semino-Mora C, Lee HS, Sivakumar K, Goldfarb LG. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med. 2000;342:770–80.
Goudeau B, Rodrigues-Lima F, Fischer D, Casteras-Simon M, Sambuughin N, de Visser M, et al. Variable pathogenic potentials of mutations located in the desmin alpha-helical domain. Hum. Mut. 2006;27:906–13.
Acknowledgements
This work was supported in part by Grants-in-Aid for Scientific Research (KAKENHI, 26460407, 15K15095 and 16H05296) from Japan Society for the Promotion of Science (JSPS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
Haruhito Harada and Takeharu Hayashi contributed equally to this work
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Harada, H., Hayashi, T., Nishi, H. et al. Phenotypic expression of a novel desmin gene mutation: hypertrophic cardiomyopathy followed by systemic myopathy. J Hum Genet 63, 249–254 (2018). https://doi.org/10.1038/s10038-017-0383-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-017-0383-x
This article is cited by
-
Recessive DES cardio/myopathy without myofibrillar aggregates: intronic splice variant silences one allele leaving only missense L190P-desmin
European Journal of Human Genetics (2019)
-
Genetic background of Japanese patients with pediatric hypertrophic and restrictive cardiomyopathy
Journal of Human Genetics (2018)
-
Molecular insights into cardiomyopathies associated with desmin (DES) mutations
Biophysical Reviews (2018)
