
Abstract
Only two partial deletions longer than 655 nucleotides had been reported for the RHD gene, constrained within the gene and causing DEL phenotypes. Using a combination of quantitative PCR and long-range PCR, we examined three distinct deletions affecting parts of the RHD gene in three blood donors. Their RHD nucleotide sequences and exact boundaries of the breakpoint regions were determined. DEL phenotypes were caused by a novel 18.4 kb deletion and a previously published 5.4 kb deletion of the RHD gene; a D-negative phenotype was caused by a novel 7.6 kb deletion. Examination of the deletion-flanking regions suggested microhomology-mediated end-joining, replication slippage, and non-homologous end-joining, respectively, as the most likely mechanisms for the three distinct deletions. We described two new deletions affecting parts of the RHD gene, much longer than any previously reported partial deletion: one was the first deletion observed at the 5′ end of the RHD gene extending into the intergenic region, and the other the second deletion observed at its 3′ end. Large deletions present at either end are a mechanism for a much reduced RhD protein expression or its complete loss. Exact molecular characterization of such deletions is instrumental for accurate RHD genotyping.
Similar content being viewed by others


Introduction
Blood groups are antigenic molecules (proteins, carbohydrates, glycoproteins, or glycolipids) present on the surface of red blood cells (RBCs); which may also be present on platelets, lymphocytes, and body tissues. Many of these antigens are clinically significant and can lead to adverse reactions in transfused patients, hemolytic disease of the fetus and newborn in pregnant women, and graft loss in organ transplant recipients [1].
Rh is the fourth blood group system (ISBT 004) and consists mainly of the D, C, E, c, and e antigens carried on two non-glycosylated hydrophobic transmembrane proteins (RhD and RhCE) [2]. It is the most complex blood group system due to the highly homologous and polymorphic genes, RHD (MIM # 111680) and RHCE (MIM # 111700) [3]. The RHD gene is located on the short arm of chromosome 1 at position 1p36.11, ~62.3 kb long and flanked by two highly homologous, 9 kb long DNA segments, the upstream and downstream Rhesus boxes [4].
Large deletions have previously been reported for blood group systems other than Rh, such as the H-deficient Bombay phenotype [5], 4-α-galactosyltransferase-deficient p phenotype [6], glucosaminyl (N-acetyl) transferase 2-deficient i phenotype [7], and XK protein-deficient Kx null phenotype [8]. A total of 43 deletions in the RHD gene [9] and 12 deletions in the RHCE gene [10] have been identified, most of them shorter than 655 nucleotides [11, 12]. Previously reported deletions of 1013 bp encompassing exon 9 of the RHD gene [13, 14] and 2.5 kb encompassing exons 4, 5, and 6 are technical errors and obsolete [15,16,17].
For the RHCE gene, two large partial gene deletions have been reported. One included exon 2 and extended beyond exon 10 [18], and another large internal deletion encompassed exons 2 through 8 [19]. A large chromosomal deletion involving both the RHD and RHCE genes, and the nearby D1S80 variable number of tandem repeats locus has been reported [20]. Besides the common whole RHD gene deletion [4], two large partial RHD gene deletions are known: the RHD allele c.1074–649_1153+266del995 represents a 995 nucleotide deletion encompassing 649 nucleotides of intron 7, all 80 nucleotides of exon 8 and 266 nucleotides of intron 8 [21], five transcripts with missing exon 8 were reported including the longest with 170 nucleotides of intron 7 attached to normal exon 9 and 10 with an open reading frame; and the RHD allele c.1228–4061_1254+1317del5405 represents a 5405 bp deletion encompassing 4061 nucleotides of intron 9, all 27 nucleotides of the exon 10 and 1317 nucleotides of 3′ UTR [22], a single transcript was reported involving RHD exons 1 to 9 followed by a sequence tract of RHD intron 9, termed pseudo-exon 10. These two partial RHD gene deletions still express some D protein representing a DEL phenotype. The RHD exon 10 deletion, common in France [22] and Northern Germany, was subsequently found in a transfused red cell product that caused a secondary anti-D immunization [23].
In the present study, we describe three large RHD genomic deletions, two of which were novel, affecting stretches of 18.4, 5.4, and 7.6 kb in length and causing DEL or D-negative phenotypes. On the basis of analysis of the breakpoints, we discuss distinct mechanisms for the three deletions.
Materials and Methods
Study subjects
Two samples were collected as part of our screening at NIH of D-negative blood donors for the presence of RHD gene [3], another sample came from a blood donor in Springe, Germany. As described previously [24], RHD screens are a standard operating procedure in a growing number of blood centers, where this red cell-genotyping application for D-negative blood donations is covered by the donor consent form. EDTA–anticoagulated whole-blood samples were used for serology and DNA studies, and RNA tubes (PAXgene; PreAnalytiX, Hombrechtikon, Switzerland) were maintained at room temperature for up to 2 h, frozen and stored at −20 °C before RNA isolation. The analysis of the three donors was performed as case studies. We are planning to eventually report the cumulative data on RHD-positive samples found among our D-negative donors at NIH since 2009 including the current two donors.
Immunohematology
Hemagglutination tests were performed by standard tube and anti-IgG gel matrix testing with licensed reagents (Ortho, Raritan, NJ). An adsorption/elution method with human polyclonal anti-D was applied to test for the presence of a DEL phenotype [24]. Several RhD typing kits with 16 monoclonal anti-Ds were used to establish the epitope patterns as described previously [25]. Antibody screening and direct antiglobulin tests were negative for all three donors.
DNA and mRNA isolation
Genomic DNA was isolated from the buffy coat (Qiagen EZ1 DNA blood kit on the BioRobot EZ1; Qiagen, Valencia, CA) and mRNA from RNA tubes (Dynabeads mRNA DIRECT kit; Invitrogen, Carlsbad, CA). Primers were designed using software online (Primer3 [26], Supplementary Table S1 [4, 22, 27,28,29]).
RACE and cDNA sequencing
The 5′- and 3′-rapid amplification of cDNA ends (RACE) method was applied to the isolated mRNA (GeneRacer kit; Invitrogen) and reverse transcribed to cDNA using the Oligo(dT)-adapter primer (GeneRacer kit and SuperScript III First-Strand Synthesis SuperMix; Invitrogen). The resultant cDNA was then used as a template to obtain the 5′ and 3′ cDNA ends for nested polymerase chain reaction (PCR) amplification (GeneRacer primers included in the GeneRacer kit) with RHD or TMEM50A cDNA primers (Supplementary Table S1). No cDNA analysis was done for sample 2, because the cDNA sequence for this RHD deletion had been published before [22].
The PCR amplicons were purified and sequenced (BigDye Terminator v3.1; Applied Biosystems, Carlsbad, CA) as described previously [30]. Nucleotide sequences were aligned (CodonCode Aligner; CodonCode, Dedham, MA) to NCBI RefSeq NM_016124.4 and nucleotide positions defined using the first nucleotide of the coding sequence of RefSeq NM_016124.4 (RHD isoform 1).
Detection of deletion breakpoint
For the novel exon 1 deletion, copy-number analysis was done using a real-time PCR-walking approach on genomic DNA from the 5′-end of upstream Rhesus box to 3′-end of RHD intron 1 to identify the breakpoint region (sample 1). Amplification to detect the breakpoints was performed using a long-range PCR (LongAmp Taq DNA polymerase; New England Biolabs, Ipswich, MA) and the nucleotides were sequenced (Supplementary Table S1). PCR reactions, described by Fichou et al. [22], confirmed the known 5405 nucleotide deletion encompassing exon 10 of the RHD gene (sample 2). For the novel exon 10 deletion, a single long-range PCR reaction was developed to amplify the breakpoint and nucleotide sequencing allowed the precise mapping of the breakpoint (sample 3). The PCR products were separated on 1% agarose gel and sequenced [30] using primers encompassing each deletion junction (Supplementary Table S1).
RHD sequencing
The RHD gene was sequenced as previously described [27, 31]. The nucleotide sequences of all 10 exons as well as the adjacent intronic regions including the 5′ and 3′ untranslated regions (UTR) were determined. Zygosity testing for the RHD gene was done by restriction fragment length polymorphism (RFLP) [4] and quantitative fluorescence polymerase chain reaction [32].
RHD sequence analysis
Nucleotide sequences were aligned and compared with the NCBI RefSeq NG_007494.1. All variations are described according to current mutation nomenclature guidelines [33], ascribing the A of the first ATG translational initiation codon as nucleotide + 1 in the mRNA coding region of RHD (RefSeq NM_016124.4).
Bioinformatics analysis
To examine the potential mechanism for the deletion, 300 bps upstream and downstream from the deletion breakpoint were analyzed for distinct repetitive elements using the RepeatMasker track in the UCSC Genome Browser [34]. In cases where repetitive elements flanked both the deletion breakpoints, MUSCLE was used to determine the percentage of sequence identity between the elements [35].
Results
We analyzed two novel and one previously known partial deletion of the RHD gene in three blood donors (Table 1). Samples 1 and 2 had a DEL phenotype, whereas sample 3 was D-negative, as reproducibly confirmed by adsorption/elution testing [24]. All three samples were negative in serologic testing with 16 different monoclonal anti-Ds (Supplementary Table S2).
RHD genetic variations
We were able to amplify all RHD exons, except exon 1 (sample 1) and exon 10 (samples 2 and 3), indicating alterations at either the 5′- or 3′-ends of the RHD gene. Amplification and sequencing of regions surrounding exon 1 and 10 using long-range PCR revealed large deletions in the RHD gene. The sequencing for amplified exons and adjacent intronic regions was identical with the RHD reference sequence (NG_007494.1).
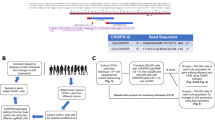
The exact breakpoints could be determined within 5 and 3 nucleotides in overlapping sequences (GAATG and AGG [22]) at the breakpoint region for the RHDex1del type 1 (sample 1, Fig. 1a) and RHDex10del type 1 alleles (sample 2, Fig. 1b). The exact nucleotides involved at the breakpoint were identified for the RHDex10del type 2 allele (sample 3, Fig. 1c).

Molecular structures of large deletions in the RHD gene observed in this study. The genomic structures of three partial gene deletions are shown (a–c). The RHD gene comprises 10 exons (black bars) flanked by the upstream (blue) and downstream Rhesus boxes (red). The nucleotide sequences of the breakpoint regions harbors microhomologies or non-templated nucleotides (bold letters, box) and are shown along with their electropherograms. The chromosomal positions flanking the three deletions are indicated for genomic DNA (NC_000001.11)
Sample 1 had an 18.4 kb deletion encompassing the upstream Rhesus box, 5′ UTR, exon 1, and part of intron 1 of the RHD gene [NC_000001.11(NG_007494.1):c.(1–15149_1–15153)_(148+3154_148+3158)del]. Sample 2 harbored the known 5.4 kb deletion encompassing intron 9, exon 10, and part of 3′ UTR of the RHD gene [NC_000001.11(NG_007494.1):c.(1227+2872_1227+2874)_(1254+1315_1254+1317)del]. Sample 3 had a 7.6 kb deletion encompassing intron 9, exon 10, and part of 3’ UTR of the RHD gene [NC_000001.11(NG_007494.1):c.1227+2108_1254+2785del].
Repetitive elements analysis
In sample 1, alignment of the 5′- and 3′-end regions bordering the deletion breakpoints showed 5-bp microhomology between the two regions (Fig. 1a). In sample 2, only one repetitive element, AluSx1, was detected upstream of the 5′ end. Alignment of the 5′- and 3′-end regions bordering the deletion breakpoints showed 3-bp microhomology (Fig. 1b). In sample 3, a FRAM element located at the 5′ breakpoint and AluYh3 element located around the 3′ breakpoint was identified. Alignment of the 5′- and 3′-end regions bordering the deletion breakpoints showed an insertion of 2 non-templated nucleotides (TT) at the junction (Fig. 1c). Sample 1 also harbored an AluSg element at the 5′ end and AluSx element at the 3′ end of the breakpoint (Fig. 2).

Proposed mechanism for the generation of the RHDex1del type 1 deletion. Schematic representation of the secondary structure formed by the two inverted Alu repeats (a). The stem-loop formation may have facilitated replication slippage, which resulted in the deletion of the upstream Rhesus box and RHD exon 1. Sequence comparison between AluSg in the intergenic region and AluSx in intron 1 (b). AluSx is shown in the reverse orientation to mimic its orientation in the putative secondary structure. Complementary nucleotides (underlined) represent ~80% of all positions
Effect on cDNA and protein structure
We determined the cDNAs of the two novel deletions (Fig. 3). For the RHDex1del type 1 allele, we detected an mRNA transcript of 478 nucleotides encompassing the 3′ end of exon 2 to the 5′ end of exon 5 (KX584098) using 5′ RACE analysis. Bioinformatic analysis indicated a potential translation start site in RHD exon 2 (Fig. 3a).

Schematic representation of transcripts observed in three partial RHD gene deletions. The RHD genes (upper model in a–d) depicted by exons (rectangles) and introns (lines). The resulting mRNA transcripts (lower model, a–d) are symbolized as a concatenation of exons. The start (blue) and stop codons (red) are underlined. RHDex1del type 1 mRNA starts at c.186 in exon 2 and extends into 3′ UTR a. RHDex10del type 2 short mRNA includes RHD exons 1 to 7, part of intron 7, exon 8, and part of intron 8 with a stop codon in exon 8 (b). RHDex10del type 2 long mRNA includes exons 1 to 9 of RHD —with a stop codon immediately after exon 9—the 5′ UTR, all 6 exons and 3′ UTR of TMEM50A. The two inserted nucleotides (UU) are highlighted at the junction (pink box) (c). RHDex10del type 1 mRNA includes exon 1 to 9 followed by a sequence tract of RHD intron 9 (pseudo-exon 10) due to the activation of a cryptic splice acceptor site (d). As RHDex1del type 1 involves the 5′ end, splicing is not affected. The splice variants are shown for the other two partial RHD gene deletions
For the RHDex10del type 2 allele, we detected two different mRNA transcripts using 3′ RACE analysis: (1) a 421 nucleotides short mRNA including at least exon 7, 170 nucleotides of intron 7, exon 8 and 107 nucleotides of intron 8 with a stop codon in exon 8 (KX619611) (Fig. 3b), and (2) a 1963 nucleotides hybrid RHD-TMEM50A long mRNA encompassing exons 1 to 9 of RHD gene with a stop codon just after RHD exon 9 and the 5′ UTR, coding sequence and 3′ UTR of the TMEM50A gene (KX584096) (Fig. 3c). Neither cDNA was expected to express any RhD protein in the membrane.
For comparison, the previously reported transcript [22] for the RHDex10del type 1 allele caused the replacement of the last eight amino acids of the wild-type RhD protein by four different amino acids (Fig. 3d). We modeled the two proteins based on the observed cDNAs of the RHDex1del type 1 and RHDex10del type 1 alleles and the previously predicted RhD protein model [36] (Supplementaruy Fig. S1). Sample 1 with the RHDex1del type 1 allele tested negative for the G-antigen by adsorption/elution technique [24].
Discussion
We reported two new deletions and confirmed a previously known deletion [22] affecting 5.4–18.4 kb of the RHD gene (Table 1). These were the first observation of a deletion at the 5′ end of the RHD gene and the second observation of a deletion at its 3′ end (Fig. 1). The two novel partial deletions were the largest known for the RHD gene and doubled the number of observed deletions longer than 655 nucleotides (Fig. 4).

Molecular structures of large deletions in the RHD gene. The genomic structures of all 4 partial gene deletions are shown. The RHD gene comprises 10 exons (black bars) flanked by the upstream (blue) and downstream Rhesus boxes (red). The positions and sizes of four deletions are indicated (green arrow)
Within a gene sequence, a high proportion of Alu repeats may promote gross gene rearrangements including partial gene deletions [37]. The core 26-bp sequence within the Alu repeat has been reported to be particularly recombinogenic [38]. In the present study, at least one breakpoint of each deletion mapped within or very close to Alu repeats, which straddled both breakpoints in the deletion of sample 1 (Fig. 2) and sample 3 and 1 breakpoint of sample 2.
An AluSg element and an AluSx element occurred near the proximal and distal breakpoints, respectively, in the RHDex1del type 1 (sample 1). A stem-loop may form during DNA replication (Fig. 2a) because the Alu repeats have 80% complementarity (Fig. 2b), bringing the distant GAATG repeats in close proximity to each other. The stem-loop can also pause the progression of replication fork, releasing the 3′ end of nascent leading strand, which can align at the downstream GAATG repeat causing the deletion (Fig. 2a). These molecular features were consistent with the fork stalling and template switching [39]/microhomology-mediated break-induced replication [39, 40] mechanism [41].
An AluSx1 element occurred near the proximal breakpoint region of RHDex10del type 1 (sample 2). Although no long stretches of continuous homology were detected around the distal breakpoint, such Alu sequences found in the vicinity of single breakpoints could still mediate the corresponding rearrangement by non-homologous recombination [38]. However, the presence of short direct repeat, AGG, flanking both the breakpoints implied classical replication slippage as the most likely mechanism for this deletion [42, 43].
A FRAM element was located at the proximal breakpoint of RHDex10del type 2 (sample 3) and also an AluYh3 element around the distal breakpoint featuring several recombination motifs GCG and GAS [44]. The presence of an insertion of 2 bp of non-template DNA (Fig. 1c) implied classical non-homologous end-joining [45] as the most likely mechanism responsible for this deletion [46, 47].
The RHDex1del type 1 expressed a DEL phenotype in sample 1, which correlated well with previous observations of many RHD exon 1 variations being associated with DEL phenotypes [9, 24, 48,49,50,51]. Deletion of amino acids 1 to 82 at the N-terminal end of the RhD protein indicated the loss of first and second extracellular loops from the mature protein (Supplementary Fig. S1A). This RhD model predicted the absence of the G-antigen, because the residue Ser103 in the second extracellular loop is known to be involved in the conformation-dependent G-antigen formation [52, 53]. An adsorption-elution with anti-G was negative, thus confirming the predicted loss of G-antigen expression.
The RHDex10del type 1 allele has previously been characterized as a serologic weak D phenotype [22], but was observed as a DEL phenotype in sample 2. The deletion was confirmed using published primers [22]. Variable strength of D-antigen expression, such as DEL and weak D, have been observed before in other RHD variants and can be caused by different sensitivities of the serologic techniques.
Sample 3 (RHDex10del type 2), lacking all eight amino acids encoded by RHD exon 10, was found to be negative for the D-antigen by adsorption/elution tests. Amino acid mutations affecting the C-terminal cytoplasmic positions 391 to 417 [54] have often been associated with a reduced expression of the protein in the RBC membrane [55,56,57]. Aromatic and hydrophobic C-terminal amino acids are important in mediating efficient transport of membrane proteins by interacting with COPII coat components [58] and also affect the interaction of RhD protein with the RBC cytoskeleton, specifically ankyrin-R [59, 60]. However, as exon 9 of RhD protein has the relevant amino acids V, F, and W at positions 406, 407, and 408, the complete lack of RhD expression in the RBC membrane was surprising. More sensitive techniques, such as mass-spectrometry, may be used to exclude the expression of any miniscule amounts of RhD protein present in the RBC membrane.
Many RHD alleles are associated with frequent anti-D alloimmunization, especially in chronically transfused patients, such as involving hemoglobinopathies [61, 62], or pregnant women. We defined the breakpoints of two novel deletions and suggested three possible mechanisms based on the sequences at and around the breakpoints for three distinct deletions. The identification of the deletion breakpoints allows designing allele-specific PCR assays and targeted screening for these deletions. Knowledge of the exact sequence details will aid in identifying the clinically relevant RHD alleles occurring in patient samples, especially when applying high throughput technologies, such as next-generation sequencing [63].
Change history
01 May 2018
The authors of the above paper noticed an error in publication. In Results section, under sub-section RHD genetic variations, the deletion nomenclature for Sample 1 was incorrectly given as [NC_000001.11(NG_007494.1):c.(1–15149_1–15153)_(148+3154_148+3158)del].
References
Poole J, Daniels G. Blood group antibodies and their significance in transfusion medicine. Transfus Med Rev. 2007;21:58–71.
Cartron JP. Defining the Rh blood group antigens. Biochemistry and molecular genetics. Blood Rev. 1994;8:199–212.
Flegel WA. Molecular genetics and clinical applications for RH. Transfus Apher Sci. 2011;44:81–91.
Wagner FF, Flegel WA. RHD gene deletion occurred in the Rhesus box. Blood. 2000;95:3662–8.
Koda Y, Soejima M, Johnson PH, Smart E, Kimura H. An Alu-mediated large deletion of the FUT2 gene in individuals with the ABO-Bombay phenotype. Hum Genet. 2000;106:80–5.
Westman JS, Hellberg A, Peyrard T, Thuresson B, Olsson ML. Large deletions involving the regulatory upstream regions of A4GALT give rise to principally novel P1PK-null alleles. Transfusion. 2014;54:1831–5.
Borck G, Kakar N, Hoch J, Friedrich K, Freudenberg J, Nürnberg G, et al. An Alu repeat-mediated genomic GCNT2 deletion underlies congenital cataracts and adult i blood group. Hum Genet. 2012;131:209–16.
Danek A, Rubio JP, Rampoldi L, Ho M, Dobson-Stone C, Tison F, et al. McLeod neuroacanthocytosis: genotype and phenotype. Ann Neurol. 2001;50:755–64.
Wagner FF, Flegel WA. The Rhesus site. Transfus Med Hemother. 2014;41:357–63.
Patnaik SK, Helmberg W, Blumenfeld OO. BGMUT database of allelic variants of genes encoding human blood group antigens. Transfus Med Hemother. 2014;41:346–51.
Apoil P-A, Blancher A. Rh gene evolution in primates: study of intron sequences. Mol Biol Evol. 2000;17:127–36.
Arce MA, Thompson ES, Wagner S, Coyne KE, Ferdman BA, Lublin DM. Molecular cloning of RhD cDNA derived from a gene present in RhD-positive, but not RhD-negative individuals. Blood. 1993;82:651–5.
Chang JG, Wang JC, Yang TY, Tsan KW, Shih MC, Peng CT, et al. Human RhD el is caused by a deletion of 1,013 bp between introns 8 and 9 including exon 9 of RHD Gene. Blood. 1998;92:2602–4.
Luettringhaus TA, Cho D, Ryang DW, Flegel WA. An easy RHD genotyping strategy for D-East Asian persons applied to Korean blood donors. Transfusion. 2006;46:2128–37.
Mouro I, Le Van Kim C, Rouillac C, van Rhenen DJ, Le Pennec PY, Bailly P, et al. Rearrangements of the blood group RhD gene associated with the DVI category phenotype. Blood. 1994;83:1129–35.
Avent ND, Liu W, Jones JW, Scott ML, Voak D, Pisacka M, et al. Molecular analysis of Rh transcripts and polypeptides from individuals expressing the DVI variant phenotype: an RHD gene deletion event does not generate All DVIccEe phenotypes. Blood. 1997;89:1779–86.
Huang CH. Human DVI category erythrocytes: correlation of the phenotype with a novel hybrid RhD-CE-D gene but not an internally deleted RhD gene. Blood. 1997;89:1834–5.
Blunt T, Steers F, Daniels G, Carritt B. Lack of RH C/E expression in the Rhesus D—phenotype is the result of a gene deletion. Ann Hum Genet. 1994;58:19–24.
Huang CH, Reid ME, Chen Y. Identification of a partial internal deletion in the RH locus causing the human erythrocyte D—phenotype. Blood. 1995;86:784–90.
Okuda H, Fujiwara H, Omi T, Iwamoto S, Kawano M, Ishida T, et al. A Japanese propositus with D—phenotype characterized by the deletion of both the RHCE gene and D1S80 locus situated in chromosome 1p and the existence of a new CE-D-CE hybrid gene. J Hum Genet. 2000;45:142–53.
Richard M, Perreault J, Constanzo-Yanez J, Khalifé S, St-Louis M. A new DEL variant caused by exon 8 deletion. Transfusion. 2007;47:852–7.
Fichou Y, Chen JM, Le Maréchal C, Jamet D, Dupont I, Chuteau C, et al. Weak D caused by a founder deletion in the RHD gene. Transfusion. 2012;52:2348–55.
Just B, Deitenbeck R. Sekundäre anti-D-Immunisierung bei RhD-negativem Empfänger, verursacht durch ein Del-Erythrozytenkonzentrat. Transfusionsmedizin. 2014;4:134–8.
Flegel WA, von Zabern I, Wagner FF. Six years’ experience performing RHD genotyping to confirm D-red blood cell units in Germany for preventing anti-D immunizations. Transfusion. 2009;49:465–71.
Srivastava K, Polin H, Sheldon SL, Wagner FF, Grabmer C, Gabriel C, et al. The DAU cluster: a comparative analysis of 18 RHD alleles, some forming partial D antigens. Transfusion. 2016;56:2520-31.
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012;40:e115.
Fasano RM, Monaco A, Meier ER, Pary P, Lee-Stroka AH, Otridge J, et al. RH genotyping in a sickle cell disease patient contributing to hematopoietic stem cell transplantation donor selection and management. Blood. 2010;116:2836–8.
Wagner FF, Gassner C, Müller TH, Schönitzer D, Schunter F, Flegel WA. Three molecular structures cause Rhesus D category VI phenotypes with distinct immunohematologic features. Blood. 1998;91:2157–68.
Hromadnikova I, Vechetova L, Vesela K, Benesova B, Doucha J, Kulovany E, et al. Non-invasive fetal RHD exon 7 and exon 10 genotyping using real-time PCR testing of fetal DNA in maternal plasma. Fetal Diagn Ther. 2005;20:275–80.
Srivastava K, Almarry NS, Flegel WA. Genetic variation of the whole ICAM4 gene in Caucasians and African Americans. Transfusion. 2014;54:2315–24.
Wagner FF, Gassner C, Müller TH, Schönitzer D, Schunter F, Flegel WA. Molecular basis of weak D phenotypes: presented at the 25th congress of the international society of blood transfusion held in Oslo on June 29, 1998 and published in abstract form in Vox Sang 74:55, 1998 (suppl). Blood. 1999;93:385–93.
Pirelli KJ, Pietz BC, Johnson ST, Pinder HL, Bellissimo DB. Molecular determination of RHD zygosity: predicting risk of hemolytic disease of the fetus and newborn related to anti-D. Prenat Diagn. 2010;30:1207–12.
Wildeman M, van Ophuizen E, den Dunnen JT, Taschner PE. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum Mutat. 2008;29:6–13.
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Flegel WA, von Zabern I, Doescher A, Wagner FF, Strathmann KP, Geisen C, et al. D variants at the RhD vestibule in the weak D type 4 and Eurasian D clusters. Transfusion. 2009;49:1059–69.
Batzer MA, Deininger PL. Alu repeats and human genomic diversity. Nat Rev Genet. 2002;3:370–9.
Rudiger NS, Gregersen N, Kielland-Brandt MC. One short well conserved region of Alu-sequences is involved in human gene rearrangements and has homology with prokaryotic chi. Nucleic Acids Res. 1995;23:256–60.
Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet. 2009;41:849–53.
Segar MW, Sakofsky CJ, Malkova A, Liu Y. MMBIRFinder: a tool to detect microhomology-mediated break-induced replication. IEEE/ACM Trans Comput Biol Bioinform. 2015;12:799–806.
Carvalho CM, Lupski JR. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet. 2016;17:224–38.
Magnani C, Cremonesi L, Giunta A, Magnaghi P, Taramelli R, Ferrari M. Short direct repeats at the breakpoints of a novel large deletion in the CFTR gene suggest a likely slipped mispairing mechanism. Hum Genet. 1996;98:102–8.
Chen JM, Chuzhanova N, Stenson PD, Ferec C, Cooper DN. Meta-analysis of gross insertions causing human genetic disease: novel mutational mechanisms and the role of replication slippage. Hum Mutat. 2005;25:207–21.
Abeysinghe SS, Chuzhanova N, Krawczak M, Ball EV, Cooper DN. Translocation and gross deletion breakpoints in human inherited disease and cancer I: nucleotide composition and recombination-associated motifs. Hum Mutat. 2003;22:229–44.
Deriano L, Roth DB. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu Rev Genet. 2013;47:433–55.
Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–6.
Perry GH, Ben-Dor A, Tsalenko A, Sampas N, Rodriguez-Revenga L, Tran CW, et al. The fine-scale and complex architecture of human copy-number variation. Am J Hum Genet. 2008;82:685–95.
Li Q, Ye LY, Guo ZH, Qian M, Zhu ZY. Study on the molecular background of Del phenotype in Chinese population. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2006;23:486–91.
Li Q, Hou L, Guo ZH, Ye LY, Yue DQ, Zhu ZY. Molecular basis of the RHD gene in blood donors with DEL phenotypes in Shanghai. Vox Sang. 2009;97:139–46.
Muller SP, Bartels I, Stein W, Emons G, Gutensohn K, Kohler M, et al. The determination of the fetal D status from maternal plasma for decision making on Rh prophylaxis is feasible. Transfusion. 2008;48:2292–301.
Wagner FF, Mardt I, Bittner R, Doscher A. RHD PCR of blood donors in Northern Germany: use of adsorption/elution to determine D antigen status (abstract). Vox Sanguinis. 2012;103:15.
Faas BH, Beckers EA, Simsek S, Overbeeke MA, Pepper R, van Rhenen DJ, et al. Involvement of Ser103 of the Rh polypeptides in G epitope formation. Transfusion. 1996;36:506–11.
Lomas C, Mougey R. Rh antigen D: variable expression in DVI phenotypes; a possible subdivision of category VI by a low frequency antigen (abstract). Transfusion. 1989;29:14S.
Wienzek-Lischka S, Flegel WA. Zur klinischen Bedeutung des Antigen D und seiner Varianten. Transfusionsmedizin. 2016;6:57–64.
Nicolas V, Mouro-Chanteloup I, Lopez C, Gane P, Gimm A, Mohandas N, et al. Functional interaction between Rh proteins and the spectrin-based skeleton in erythroid and epithelial cells. Transfus Clin Biol. 2006;13:23–28.
Silvy M, Simon S, Gouvitsos J, Di Cristofaro J, Ferrera V, Chiaroni J, et al. Weak D and DEL alleles detected by routine SNaPshot genotyping: identification of four novel RHD alleles. Transfusion. 2011;51:401–11.
Vege S, Copeland TR, Nickle PA, Westhoff CM. RHD exon consensus splice-site changes, 344A>G and 1228T>G, associated with weak D expression. Transfusion. 2009;49:SP172.
Nufer O, Guldbrandsen S, Degen M, Kappeler F, Paccaud JP, Tani K, et al. Role of cytoplasmic C-terminal amino acids of membrane proteins in ER export. J Cell Sci. 2002;115:619–28.
Nicolas V, Le Van Kim C, Gane P, Birkenmeier C, Cartron JP, Colin Y, et al. Rh-RhAG/ankyrin-R, a new interaction site between the membrane bilayer and the red cell skeleton, is impaired by Rh(null)-associated mutation. J Biol Chem. 2003;278:25526–33.
Dahl KN, Westhoff CM, Discher DE. Fractional attachment of CD47 (IAP) to the erythrocyte cytoskeleton and visual colocalization with Rh protein complexes. Blood. 2003;101:1194–9.
Denomme GA, Wagner FF, Fernandes BJ, Li W, Flegel WA. Partial D, weak D types, and novel RHD alleles among 33,864 multiethnic patients: implications for anti-D alloimmunization and prevention. Transfusion. 2005;45:1554–60.
Rujirojindakul P, Flegel WA. Applying molecular immunohaematology to regularly transfused thalassaemic patients in Thailand. Blood Transfus. 2014;12:28–35.
Johansen Taber KA, Dickinson BD, Wilson M. The promise and challenges of next-generation genome sequencing for clinical care. JAMA Intern Med. 2014;174:275–80.
Acknowledgements
We thank Harvey Gordon Klein for critical review of the manuscript; Sharon Dolores Adams for sample coordination; the staff of HLA laboratory for DNA extraction; and Elizabeth Jane Furlong for English edits. This work was supported by the Intramural Research Program (Project ID Z99 CL999999) of the NIH Clinical Center.
Author information
Authors and Affiliations
Contributions
All authors contributed to the design of the study, performed experiments, and discussed data; K.S., D.A.S., and W.A.F. analyzed the alleles; K.S. and W.A.F. wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Data access and submission
All authors had full access to the data and approved of the decision to submit.
Web resource
ISBT website (http://www.isbtweb.org/working-parties/red-cell-immunogenetics-and-blood-group-terminology/) accessed on 16 May 2017. The human RhesusBase version 2.0 (http://www.rhesusbase.info/) accessed on 16 May 2017. The human RhesusBase, RHD large deletion listing (http://www.rhesusbase.info/M_RHDlargedeletion.htm) accessed on 16 May 2017. UCSC browser (https://genome.ucsc.edu/).
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Srivastava, K., Stiles, D.A., Wagner, F.F. et al. Two large deletions extending beyond either end of the RHD gene and their red cell phenotypes. J Hum Genet 63, 27–35 (2018). https://doi.org/10.1038/s10038-017-0345-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-017-0345-3
This article is cited by
-
DEL variants: review of molecular mechanisms, clinical consequences and molecular testing strategy
Functional & Integrative Genomics (2023)
-
DEL in China: the D antigen among serologic RhD-negative individuals
Journal of Translational Medicine (2021)
