
Abstract
Children of obese mothers are at increased risk of developmental adversities. Maternal obesity is linked to an inflammatory in utero environment, which, in turn, is associated with neurodevelopmental impairments in the offspring. This is an integrated mechanism review of animal and human literature related to the hypothesis that maternal obesity causes maternal and fetal inflammation, and that this inflammation adversely affects the neurodevelopment of children. We propose integrative models in which several aspects of inflammation are considered along the causative pathway linking maternal obesity with neurodevelopmental limitations.
Similar content being viewed by others


Main
The prevalence of obesity in 2011–2012 was 35% among adult women in the United States, with no significant change compared with 2002–2004 (1). The prevalence of obesity among adult women in Europe was 12% in 2010 (2). Nearly 30% (2.1 billion people) of the world’s population today are estimated to be overweight or obese (3). This prevalence is projected to continue to increase, presenting a major public health epidemic in both the developing and developed world (4).
Just before pregnancy, almost two out of three women (64%) in the United States are either overweight or obese (5). Prepregnancy overweight and obesity are associated with gestational diabetes, pre-eclampsia, labor complications, and maternal hypertension during pregnancy (6). Maternal obesity is also associated with chorioamnionitis and pregnancy-related infection, such as group B streptococcal disease (7).
In addition to these potential risks to the mother, maternal obesity also may have life-long repercussions for her offspring. For example, children of obese mothers are prone to obesity (8,9), metabolic syndrome (10), neural tube defects (11), and cognitive impairment (12,13,14,15,16).
The causal pathway by which a mother’s obesity contributes to adverse neurodevelopmental outcomes among her offspring remains to be elucidated (12,17). Prepregnancy BMI studies focusing on pediatric outcomes are challenging because it is possible that characteristics and/or exposures associated with both prepregnancy BMI and neurodevelopmental outcomes influence the postnatal environment. In this review, we offer evidence from laboratory and human studies in support of the hypothesis that maternal obesity influences fetal, neonatal, and later developmental outcomes by increasing the risk of systemic and (18,19) and brain (20,21) inflammation.
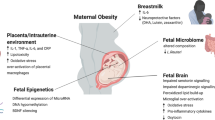
We first address the reported associations between maternal obesity and long-term neurodevelopment in offspring. In the subsequent sections, we outline an explanatory model ( Figure 1 ) in which a series of inflammatory processes are hypothesized to account for neurodevelopmental limitations.

Proposed mechanistic framework outlining inter-relationships between maternal obesity, inflammation, and neurodevelopmental deficits. Numbers along arrows correspond to numbers identifying sections in this article.
Integrated Mechanism Review
We used an integrated mechanism review method as outlined by Dammann and Gressens (22) to propose a hypothesis that explains the causal pathway for why children of obese mothers are at increased risk of developmental adversities. As suggested for integrated mechanism review, we first identified basic, clinical, and epidemiologic research on the neurodevelopment of children born to obese mothers. We then developed a graphic to illustrate possible causal models to explain the elevated risk of undesirable outcomes among the children of obese mothers. Finally, we reviewed available studies that could be used to support (or reject) each proposed incremental pathway of our explanatory model. The following sections summarize our findings.
Results of Maternal Obesity and Neuropsychological Outcomes
Children of mothers who were overweight or obese during pregnancy were at elevated risk of four major categories of neurodevelopmental deficits, including cognitive and intelligence deficits, attention deficit hyperactivity disorder (ADHD), autism, and psychoses ( Table 1 ). The most studied area was the relationship between the weight status of the mother before or during pregnancy and cognition/intelligence of the offspring.
Cognitive Deficits and Intelligence
By and large, children born to obese mothers have lower mental development scores than their peers born to normal weight women (14,16,23,24,25,26,27,28,29,30,31,32,33). Children whose mother was overweight near the time of the pregnancy tend to have mental development scores that are intermediate between obese and healthy weight mothers (16). No association was found between maternal prepregnancy BMI and motor skills (25,27,30).
ADHD
Maternal prepregnancy overweight and obesity were associated with child inattention and related symptoms at the age of 5 y (34), and with ADHD symptoms in children aged 7–12 y (35), and 9–17 y (36). The authors of a study of 7-y-old children that found a 2.8-fold increase in the prevalence of ADHD among children of obese compared to those of nonobese mothers attributed some of what they found to impaired executive function (37).
Autism
Two studies found that autistic children were more likely than their nonautistic peers to have a mother who was obese before the pregnancy (29), or had a prepregnancy weight ≥ 90 kg (38). Only one study showed a weak association between maternal obesity and ASD risk (39).
Psychoses
Of four studies that evaluated the relationship between maternal BMI and the risk of schizophrenia in the offspring, two found that the adult children born to obese women were two- to threefold more likely to be given a diagnosis of schizophrenia than the adult offspring of normal weight women (40,41). Another reported a progressive increase in the risk of schizophrenia in offspring was associated with each unit increase of maternal BMI (42). Only one of the four studies reported no relationship between maternal obesity and psychoses among their offspring. The BMI of mothers of adults with schizophrenia did not differ from the BMI of mothers of adults without schizophrenia (43). However, a review of the four studies raised the possibility that “the discrepant findings from one study could be attributable to sample characteristics and other factors” (44).
Integrated Explanatory Model
In Figure 1 , we illustrate the possible interrelationships between maternal obesity (on the left) and neurodevelopmental deficits of the offspring (on the right). Arrows indicate proposed pathways between exposures and outcomes. The numbers with each arrow in the figure refer to the following subsections and provide the available evidence to support each link in the model.
Not all intermediate steps in the proposed pathways are addressed in the model. For example, section 7 (Maternal systemic inflammation and fetal brain inflammation) appears to bypass the involvement of the uterus. We include such by-pass sections when the reports deal with an exposure (e.g., lipopolysaccharide administered into the peritoneal cavity) and an outcome (expression of the proinflammatory cytokines in the fetal rat brain), and do not report on, or discuss the likely intermediate steps.
1. Maternal Obesity and Maternal Systemic Inflammation
Obesity can contribute to chronic systemic inflammation (45,46), as can pregnancy (47,48). Beyond their many other functions, cytokines serve as signaling molecules between the immune and nervous systems (49). C-reactive protein is an acute phase protein that promotes further inflammation, while leptin is an adipokine associated not only with satiety and energy homeostasis, but also with inflammation (50,51,52,53). The systemic responses to pregnancy, including IL-6, C-reactive protein, and leptin, were exaggerated in women with prepregnancy obesity (54,55,56). At 4 wk of gestation, obese women had higher levels of C-reactive protein compared to normal weight pregnant women (57,58).
2. Maternal Systemic Inflammation and Long-Term Neurodevelopmental Deficits
Children of women who had high circulating levels of TNF-α (59) and IL-8 (60) during pregnancy were at increased risk of schizophrenia. Other inflammatory phenomena during fetal development might also contribute to the occurrence of autism (61). In a mouse model, systemic maternal inflammation with lipopolysaccharide combined with neonatal hyperoxic exposure appears to decrease oligodendrocyte numbers in the cerebral cortex and hippocampus in adulthood (62).
3. Maternal Obesity and Intrauterine Inflammation
Compared to the placentas of their lean peers, the placentas of obese women tend to have increased CD68+ and CD14+ cells, along with increased expression of the proinflammatory cytokines IL-1, TNF-α, IL-6, and C-reactive protein (54). Histologic inflammation was much more common in the placentas of obese women than in the placentas of normal-weight women (63). The higher a pregnant woman’s BMI, the higher her blood concentrations of cytokines and activation of placental p38-MAPK and STAT3 inflammatory pathways activated by MCP-1 and TNF-α (64).
4. Maternal Obesity and Systemic Inflammation in the Offspring
Preterm newborns of overweight and obese women were more likely than their peers born to women with lower BMIs to have systemic inflammation, but only among those delivered for maternal or fetal indications (65). The association was more prominent for protein elevations observed on two or more days than for elevations present on 1 d only, especially among the infants of overweight women. These findings suggest that mother’s prepregnancy overweight or obesity can contribute to a prolonged proinflammatory state in very preterm infants delivered for maternal or fetal indications.
At the age of 12 y, children born to obese mothers tended to have higher blood levels of C-reactive protein, but not IL-6, TNF-α, or adiponectin, than children born to nonobese mothers (19). In a study of adults (mean age 57 y), those with two obese parents (vs. none or one parent) had higher levels of C-reactive protein, but not IL-6, TNF, or adiponectin (18). Since the children of obese adults are at increased risk of becoming obese themselves (66), these reports support the hypothesis that maternal obesity increases susceptibility to an inflammatory state in the offspring perhaps even before the onset of obesity, suggesting that inflammation may be a precursor to the development of obesity rather than a consequence.
5. Maternal Obesity and Neuroinflammation in the Offspring
In a rat model, maternal high-fat-diet consumption (a correlate/contributor to maternal obesity) appears to sensitize offspring to the brain inflammation effects of their own high fat diet (67). These effects include increased reactivity of astrocytes and microglia, as well as increased levels of IL-6 and impaired water maze performance. Proinflammatory cytokines, including IL-1β and IL-1receptor 1, as well as markers of microglia activation, were upregulated in the hypothalamus of fetal macaques whose mothers received a high-fat-diet during pregnancy (68). The 90-d-old offspring of rats fed hydrogenated vegetable trans-fats, had increased IL-6, TNF-α, and IL1-β levels in their hypothalamus, compared to rats born to dams fed standard chow (69). Offspring of dams fed a high-saturated-fat or a high-trans-fat diet tended to have more microglial activation markers, TLR4 mRNA expression, and higher IL-1β levels in the hippocampus at birth compared to offspring of control-fed dams (70). The pups also displayed impaired spatial learning.
6. Maternal Obesity and Fetal/Neonatal Brain Damage
Four studies evaluated the relationship between maternal obesity and cerebral palsy. One study found that children of overweight and obese mothers had a 3.5-fold increased risk of cerebral palsy (71). Another study reported that maternal obesity was associated with a 30% increased risk of having a child with cerebral palsy. This risk was even higher among infants born to a morbidly-obese mother (72). Two studies found no association between maternal weight and cerebral palsy (73,74).
In a rat model, maternal obesity during pregnancy is associated with diminished proliferation and neuronal maturation of stem-like cells in the cerebral cortex of the pups’ brains, possibly resulting in impaired neurodevelopment at a later age (75). At postnatal day 21, the hypothalamic tissue of pups born to high-fat-diet dams shows upregulation of the toll-like receptor 4 (TLR4) signaling cascade, as well as increased phosphorylation of c-Jun N-terminal kinase 1 (JNK1) and IκB kinase-β (IKKβ) (76). Although murine responses to inflammatory stresses do not correlate well with human responses (77), activation of TLR4, JNK, and IKKβ can promote the synthesis and release of proinflammatory cytokines (78,79,80).
7. Maternal Systemic Inflammation and Fetal/Neonatal Brain Inflammation
When injected into the peritoneal cavity of pregnant rodents, lipopolysaccharide, an endotoxin synthesized by Gram-negative bacteria, increased the expression of TNF-α and IL-1β mRNA, in the fetal rat brain within hours, and promoted the presence of glial fibrillary acidic protein-positive astrocytes in the brain accompanied by decreased myelin basic protein (81).
8. Intrauterine Inflammation and Fetal/Neonatal Brain Damage
Placenta inflammation (chorioamnionitis) can induce a systemic fetal inflammatory response that contributes to white matter injury in the fetal brain (82,83). Inflammation in the placenta is also associated with neonatal brain damage (84,85,86). The inflammation signal, likely transmitted across the blood–brain barrier, initiates a neuroinflammatory response that has been offered as an explanation for later neurodevelopmental complications including cerebral palsy, autism, schizophrenia, and cognitive impairments (82,87,88). The authors of a review that found no support for an association between chorioamnionitis and central nervous system impairment in humans born preterm raised the possibility “that inflammation enhances maturation of the preterm infant and therefore has protective effects balancing its potential harmful effects” (89). Preconditioning might also be invoked to explain some of these inconsistent findings (v.i., 5.1 Systemic inflammation, preconditioning and sensitization).
9. Intrauterine Inflammation and Fetal/Neonatal Systemic Inflammation
Preterm newborns whose umbilical cord was inflamed (funisitis) tended to have higher blood concentrations of inflammation-related proteins, including C-reactive protein, MPO, IL1β, IL8, TNF-α, ICAM3, and MMP on postnatal day 7 than their peers without funisitis (90).
10. Intrauterine Inflammation and Neurodevelopmental Deficits
Brain damage in both preterm and term mice following intrauterine inflammation is accompanied by increased TNF-α expression and alterations of other gene pathways in the brain thought to influence neurobehavioral, motor, and psychosocial behavior (91). Placenta inflammation is also associated with a decreased number of dendritic processes in the offspring’s brain, resulting in impaired learning and memory (91,92). Additionally, intrauterine inflammation in pregnant sheep resulted in microglial activation and macrophage infiltration in the fetal brain (93). Among humans born before the 28th week of gestation, placenta inflammation and the presence of microorganisms in the placenta were not associated with low Bayley Scales Mental Development Index scores at the age of 2 y (24).
11. Fetal/Neonatal Brain Inflammation and Fetal/Neonatal Brain Damage
Experimental and epidemiological studies document that perinatal inflammation can be a risk factor for abnormalities in brain structure and function (61,85,88,94). Cytokines such as TNF-α released during intrauterine inflammation are a possible cause of brain damage observed in animal studies linking preterm birth and periventricular white matter damage (61,85,94). Five mechanisms of cytokine-induced brain injury have been proposed (95). The first mechanism is the direct effect of cytokines (IL-6) on the cerebral circulation (96). Second, inflammation promotes coagulation which in turn can increase the risk of brain damage via vessel obstruction (90) or enhanced inflammation (97,98). Third, activated microglia can cause direct toxic effects on oligodendrocytes and myelin, in part, via microglial production of cytokines (99), which leads to neuronal loss and impaired neuronal guidance (99,100), as well as inhibition of oligodendrocyte maturation (101). Fourth, the activation of microglia leads to production of free radicals, which contribute to oligodendrocyte death via oxidative stress (99,102). Fifth, inflammation can promote excitotoxic mechanisms resulting in damage to both neurons and oligodendrocytes (103). All of these mechanisms are enhanced by the increased permeability of the developing blood–brain barrier that has been documented following intraperitoneal injection of lipopolysaccharide (104).
12. Fetal/Neonatal Brain Damage and Fetal/Neonatal Systemic Inflammation
The systemic inflammation that follows brain damage has two explanations (105). One postulates that what is seen in the blood is merely persistence of the inflammation that caused the brain damage. Another postulates that what is seen in the blood is the “spill over” of the inflammation in the brain. What is impressive is how long the local and systemic inflammation can continue (106).
13. Fetal/Neonatal Systemic Inflammation and Fetal/Neonatal Brain Damage
Among infants born before 28 wk gestation, elevated levels of inflammation-related proteins in blood collected on both postnatal days 7 and 14 were associated with ventriculomegaly on an ultrasound scan of the brain when the very preterm newborn was in the intensive care nursery (107). Because the ventriculomegaly is not accompanied by macrocephaly, the ventricular enlargement is usually attributed to processes that lead to hydrocephalus ex vacuo.
14. Fetal/Neonatal Brain Inflammation and Clinical Neurodevelopmental Deficits
Rats that have increased reactivity of astrocytes and microglia, as well as increased levels of IL-6 in the brain, are more likely than others to have impaired retention of what they learned in a water maze (67). Compared to control rat pups, those with elevated IL-1β levels in the hippocampus at birth displayed impaired spatial learning (70).
15. Fetal/Neonatal Brain Damage and Neurodevelopmental Deficits
Fetal brain damage is linked with neurodevelopmental deficits in early childhood (108). Reductions and other modifications in total and regional volume of the cerebellum, as well as central and occipital regions of the cerebrum at term-equivalent age predict neurodevelopmental impairment in early childhood (109). Indicators of cerebral white matter damage also convey information about heightened risk of developmental disorders (110,111,112).
16. Fetal/Neonatal Systemic Inflammation and Neurodevelopmental Deficits
Newborn mice injected with IL-1β twice-daily over 5 d, had later memory deficits (113). Among infants born before 32 wk gestation, those who had elevated levels of proinflammatory and modulatory cytokines in blood obtained during the first 72 postnatal hours were at increased risk of cerebral palsy and less severe motor limitations at the age of 2 y (114). Among infants born before 28 wk gestation, elevated levels of inflammation-related proteins in blood collected on both postnatal days 7 and 14 were associated with impaired mental and motor development (115) as well as microcephaly (116) at the age of 2 y.
Other Mechanisms Explaining link with Neurodevelopmental Outcomes
Systemic Inflammation, Preconditioning, and Sensitization
Exposing the brain of a fetus to a subdamaging stimulus can protect against a subsequent insult (117,118). This phenomenon has been given two names, preconditioning, and tolerance. The same type of subdamaging stimulus can also sensitize the perinatal brain to a subsequent subdamaging insult (119,120). Among the characteristics of the subdamaging exposure that influence whether the result is preconditioning (tolerance) or sensitization are duration of the interval between first and second exposures (120) and postnatal age (121). The mechanisms that mediate preconditioning mechanisms in the immature brain likely differ from those observed in the adult (122). Figure 1 shows that maternal obesity associated with the increased risk of brain damage is linked to systemic inflammation. Due to this systemic inflammation, the preterm newborn is exposed to subdamaging stimuli within a narrow time range, resulting in greater injury explaining the higher risk for adverse neurodevelopmental outcomes in these children.
Confounding Factors
It is possible that characteristics and/or exposures associated with both maternal obesity and systemic inflammation, and not the obesity per se, explain the links with neurodevelopmental outcomes. One of these confounding factors might be maternal distress, as obesity has been related to mental health problems, which have been linked to developmental limitations (123).
In a study that adjusted for maternal stress and depression symptoms during and after pregnancy, the association between prenatal maternal obesity and ADHD symptoms and emotional problems did not change (34). This suggests that maternal obesity and maternal distress may possibly act through separate mechanisms influencing fetal brain development.
Studies of the relationship between maternal obesity and the child’s development need to address the potential confounding due to variables that are related to both the mother’s obesity and her child’s development. For example, obese women and their families are more likely than others to have low socioeconomic status (124), micronutrient deficiencies (125), emotional distress (126,127), child behavior problems (36,42), and mental health dysfunctions in general (128). Another confounder of maternal obesity is paternal obesity, which has been associated with autistic disorder and Asperger (39).
It is also possible that maternal obesity increases the risk of neurodevelopmental limitations in the offspring via epigenetic phenomena (129,130,131,132).
Conclusion
We offer support for the claim that the contribution of maternal obesity to adverse brain development is achieved in part via inflammatory phenomena. The majority of data delineating the role of systemic inflammation in this association included animal and basic research studies. Therefore, studies of humans are needed that follow offspring from infancy into adulthood measuring their neurodevelopment in cognition and mental illnesses, as well as inflammatory phenomena. With these studies, the full impact of being exposed to maternal obesity in utero can be better understood. Attempts to better control maternal obesity could lead to important benefits for the cognitive and psychiatric functioning of offspring. The exact mechanisms remain unknown. We, however, offer a model with several pathways with support for each component of these pathways.
Statement of Financial Support
Preparation of this report was supported by The National Institute of Neurological Disorders and Stroke (5U01NS040069-05; 2R01NS040069-06A2), The National Eye Institute in (1-R01-EY021820-01), and the National Institute of Child Health and Human Development (5P30HD018655-28). The various branches of the National Institutes of Health that made our work possible are located in Rockville and Bethesda, Maryland, USA.
Disclosure
The authors declare that there are no conflicts of interest.
References
Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011-2012. JAMA 2014;311:806–14.
Gallus S, Lugo A, Murisic B, Bosetti C, Boffetta P, La Vecchia C. Overweight and obesity in 16 European countries. Eur J Nutr. 2014 Aug 5:1–11.
Ng M, Fleming T, Robinson M, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014;384:766–81.
Kelly T, Yang W, Chen CS, Reynolds K, He J. Global burden of obesity in 2005 and projections to 2030. Int J Obes (Lond) 2008;32:1431–7.
Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999-2008. JAMA 2010;303:235–41.
Rowlands I, Graves N, de Jersey S, McIntyre HD, Callaway L. Obesity in pregnancy: outcomes and economics. Semin Fetal Neonatal Med 2010;15:94–9.
Håkansson S, Källen K. High maternal body mass index increases the risk of neonatal early onset group B streptococcal disease. Acta Paediatr 2008;97:1386–9.
Lawlor DA, Smith GD, O’Callaghan M, et al. Epidemiologic evidence for the fetal overnutrition hypothesis: findings from the mater-university study of pregnancy and its outcomes. Am J Epidemiol 2007;165:418–24.
Sen S, Carpenter AH, Hochstadt J, et al. Nutrition, weight gain and eating behavior in pregnancy: a review of experimental evidence for long-term effects on the risk of obesity in offspring. Physiol Behav 2012;107:138–45.
Boney CM, Verma A, Tucker R, Vohr BR. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics 2005;115:e290–6.
Rasmussen SA, Chu SY, Kim SY, Schmid CH, Lau J. Maternal obesity and risk of neural tube defects: a metaanalysis. Am J Obstet Gynecol 2008;198:611–9.
Van Lieshout RJ, Taylor VH, Boyle MH. Pre-pregnancy and pregnancy obesity and neurodevelopmental outcomes in offspring: a systematic review. Obes Rev 2011;12:e548–59.
Brion MJ, Zeegers M, Jaddoe V, et al. Intrauterine effects of maternal prepregnancy overweight on child cognition and behavior in 2 cohorts. Pediatrics 2011;127:e202–11.
Tanda R, Salsberry PJ, Reagan PB, Fang MZ. The impact of prepregnancy obesity on children’s cognitive test scores. Matern Child Health J 2013;17:222–9.
Basatemur E, Gardiner J, Williams C, Melhuish E, Barnes J, Sutcliffe A. Maternal prepregnancy BMI and child cognition: a longitudinal cohort study. Pediatrics 2013;131:56–63.
Casas M, Chatzi L, Carsin AE, et al. Maternal pre-pregnancy overweight and obesity, and child neuropsychological development: two Southern European birth cohort studies. Int J Epidemiol 2013;42:506–17.
Van Lieshout RJ. Role of maternal adiposity prior to and during pregnancy in cognitive and psychiatric problems in offspring. Nutr Rev 2013;71:Suppl 1:S95–101.
Lieb W, Pencina MJ, Lanier KJ, et al. Association of parental obesity with concentrations of select systemic biomarkers in nonobese offspring: the Framingham Heart Study. Diabetes 2009;58:134–7.
Leibowitz KL, Moore RH, Ahima RS, et al. Maternal obesity associated with inflammation in their children. World J Pediatr 2012;8:76–9.
Chen H, Simar D, Morris MJ. Hypothalamic neuroendocrine circuitry is programmed by maternal obesity: interaction with postnatal nutritional environment. PLoS One 2009;4:e6259.
Chen H, Simar D, Lambert K, Mercier J, Morris MJ. Maternal and postnatal overnutrition differentially impact appetite regulators and fuel metabolism. Endocrinology 2008;149:5348–56.
Dammann O, Gressens P. Integrated mechanism reviews. Pediatr Res 2012;71:530–1.
Kerstjens JM, de Winter AF, Sollie KM, et al. Maternal and pregnancy-related factors associated with developmental delay in moderately preterm-born children. Obstet Gynecol 2013;121:727–33.
Helderman JB, O’Shea TM, Kuban KC, et al.; ELGAN study Investigators. Antenatal antecedents of cognitive impairment at 24 months in extremely low gestational age newborns. Pediatrics 2012;129:494–502.
Hinkle SN, Schieve LA, Stein AD, Swan DW, Ramakrishnan U, Sharma AJ. Associations between maternal prepregnancy body mass index and child neurodevelopment at 2 years of age. Int J Obes (Lond) 2012;36:1312–9.
Craig WY, Palomaki GE, Neveux LM, Haddow JE. Maternal body mass index during pregnancy and offspring neurocognitive development. Obstet Med. 2013:20–5.
Neggers YH, Goldenberg RL, Ramey SL, Cliver SP. Maternal prepregnancy body mass index and psychomotor development in children. Acta Obstet Gynecol Scand 2003;82:235–40.
Heikura U, Taanila A, Hartikainen AL, et al. Variations in prenatal sociodemographic factors associated with intellectual disability: a study of the 20-year interval between two birth cohorts in northern Finland. Am J Epidemiol 2008;167:169–77.
Krakowiak P, Walker CK, Bremer AA, et al. Maternal metabolic conditions and risk for autism and other neurodevelopmental disorders. Pediatrics 2012;129:e1121–8.
Hinkle SN, Sharma AJ, Kim SY, Schieve LA. Maternal prepregnancy weight status and associations with children’s development and disabilities at kindergarten. Int J Obes (Lond) 2013;37:1344–51.
Huang L, Yu X, Keim S, Li L, Zhang L, Zhang J. Maternal prepregnancy obesity and child neurodevelopment in the Collaborative Perinatal Project. Int J Epidemiol 2014;43:783–92.
Gage SH, Lawlor DA, Tilling K, Fraser A. Associations of maternal weight gain in pregnancy with offspring cognition in childhood and adolescence: findings from the Avon Longitudinal Study of Parents and Children. Am J Epidemiol 2013;177:402–10.
Bliddal M, Olsen J, Støvring H, et al. Maternal pre-pregnancy BMI and intelligence quotient (IQ) in 5-year-old children: a cohort based study. PLoS One 2014;9:e94498.
Rodriguez A. Maternal pre-pregnancy obesity and risk for inattention and negative emotionality in children. J Child Psychol Psychiatry 2010;51:134–43.
Rodriguez A, Miettunen J, Henriksen TB, et al. Maternal adiposity prior to pregnancy is associated with ADHD symptoms in offspring: evidence from three prospective pregnancy cohorts. Int J Obes (Lond) 2008;32:550–7.
Chen Q, Sjölander A, Långström N, et al. Maternal pre-pregnancy body mass index and offspring attention deficit hyperactivity disorder: a population-based cohort study using a sibling-comparison design. Int J Epidemiol 2014;43:83–90.
Buss C, Entringer S, Davis EP, et al. Impaired executive function mediates the association between maternal pre-pregnancy body mass index and child ADHD symptoms. PLoS One 2012;7:e37758.
Dodds L, Fell DB, Shea S, Armson BA, Allen AC, Bryson S. The role of prenatal, obstetric and neonatal factors in the development of autism. J Autism Dev Disord 2011;41:891–902.
Surén P, Gunnes N, Roth C, et al. Parental obesity and risk of autism spectrum disorder. Pediatrics 2014;133:e1128–38.
Schaefer CA, Brown AS, Wyatt RJ, et al. Maternal prepregnant body mass and risk of schizophrenia in adult offspring. Schizophr Bull 2000;26:275–86.
Wahlbeck K, Forsén T, Osmond C, Barker DJ, Eriksson JG. Association of schizophrenia with low maternal body mass index, small size at birth, and thinness during childhood. Arch Gen Psychiatry 2001;58:48–52.
Kawai M, Minabe Y, Takagai S, et al. Poor maternal care and high maternal body mass index in pregnancy as a risk factor for schizophrenia in offspring. Acta Psychiatr Scand 2004;110:257–63.
Jones PB, Rantakallio P, Hartikainen AL, Isohanni M, Sipila P. Schizophrenia as a long-term outcome of pregnancy, delivery, and perinatal complications: a 28-year follow-up of the 1966 north Finland general population birth cohort. Am J Psychiatry 1998;155:355–64.
Khandaker GM, Dibben CR, Jones PB. Does maternal body mass index during pregnancy influence risk of schizophrenia in the adult offspring? Obes Rev 2012;13:518–27.
Shah TJ, Leik CE, Walsh SW. Neutrophil infiltration and systemic vascular inflammation in obese women. Reprod Sci 2010;17:116–24.
Howe LR, Subbaramaiah K, Hudis CA, Dannenberg AJ. Molecular pathways: adipose inflammation as a mediator of obesity-associated cancer. Clin Cancer Res 2013;19:6074–83.
McElrath TF, Fichorova RN, Allred EN, et al.; ELGAN Study Investigators. Blood protein profiles of infants born before 28 weeks differ by pregnancy complication. Am J Obstet Gynecol 2011;204:418.e1–418.e12.
Friis CM, Roland P, Marie C, Godang K, Ueland T, Tanbo T, et al. Adiposity‐related inflammation: Effects of pregnancy. Obesity. 2013;21:124–30.
Potvin S, Stip E, Sepehry AA, Gendron A, Bah R, Kouassi E. Inflammatory cytokine alterations in schizophrenia: a systematic quantitative review. Biol Psychiatry 2008;63:801–8.
Hwang HS, Kwon JY, Kim MA, Park YW, Kim YH. Maternal serum highly sensitive C-reactive protein in normal pregnancy and pre-eclampsia. Int J Gynaecol Obstet 2007;98:105–9.
Alexe DM, Syridou G, Petridou ET. Determinants of early life leptin levels and later life degenerative outcomes. Clin Med Res 2006;4:326–35.
Hauguel-de Mouzon S, Lepercq J, Catalano P. The known and unknown of leptin in pregnancy. Am J Obstet Gynecol. 2006;194:1537–45.
Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med 1999;340:448–54.
Challier JC, Basu S, Bintein T, et al. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta 2008;29:274–81.
Ramsay JE, Ferrell WR, Crawford L, Wallace AM, Greer IA, Sattar N. Maternal obesity is associated with dysregulation of metabolic, vascular, and inflammatory pathways. J Clin Endocrinol Metab 2002;87:4231–7.
Madan JC, Davis JM, Craig WY, et al. Maternal obesity and markers of inflammation in pregnancy. Cytokine 2009;47:61–4.
Sacks GP, Seyani L, Lavery S, Trew G. Maternal C-reactive protein levels are raised at 4 weeks gestation. Hum Reprod 2004;19:1025–30.
Stewart FM, Freeman DJ, Ramsay JE, Greer IA, Caslake M, Ferrell WR. Longitudinal assessment of maternal endothelial function and markers of inflammation and placental function throughout pregnancy in lean and obese mothers. J Clin Endocrinol Metab 2007;92:969–75.
Buka SL, Tsuang MT, Torrey EF, Klebanoff MA, Wagner RL, Yolken RH. Maternal cytokine levels during pregnancy and adult psychosis. Brain Behav Immun 2001;15:411–20.
Brown AS, Hooton J, Schaefer CA, et al. Elevated maternal interleukin-8 levels and risk of schizophrenia in adult offspring. Am J Psychiatry 2004;161:889–95.
Meyer U, Feldon J, Dammann O. Schizophrenia and autism: both shared and disorder-specific pathogenesis via perinatal inflammation? Pediatr Res 2011;69(5 Pt 2):26R–33R.
Graf AE, Haines KM, Pierson CR, et al. Perinatal inflammation results in decreased oligodendrocyte numbers in adulthood. Life Sci 2014;94:164–71.
Bar J, Schreiber L, Saruhanov E, Ben-Haroush A, Golan A, Kovo M. Placental histopathological findings in obese and nonobese women with complicated and uncomplicated pregnancies. Arch Gynecol Obstet 2012;286:1343–7.
Aye IL, Lager S, Ramirez VI, et al. Increasing maternal body mass index is associated with systemic inflammation in the mother and the activation of distinct placental inflammatory pathways. Biol Reprod 2014;90:129.
van der Burg JW, Allred EN, McElrath TF, et al. Is maternal obesity associated with sustained inflammation in extremely low gestational age newborns? Early Hum Dev 2013;89:949–55.
Patro B, Liber A, Zalewski B, Poston L, Szajewska H, Koletzko B. Maternal and paternal body mass index and offspring obesity: a systematic review. Ann Nutr Metab 2013;63:32–41.
White CL, Pistell PJ, Purpera MN, et al. Effects of high fat diet on Morris maze performance, oxidative stress, and inflammation in rats: contributions of maternal diet. Neurobiol Dis 2009;35:3–13.
Grayson BE, Levasseur PR, Williams SM, Smith MS, Marks DL, Grove KL. Changes in melanocortin expression and inflammatory pathways in fetal offspring of nonhuman primates fed a high-fat diet. Endocrinology 2010;151:1622–32.
Pimentel GD, Lira FS, Rosa JC, et al. Intake of trans fatty acids during gestation and lactation leads to hypothalamic inflammation via TLR4/NFκBp65 signaling in adult offspring. J Nutr Biochem 2012;23:265–71.
Bilbo SD, Tsang V. Enduring consequences of maternal obesity for brain inflammation and behavior of offspring. FASEB J 2010;24:2104–15.
Walstab J, Bell R, Reddihough D, Brennecke S, Bessell C, Beischer N. Antenatal and intrapartum antecedents of cerebral palsy: a case-control study. Aust N Z J Obstet Gynaecol 2002;42:138–46.
Crisham Janik MD, Newman TB, Cheng YW, Xing G, Gilbert WM, Wu YW. Maternal diagnosis of obesity and risk of cerebral palsy in the child. J Pediatr 2013;163:1307–12.
Blair E, Stanley F. When can cerebral palsy be prevented? The generation of causal hypotheses by multivariate analysis of a case-control study. Paediatr Perinat Epidemiol 1993;7:272–301.
O’Callaghan ME, MacLennan AH, Gibson CS, et al.; Australian Collaborative Cerebral Palsy Research Group. Epidemiologic associations with cerebral palsy. Obstet Gynecol 2011;118:576–82.
Stachowiak EK, Oommen S, Vasu VT, et al. Maternal obesity affects gene expression and cellular development in fetal brains. Nutr Neurosci 2013;16:96–103.
Rother E, Kuschewski R, Alcazar MA, et al. Hypothalamic JNK1 and IKKβ activation and impaired early postnatal glucose metabolism after maternal perinatal high-fat feeding. Endocrinology 2012;153:770–81.
Seok J, Warren HS, Cuenca AG, et al.; Inflammation and Host Response to Injury, Large Scale Collaborative Research Program. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA 2013;110:3507–12.
Caron JE, La Pine TR, Augustine NH, Martins TB, Hill HR. Multiplex analysis of toll-like receptor-stimulated neonatal cytokine response. Neonatology 2010;97:266–73.
Karin M, Gallagher E. From JNK to pay dirt: jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life 2005;57:283–95.
Verhelst K, Verstrepen L, Carpentier I, Beyaert R. IκB kinase ɛ (IKKɛ): a therapeutic target in inflammation and cancer. Biochem Pharmacol 2013;85:873–80.
Cai Z, Pan ZL, Pang Y, Evans OB, Rhodes PG. Cytokine induction in fetal rat brains and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatr Res 2000;47:64–72.
Kuypers E, Ophelders D, Jellema RK, Kunzmann S, Gavilanes AW, Kramer BW. White matter injury following fetal inflammatory response syndrome induced by chorioamnionitis and fetal sepsis: lessons from experimental ovine models. Early Hum Dev 2012;88:931–6.
Balakrishnan B, Dai H, Janisse J, Romero R, Kannan S. Maternal endotoxin exposure results in abnormal neuronal architecture in the newborn rabbit. Dev Neurosci 2013;35:396–405.
Leviton A. Preterm birth and cerebral palsy: is tumor necrosis factor the missing link? Dev Med Child Neurol 1993;35:553–8.
Dammann O, Leviton A. Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res 1997;42:1–8.
Patrick LA, Smith GN. Proinflammatory cytokines: a link between chorioamnionitis and fetal brain injury. J Obstet Gynaecol Can 2002;24:705–9.
Malaeb S, Dammann O. Fetal inflammatory response and brain injury in the preterm newborn. J Child Neurol 2009;24:1119–26.
Hagberg H, Gressens P, Mallard C. Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Ann Neurol 2012;71:444–57.
Ylijoki M, Ekholm E, Haataja L, Lehtonen L ; PIPARI study group. Is chorioamnionitis harmful for the brain of preterm infants? A clinical overview. Acta Obstet Gynecol Scand 2012;91:403–19.
Leviton A, Hecht JL, Allred EN, Yamamoto H, Fichorova RN, Dammann O ; ELGAN Study Investigators. Persistence after birth of systemic inflammation associated with umbilical cord inflammation. J Reprod Immunol 2011;90:235–43.
Elovitz MA, Brown AG, Breen K, Anton L, Maubert M, Burd I. Intrauterine inflammation, insufficient to induce parturition, still evokes fetal and neonatal brain injury. Int J Dev Neurosci 2011;29:663–71.
Burd I, Bentz AI, Chai J, et al. Inflammation-induced preterm birth alters neuronal morphology in the mouse fetal brain. J Neurosci Res 2010;88:1872–81.
Hutton LC, Castillo-Melendez M, Smythe GA, Walker DW. Microglial activation, macrophage infiltration, and evidence of cell death in the fetal brain after uteroplacental administration of lipopolysaccharide in sheep in late gestation. Am J Obstet Gynecol 2008;198:117.e1–11.
Dammann O, O’Shea TM. Cytokines and perinatal brain damage. Clin Perinatol 2008;35:643–63, v.
Galinsky R, Polglase GR, Hooper SB, Black MJ, Moss TJ. The consequences of chorioamnionitis: preterm birth and effects on development. J Pregnancy 2013;2013:412831.
Yanowitz TD, Jordan JA, Gilmour CH, et al. Hemodynamic disturbances in premature infants born after chorioamnionitis: association with cord blood cytokine concentrations. Pediatr Res 2002;51:310–6.
Ogata J, Yamanishi H, Ishibashi-Ueda H. Review: role of cerebral vessels in ischaemic injury of the brain. Neuropathol Appl Neurobiol 2011;37:40–55.
Leviton A, Dammann O. Coagulation, inflammation, and the risk of neonatal white matter damage. Pediatr Res 2004;55:541–5.
Gavilanes AW, Strackx E, Kramer BW, et al. Chorioamnionitis induced by intraamniotic lipopolysaccharide resulted in an interval-dependent increase in central nervous system injury in the fetal sheep. Am J Obstet Gynecol 2009;200:437.e1–8.
Leviton A, Gressens P. Neuronal damage accompanies perinatal white-matter damage. Trends Neurosci 2007;30:473–8.
Feldhaus B, Dietzel ID, Heumann R, Berger R. Effects of interferon-gamma and tumor necrosis factor-alpha on survival and differentiation of oligodendrocyte progenitors. J Soc Gynecol Investig 2004;11:89–96.
Khwaja O, Volpe JJ. Pathogenesis of cerebral white matter injury of prematurity. Arch Dis Child Fetal Neonatal Ed 2008;93:F153–61.
Kaindl AM, Favrais G, Gressens P. Molecular mechanisms involved in injury to the preterm brain. J Child Neurol 2009;24:1112–8.
Stolp HB, Dziegielewska KM, Ek CJ, et al. Breakdown of the blood-brain barrier to proteins in white matter of the developing brain following systemic inflammation. Cell Tissue Res 2005;320:369–78.
Anthony DC, Couch Y, Losey P, Evans MC. The systemic response to brain injury and disease. Brain Behav Immun 2012;26:534–40.
Winerdal M, Winerdal ME, Kinn J, Urmaliya V, Winqvist O, Adén U. Long lasting local and systemic inflammation after cerebral hypoxic ischemia in newborn mice. PLoS One 2012;7:e36422.
Leviton A, Kuban K, O’Shea TM, et al. The relationship between early concentrations of 25 blood proteins and cerebral white matter injury in preterm newborns: the ELGAN study. J Pediatr 2011;158:897–903.e1–5.
Dammann O, Leviton A. Role of the fetus in perinatal infection and neonatal brain damage. Curr Opin Pediatr 2000;12:99–104.
Keunen K, Kersbergen KJ, Groenendaal F, Isgum I, de Vries LS, Benders MJ. Brain tissue volumes in preterm infants: prematurity, perinatal risk factors and neurodevelopmental outcome: a systematic review. J Matern Fetal Neonatal Med 2012;25:Suppl 1:89–100.
Woodward LJ, Edgin JO, Thompson D, Inder TE. Object working memory deficits predicted by early brain injury and development in the preterm infant. Brain 2005;128(Pt 11):2578–87.
O’Shea TM, Kuban KC, Allred EN, et al.; Extremely Low Gestational Age Newborns Study Investigators. Neonatal cranial ultrasound lesions and developmental delays at 2 years of age among extremely low gestational age children. Pediatrics 2008;122:e662–9.
Kuban KC, Allred EN, O’Shea TM, et al.; ELGAN study investigators. Cranial ultrasound lesions in the NICU predict cerebral palsy at age 2 years in children born at extremely low gestational age. J Child Neurol 2009;24:63–72.
Favrais G, van de Looij Y, Fleiss B, et al. Systemic inflammation disrupts the developmental program of white matter. Ann Neurol 2011;70:550–65.
Hansen-Pupp I, Hallin AL, Hellström-Westas L, et al. Inflammation at birth is associated with subnormal development in very preterm infants. Pediatr Res 2008;64:183–8.
O’Shea TM, Allred EN, Kuban KC, et al.; Extremely Low Gestational Age Newborn (ELGAN) Study Investigators. Elevated concentrations of inflammation-related proteins in postnatal blood predict severe developmental delay at 2 years of age in extremely preterm infants. J Pediatr 2012;160:395–401.e4.
Leviton A, Kuban KC, Allred EN, Fichorova RN, O’Shea TM, Paneth N ; ELGAN Study Investigators. Early postnatal blood concentrations of inflammation-related proteins and microcephaly two years later in infants born before the 28th post-menstrual week. Early Hum Dev 2011;87:325–30.
Hagberg H, Dammann O, Mallard C, Leviton A. Preconditioning and the developing brain. Semin Perinatol 2004;28:389–95.
Kumral A, Tuzun F, Ozbal S, et al. Lipopolysaccharide-preconditioning protects against endotoxin-induced white matter injury in the neonatal rat brain. Brain Res 2012;1489:81–9.
Adén U, Favrais G, Plaisant F, et al. Systemic inflammation sensitizes the neonatal brain to excitotoxicity through a pro-/anti-inflammatory imbalance: key role of TNFalpha pathway and protection by etanercept. Brain Behav Immun 2010;24:747–58.
Eklind S, Mallard C, Arvidsson P, Hagberg H. Lipopolysaccharide induces both a primary and a secondary phase of sensitization in the developing rat brain. Pediatr Res 2005;58:112–6.
Hickey E, Shi H, Van Arsdell G, Askalan R. Lipopolysaccharide-induced preconditioning against ischemic injury is associated with changes in toll-like receptor 4 expression in the rat developing brain. Pediatr Res 2011;70:10–4.
Mallard C. Innate immune regulation by toll-like receptors in the brain. ISRN Neurol 2012;2012:701950.
Waters CS, Hay DF, Simmonds JR, van Goozen SH. Antenatal depression and children’s developmental outcomes: potential mechanisms and treatment options. Eur Child Adolesc Psychiatry 2014;23:957–71.
Wang Y, Beydoun MA. The obesity epidemic in the United States–gender, age, socioeconomic, racial/ethnic, and geographic characteristics: a systematic review and meta-regression analysis. Epidemiol Rev 2007;29:6–28.
Kimmons JE, Blanck HM, Tohill BC, Zhang J, Khan LK. Associations between body mass index and the prevalence of low micronutrient levels among US adults. MedGenMed 2006;8:59.
Carr D, Friedman MA, Jaffe K. Understanding the relationship between obesity and positive and negative affect: the role of psychosocial mechanisms. Body Image 2007;4:165–77.
Pasco JA, Williams LJ, Jacka FN, Brennan SL, Berk M. Obesity and the relationship with positive and negative affect. Aust N Z J Psychiatry 2013;47:477–82.
Magallares A, Pais-Ribeiro JL. Mental health and obesity: A meta-analysis. Applied Research in Quality of Life. 2014;9:295–308.
Lane M, Zander-Fox DL, Robker RL, McPherson NO. Peri-conception parental obesity, reproductive health, and transgenerational impacts. Trends Endocrinol Metab 2015;26:84–90.
El Hajj N, Schneider E, Lehnen H, Haaf T. Epigenetics and life-long consequences of an adverse nutritional and diabetic intrauterine environment. Reproduction 2014;148:R111–20.
van Dijk SJ, Molloy PL, Varinli H, Morrison JL, Muhlhausler BS ; Members of EpiSCOPE. Epigenetics and human obesity. Int J Obes (Lond) 2015;39:85–97.
Martínez JA, Milagro FI, Claycombe KJ, Schalinske KL. Epigenetics in adipose tissue, obesity, weight loss, and diabetes. Adv Nutr 2014;5:71–81.
Author information
Authors and Affiliations
Corresponding author
PowerPoint slides
Rights and permissions
About this article

Cite this article
van der Burg, J., Sen, S., Chomitz, V. et al. The role of systemic inflammation linking maternal BMI to neurodevelopment in children. Pediatr Res 79, 3–12 (2016). https://doi.org/10.1038/pr.2015.179
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2015.179
This article is cited by
-
Supplementation with antioxidant micronutrients in pregnant women with obesity: a randomized controlled trial
International Journal of Obesity (2024)
-
Association between telomere length and neuropsychological function at 4–5 years in children from the INMA project: a cross-sectional study
European Child & Adolescent Psychiatry (2024)
-
Is the association between mothers’ autistic traits and childhood autistic traits moderated by maternal pre-pregnancy body mass index?
Molecular Autism (2023)
-
Maternal weight, gut microbiota, and the association with early childhood behavior: the PREOBE follow-up study
Child and Adolescent Psychiatry and Mental Health (2023)
-
Maternal obesity, gestational diabetes mellitus, and diet in association with neurodevelopment of 2-year-old children
Pediatric Research (2023)
