
Abstract
We have taken a basic biologic approach to elucidate the pathophysiology of bronchopulmonary dysplasia (BPD), the chronic lung disease of prematurity, based on cell/molecular mechanisms of physiologic lung development. Stretch coordinates parathyroid hormone-related protein (PTHrP) signaling between the alveolar type II cell and the mesoderm to coordinately up-regulate key genes for the homeostatic fibroblast phenotype- including peroxisome proliferator activated receptor gamma (PPARγ), adipocyte differentiation related protein (ADRP), and leptin- and the retrograde stimulation of type II cell surfactant synthesis by leptin. Each of these paracrine interactions requires cell-specific receptors on adjacent cells derived from the mesoderm or endoderm, respectively, to serially up-regulate the signaling pathways between and within each cell-type. It is this functional compartmentation that is key to understanding how specific agonists and antagonists can predictably affect this mechanism of alveolar homeostasis. Using a wide variety of pathophysiologic insults associated with BPD- barotrauma, oxotrauma, and infection, we have found that there are type II cell and/or fibroblast cell/molecular effects generated by these insults, which can lead to the BPD phenotype. We have exploited these cell-specific mechanisms to effectively prevent and treat lung injuries using PPARγ agonists to sustain this signaling pathway. It is critically important to judiciously select physiologically and developmentally relevant interventions when treating the preterm neonate.
Similar content being viewed by others


Main
In the current reductionist era of genomics, proteomics, metabolomics, etc., it is important to bear in mind that genes can only be understood functionally within their biologic context (1). We cannot decipher complex diseases merely by identifying the genes that are associated with them without understanding how those genes relate to their pathophysiologic phenotypes. In this spirit, we will review a series of studies performed in our laboratory designed to determine the cell/molecular etiology and treatment of chronic lung disease based on an integrated model of lung development, homeostasis, and disease (2).
Lung morphogenesis and repair are characterized by a complex sequence of cell-cell interactions of endodermal and mesodermal origins, which has evolved an alveolar structure that can effectively exchange gases between the circulation and the alveolar space (3). By determining the spatio-temporal nature of this process, we can gain insights into the pathophysiology of a broad spectrum of chronic lung diseases caused by both intrinsic and extrinsic factors, bearing in mind that they descriptively funnel into just a few pathologies—epithelial, interstitial, and/or vascular.
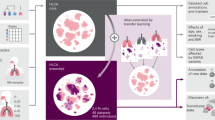
The global mechanisms that mediate mesenchymal-epithelial interactions and the plasticity of mesenchymal cells in normal lung development and remodeling provide a functional genomic model that offers insights to the cell/molecular basis for lung homeostasis (4–6), as depicted in Figure 1. Because we can effectively recapitulate the natural processes of development that determine normal lung structure, function, and homeostasis by this approach, we should be able to exploit this knowledge to understand lung pathophysiology and treat it effectively. With this concept in mind, we present a synopsis of work in our laboratory designed to identify such developmentally based mechanisms and how to use that knowledge for the safe, effective treatment of BPD, the chronic lung disease (CLD) of prematurity.

Schematic for paracrine determinants of alveolar homeostasis and disease. We have observed coordinating effects of stretch on alveolar type II cell expression of parathyroid hormone-related protein (PTHrP) (step 1), the lipofibroblast PTHrP receptor (step 2), its downstream effect on lipofibroblast ADRP expression (step 3), and triglyceride uptake (step 4), and on the interaction between lipofibroblast-produced leptin (step 5) and the alveolar type II cell leptin receptor (step 6), stimulating de novo surfactant phospholipid synthesis by alveolar type II cells (step 7). The schematic depicts lipofibroblast-to-myofibroblast transdifferentiation (step 8) due to decreased PTHrP after exposure to hyperoxia (O2), volutrauma (stretch), or infection (lipopolysaccharides). All of these effects are shown to be prevented by PPARγ agonists (step 9).
The pathophysiology of BPD involves interactions between multiple extrinsic and intrinsic factors, including oxygen free radicals, ventilator-induced lung injury, and release of inflammatory cytokines and cytotoxic enzymes such as proteases and elastases. Injury at early developmental stages leads to an arrest of both alveolar and vascular growth. The histopathological changes are characterized by fewer, larger alveoli and fewer capillaries, otherwise seen as alveolar simplification. Compared with the BPD first described by Northway in 1967, the “new” BPD is characterized by less airway injury, such as fibrosis, epithelial metaplasia, and scarring (6a).
BPD has been associated with perturbations in key factors associated with normal alveolarization, including retinoic acid (7), vascular endothelial growth factor (VEGF) (8), and parathyroid hormone-related protein (PTHrP) (9), suggesting dysregulation of the normal cellular processes of lung maturation. However, human trials of vitamin A supplementation have shown either no change or only a slight decrease in the incidence of BPD in infants delivered prematurely (10,11). Furthermore, although extensive animal data suggest an essential role of VEGF signaling in normal alveolarization, there are no human data suggesting its central involvement in BPD. We have focused on the role of PTHrP signaling in both normal and abnormal alveolar development because it is a stretch-regulated gene that is unusual among the alveolar paracrine growth factors that have been identified to mediate lung development [see Fig. 1] for a number of reasons: 1) the PTHrP knock-out is stage specific, resulting in failed alveolarization, which can be ‘rescued' with exogenous PTHrP (12); 2) unlike other such growth factors, PTHrP is expressed in the endoderm, and it binds to the mesoderm; 3) only PTHrP has been shown to act pleiotropically to integrate surfactant synthesis and alveolar capillary perfusion, i.e. alveolar homeostasis; 4) preterm newborns are PTHrP deficient (9). Below, we review the role of PTHrP in lung development, homeostasis and repair.
EPITHELIAL-MESENCHYMAL INTERACTIONS THAT ESTABLISH ALVEOLAR LUNG DEVELOPMENT
Our laboratory has used a developmental biologic approach to understand the etiology of CLD in newborns. We began with a paracrine growth factor model for the maturation of the pulmonary surfactant system based on classic mesenchymal-epithelial interactions (see Fig. 1, steps 1–7). These studies were originally fostered by Barry Smith's seminal observation (13) that glucocorticoids accelerate alveolar type II cell surfactant synthesis by stimulating fibroblast synthesis of an oligopeptide he termed fibroblast-pneumonocyte factor (FPF). It was known at that time that lung, prostate, and mammary mesodermal development were under endocrine control, and importantly, Kratochwil (14) had shown that early signals emanated from the epithelium to differentiate the immature mesenchyme in the neighboring epithelium. Moreover, Brody's laboratory had shown that the developing lung fibroblast acquired an adipocyte-like phenotype (15), termed the lipid-laden fibroblast, leaving open the question as to whether these cells might be a source of lipid substrate for surfactant synthesis by the alveolar type II cell. We discovered that co-culture of the lipid-laden fibroblasts with type II cells resulted in the trafficking of the lipid from the fibroblast to the type II cell and its highly enriched incorporation into surfactant phospholipids, particularly when treated with glucocorticoids, suggesting a specific, regulated mechanism for neutral lipid trafficking (16). Interestingly, the fibroblasts took up the neutral lipid, but did not release it unless they were in the presence of type II cells; conversely, the type II cells were unable to take up neutral lipid. These observations led to the discovery that type II cell secretion of prostaglandin E2 caused the release of neutral lipid from the fibroblasts (17), but the nature of the lipid uptake mechanism by the type II cells remained unknown. Yet, we were aware that the synthesis of pulmonary surfactant was an “on demand” system (18–20) in which increased respiration resulted in increased surfactant production, suggesting a stretch-sensitive signal from the type II cell. With this in mind, we began studying the role of PTHrP in lung development because a) it was expressed in the embryonic endoderm (21), b) its receptor was present on the adepithelial mesoderm (22), c) it had been shown to be stretch-regulated in the urinary bladder (23) and uterus (24), and distension of the lung was known to be of physiologic importance in normal lung development (25), and d) knock-out of PTHrP caused stage-specific inhibition of fetal lung alveolarization in the transition from the pseudoglandular to the canalicular stage (12).
Early functional studies of PTHrP had shown that it was a paracrine factor that stimulated surfactant phospholipid synthesis (26), and that it was stretch-regulated (27). We subsequently discovered that PTHrP stimulated neutral lipid uptake by the developing lung fibroblast, which we chose to call lipofibroblasts (28), by up-regulating ADRP, a molecule shown to be necessary for lipid uptake and storage (29). We subsequently found that ADRP was the factor necessary for the transit of neutral lipid from the lipofibroblast to the alveolar type II cell for surfactant phospholipid synthesis (30). The missing component for PTHrP regulation of lung surfactant was the lipofibroblast paracrine factor that stimulated surfactant synthesis. Reasoning that lipofibroblasts were similar to adipocytes, we hypothesized that lipofibroblasts would, like fat cells, express leptin, which would bind to the type II cell and stimulate surfactant synthesis. We found that lipofibroblasts do express leptin during rat lung development just before the onset of surfactant synthesis by the type II cell (31). Importantly, type II cells express the leptin receptor (31), thus providing a ligand-receptor signaling pathway between the lipofibroblast and type II cell. Moreover, PTHrP was discovered to stimulate leptin expression by fetal lung fibroblasts (32), thus providing a complete growth factor-mediated paracrine loop for the synthesis of pulmonary surfactant, as predicted by the PTHrP-based model of lung development.
Because the major inducers of BPD—barotrauma (33), oxotrauma (34), and infection (35)—cause alveolar type II cell injury and damage, we investigated the effects of PTHrP deprivation on the lipofibroblast phenotype, only to discover that in the absence of PTHrP, the lipofibroblast transdifferentiates to a myofibroblast, the cell-type that characterizes lung fibrosis. Furthermore, myofibroblasts cannot sustain type II cell growth and differentiation, whereas the lipofibroblast can (36), demonstrating the functional significance of these two fibroblast phenotypes for lung development; importantly, when myofibroblasts are treated with a PPARγ agonist they revert back to the lipofibroblast phenotype, including their ability to promote type II cell growth and differentiation.
These novel observations regarding the regulated development of the alveolus by paracrine growth factors have been exploited as a model for determining several cell/molecular causes of neonatal lung injury, including volutrauma, oxotrauma, and infection.
THE EFFECT OF VOLUTRAUMA ON PTHrP SIGNALING
The effective distension of the newborn lung has a profound physiologic effect on pulmonary homeostasis (37). Conversely, overdistension can almost instantaneously disrupt that homeostatic mechanism (refer to Fig. 1, step 8), resulting in volutrauma/atelectrauma (38). As indicated in the mechanism section above, the stretching of the alveolar type II cell increases the expression and production of PTHrP (27), potentially explaining why increased lung tidal volume stimulates pulmonary surfactant synthesis. And may explain why Wirtz and Dobbs (39) observed secretion of surfactant by isolated type II cells, but only after one stretching maneuver, without any effect of subsequent stretching maneuvers, perhaps due to the absence of lipid substrate from the neighboring fibroblast. In contrast to the increased synthesis of surfactant in response to mild stretch (3–5%), overdistension of the type II cell (40) results in down-regulation of PTHrP expression, simulating the consequences of volutrauma or atelectrauma. In other experiments, we found that when the type II cell contracts in the simulated microgravity created by a rotating wall bioreactor that PTHrP is also down-regulated (41), indicating that the set-point for the effect of stretch on type II cells is finely regulated and has likely evolved to effectively regulate surfactant synthesis (3).
EFFECT OF OXOTRAUMA ON PTHrP SIGNALING
In vitro.
Since lipofibroblasts play an important role in injury-repair mechanisms in the lung, we studied the effects of hyperoxia on the fibroblast phenotype in immature and relatively mature alveolar interstitial fibroblasts, and found that exposure to hyperoxia down-regulated PTHrP signaling, augmenting the transdifferentiation of pulmonary lipofibroblasts to myofibroblasts (refer to Fig. 1, step 8) (42). FRLF from e (embryonic, term = e22) 18 and e21 gestation were studied. After initial culture in Minimum Essential Medium (MEM) and 10% fetal bovine serum (FBS) in 21% O2/5% CO2 at 37°C, FRLF were maintained in MEM and 10%FBS at 37°C under control (21% O2/5% CO2) and experimental conditions (24-h exposure to 95% O2/5% CO2) at passages (P) 1 and P5. Passage 1 and 5 cells were analyzed for the expression of well-characterized lipogenic and myogenic markers (PTHrP receptor, ADRP, and αSMA) based on semi-quantitative competitive RT-PCR, triglyceride uptake, and leptin assay. Serial passaging and maintenance of cells in 21% O2 resulted in a significant spontaneous decrease in the expression of the lipogenic markers between P1 and P5. This decrease was greater for immature (e18) than for (near-mature) e21 FRLF. However, exposing cells to 95% O2 augmented the loss of the lipogenic markers and gain of the myogenic marker from P1 to P5 in comparison to cells maintained in 21% O2. This augmentation was also greater for e18 versus e21 lipofibroblasts. The changes in mRNA expression were accompanied by decreased triglyceride uptake and leptin secretion on exposure to hyperoxia. These data suggest that exposure to hyperoxia augments the transdifferentiation of pulmonary lipofibroblasts to myofibroblasts. Further, pretreatment with a PPARγ agonist, prostaglandin J2 (PGJ2), at least partially attenuated the hyperoxia-augmented lipo-to-myofibroblast transdifferentiation.
In an accompanying series of experiments (43), using [1,2-13C2]-D-glucose tracer and gas chromatography/mass spectrometry, we metabolically profiled e18 and e21 FRLF with and without hyperoxia exposure at passages 1, 4, 7, and 10. For this series of studies, glucose carbon redistribution between the nucleic acid ribose, lactate, and palmitate synthetic pathways and ADRP expression by RT-PCR were examined. Exposure to hyperoxia at each passage caused a decrease in ADRP mRNA expression. This passage-dependent transdifferentiation was accompanied by a moderate (9–20%) increase in the synthesis of nucleic acid ribose from glucose through the nonoxidative steps of the pentose cycle. E18 fibroblasts showed over an 85% decrease in the de novo synthesis of palmitate from glucose, while e21 fibroblasts showed a less pronounced 32–38% decrease in de novo lipid synthesis in hyperoxia-exposed cultures. From these data we conclude that 1) there is a maturation-dependent sensitivity to hyperoxia; 2) transdifferentiation of the lipofibroblast, as demonstrated by changes in ADRP expression, accompanied by metabolic enzyme changes affecting ribose synthesis from glucose; and 3) hyperoxia specifically inhibits lipogenesis from glucose. These molecular and metabolomic data were further complemented by genome-wide microarray analysis of RNA extracted from P1 and P10 e19 FRLF with or without exposure to hyperoxia (95% O2 for 24 h) (43a). In accord with our molecular and metabolomic data, cluster analysis of the microarray data confirmed the down-regulation of cholesterol and fatty acid synthetic genes and up-regulation of fatty acid degradation and Wnt signaling pathway genes on passaging e19 FRLF from P1 to P10, and on exposure to hyperoxia at passages 1 and 10, thereby confirming lipo-to-myofibroblast transdifferenation under these conditions. Based on these data, it appears that lipo-to-myofibroblast transdifferentiation may be an important mechanism for hyperoxia-induced lung injury and likely a key element in the pathophysiology of BPD, allowing us to speculate that induction of adipogenic transcription factors may not only prevent, but in fact may revert the myogenic fibroblast phenotype back to the adipogenic fibroblast phenotype.
In vivo.
Following our in vitro studies, as outlined above, we next determined whether upon exposure to hyperoxia, pulmonary alveolar lipo-to-myofibroblast transdifferentiation occurs in vivo, and whether treatment with a potent PPARγ (the key lipogenic fibroblast nuclear transcription factor) agonist, RGZ, would prevent this process (see Fig. 1, step 9) (45). Newborn Sprague-Dawley rat pups were exposed to hyperoxia alone (95% O2 for 24 h), or hyperoxia with RGZ (95% O2 for 24 h + RGZ, 3 mg/kg, administered intraperitoneally) conditions.
We observed a significant decrease in the expression of lipogenic markers, and a significant increase in the expression of myogenic markers in the hyperoxia alone group. Exposure of rat pups to 24 h of hyperoxia dramatically affected normal postnatal lung development. Histologic examination of lungs obtained from hyperoxic pups revealed a remarkable arrest of alveolarization compared with their normoxic littermate controls. Hyperoxia-exposed lungs demonstrated relatively large air spaces, thinned interstitia, and decreased secondary septal crests compared with air-exposed controls. In lungs from hyperoxic animals, septal thickness was reduced significantly (40%) compared with control lungs. Furthermore, quantitative analysis of alveolar number demonstrated a significant reduction (50%) in average radial alveolar counts in hyperoxic animals compared with controls. Pretreatment with RGZ virtually prevented the hyperoxia-induced changes in lung morphology, including the effects on septal thickness and radial alveolar counts. The hyperoxia-induced morphologic, molecular, and immunohistochemical changes were virtually prevented by RGZ. This is the first evidence of in vivo lipo-to-myofibroblast transdifferentiation and its virtual prevention by RGZ, prompting us to conclude that administration of PPARγ agonists may be a novel, effective strategy to prevent the hyperoxia-induced lung molecular injury that has been implicated in the pathogenesis of BPD.
THE EFFECT OF LIPOPOLYSACCHARIDE ON PTHrP SIGNALING
Because lung inflammation is a key factor that predisposes preterm infants to BPD, in a series of studies (45) we determined the effects of lipopolysaccharide (LPS) on PTHrP-driven pulmonary epithelial-mesenchymal interactions that have been shown by us to be essential in the maintenance of lung homeostasis (refer to Fig. 1). Lung explants derived from e19.5 Sprague-Dawley rat pups were treated with LPS (0–50 ng/mL) with or without a PTHrP pathway agonist, PGJ2, for up to 72 h. LPS treatment affected the expression of the key markers of the epithelial-mesenchymal paracrine loop in a dose- and time-dependent manner. There were acute (24 h), significant increases in the expression of PTHrP, PPARγ, ADRP, and surfactant protein-B (SP-B), without any significant effects on the expression of αSMA. This was followed (72 h) by significant decreases in the expression of PTHrP, PPARγ, ADRP, and SP-B, accompanied by a significant increase in the expression of αSMA, the key functional marker for BPD.
As in our in vivo hyperoxia model, treatment with a specific agonist of epithelial-mesenchymal interactions prevented the inflammation-induced molecular lung injury that is known to result in BPD. Although these data suggest a possible mechanism for the acute stimulation of lung maturation, accompanied paradoxically by BPD following intrauterine inflammation, whether this actually occurs in human BPD following intrauterine inflammation remains to be determined.
PTHrP SIGNALING IN BPD
Because PTHrP secreted by pulmonary alveolar type II cells is a key physiologic paracrine factor in maintaining alveolar homeostasis (see Fig. 1, steps 1–8), we hypothesized that its levels in the tracheal aspirates (TA) of ventilated VLBWI would predict the development of BPD (9). We examined whether TA PTHrP content during the first week of life correlated with the later development of BPD. Forty VLBWI [birth weight 943 ± 302 g (mean ± SD); gestational age 27 ± 2 wk; 21 males and 19 females], who were ventilated for respiratory distress syndrome, were studied. The TA were collected once daily until the infants were extubated, and were assayed for PTHrP. The levels of TA PTHrP were compared with the later development of BPD. PTHrP in the TA during the first week of life was significantly lower in those infants who developed BPD (12/40) than among those who did not (28/40). The PTHrP levels also correlated with the duration of mechanical ventilation needed in these infants. A PTHrP level of ≤1.32 pg/mg protein predicts the later development of BPD maximally [84.6% correct classifications (true positives + true negatives)], with a sensitivity of 76.9% and specificity of 88.5%. Using a TA PTHrP level of 1.32 pg/mg protein as the cutoff, we constructed Kaplan-Meier curves to compare the duration of ventilation needed between the two groups, i.e. ≤ and > than 1.32 pg/mg protein TA PTHrP level. Infants with TA PTHrP levels greater than 1.32 pg/mg protein were off ventilatory support significantly earlier. To determine how PTHrP levels compared with the other known predictors of BPD, such as birth weight (<1000 g), GA (<28 wk), and male gender, we performed multivariate logistic regression analysis for these 4 variables in predicting the development of BPD. Of these variables, a PTHrP level of ≤1.32 pg/mg protein was the strongest predictor of BPD, and remained so after adjusting for the other three variables, i.e. birth weight of <1000 g, GA of <28 wk, and male gender. From these data, we concluded that lower TA PTHrP content during the first week of life in ventilated VLBWI inversely correlates with prolonged ventilation and the later development of BPD.
PREVENTION OF MOLECULAR LUNG INJURIES LEADING TO BPD WITH PPARγ AGONISTS
It is clear from the work outlined above that we have systematically demonstrated the central role of epithelial-mesenchymal interactions in maintaining alveolar homeostasis in volutrauma, oxotrauma, and infection-mediated lung injury. The lipofibroblast expresses both ADRP and leptin in response to PTHrP signaling from the alveolar type II cell, resulting in direct protection of the mesoderm against oxidant injury (46), and protection against atelectasis by augmenting surfactant phospholipid (26) and protein (32) synthesis. Molecular injury to either the alveolar type II cell or the lipofibroblast down-regulates this molecular signaling pathway, causing myofibroblast transdifferentiation (see Schematic, step 8). Unlike lipofibroblasts, myofibroblasts cannot promote alveolar type II cell growth and differentiation (36), leading to the failed alveolarization characteristic of BPD (47). PPARγ is the key nuclear transcription factor that determines the lipofibroblastic phenotype, which supports alveolar type II cell growth and differentiation under the influence of factors implicated in the pathogenesis of BPD (36). We have shown that a wide variety of factors associated with failed alveolarization- volutrauma, oxotrauma, and infection- all cause myofibroblast transdifferentiation in vitro (36,42,43,45) and in vivo (44). More importantly, we have shown that PPARγ agonists such as PGJ2 and RGZ can prevent or rescue myofibroblast transdifferentiation (see Fig. 1, step 9), potentially preventing the inhibition of alveolarization in the developing lung (36,42,44,45).
CONCLUSIONS
We have taken a basic biologic approach to elucidate the pathophysiology of BPD based on cell physiologic principles. We have determined the paracrine cell/molecular mechanism by which stretch coordinates PTHrP signaling between the alveolar type II cell and the mesoderm to up-regulate key genes for the homeostatic fibroblast phenotype, including PPARγ, ADRP, and leptin, and the retrograde stimulation of alveolar type II cell surfactant phospholipid and protein synthesis by leptin. Each of these paracrine interactions requires cell-specific receptors on adjacent cells derived from the endoderm or mesoderm, respectively, i.e. PTHrP receptors on the mesoderm, and leptin receptors on the endoderm, to mediate the signaling pathways within each cell-type. It is this functional compartmentalization that is the key to understanding how specific agonists and antagonists predictably affect this mechanism of alveolar homeostasis. More importantly, it is the cell-specific molecular nature of this mechanism that we have exploited to effectively prevent and treat lung injuries that affect this signaling pathway.
This work clearly demonstrates that it is critically important to judiciously select physiologically and developmentally relevant interventions when treating the developing neonate.
Abbreviations
- ADRP:
-
adipocyte differentiation related protein
- αSMA:
-
alpha smooth muscle actin
- BPD:
-
bronchopulmonary dysplasia
- FRLF:
-
fetal rat lung fibroblast
- PGJ2:
-
prostaglandin J2
- PPARγ:
-
peroxisome proliferator activated receptor gamma
- RGZ:
-
rosiglitazone
- VLBWI:
-
very low birth weight infant
References
Newman SA 2002 Developmental mechanisms: putting genes in their place. J Biosci 27: 97–104
Torday J, Rehan VK 2007 The evolutionary continuum from lung development to homeostasis and repair. Am J Physiol Lung Cell Mol Physiol 292: L608–L611
Torday JS, Rehan VK 2004 Deconvoluting lung evolution using functional/comparative genomics. Am J Respir Cell Mol Biol 31: 8–12
Warburton D, Bellusci S 2004 The molecular genetics of lung morphogenesis and injury repair. Paediatr Respir Rev 5: S283–S287
Desai TJ, Cardoso WV 2002 Growth factors in lung development and disease: friends or foe?. Respir Res 3: 2
Shannon JM, Hyatt BA 2004 Epithelial-mesenchymal interactions in the developing lung. Annu Rev Physiol 66: 625–645
Northway WH Jr, Rosan RC, Porter DY 1967 Pulmonary disease following respirator therapy of hyaline-membrane disease Bronchopulmonary dysplasia. N Engl J Med 16; 276(7): 357–368
Zachman RD 1995 Role of vitamin A in lung development. J Nutr 125: 1634S–1638S
Thebaud B, Ladha F, Michelakis ED, Sawicka M, Thurston G, Eaton F, Hashimoto K, Harry G, Haromy A, Korbutt G, Archer SL 2005 Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury: evidence that angiogenesis participates in alveolarization. Circulation 112: 2477–2486
Rehan VK, Torday JS 2006 Lower parathyroid hormone-related protein content of tracheal aspirates in very low birth weight infants who develop bronchopulmonary dysplasia. Pediatr Res 60: 216–220
Pearson E, Bose C, Snidow T, Ransom L, Young T, Bose G, Stiles A 1992 Trial of vitamin A supplementation in very low birth weight infants at risk for bronchopulmonary dysplasia. J Pediatr 121: 420–427
Tyson JE, Wright LL, Oh W, Kennedy KA, Mele L, Ehrenkranz RA, Stoll BJ, Lemons JA, Stevenson DK, Bauer CR, Korones SB, Fanaroff AA 1999 Vitamin A supplementation for extremely-low-birth-weight infants. National Institute of Child Health and Human Development Neonatal Research Network. N Engl J Med 340: 1962–1968
Rubin LP, Kovacs CS, De Paepe ME, Tsai SW, Torday JS, Kronenberg HM 2004 Arrested pulmonary alveolar cytodifferentiation and defective surfactant synthesis in mice missing the gene for parathyroid hormone-related protein. Dev Dyn 230: 278–289
Smith BT 1979 Lung maturation in the fetal rat: acceleration by injection of fibroblast-pneumonocyte factor. Science 204: 1094–1095
Heuberger B, Fitzka I, Wasner G, Kratochwil K 1982 Induction of androgen receptor formation by epithelium-mesenchyme interaction in embryonic mouse mammary gland. Proc Natl Acad Sci U S A 79: 2957–2961
Vaccaro C, Brody JS 1978 Ultrastructure of developing alveoli. I. The role of the interstitial fibroblast. Anat Rec 192: 467–479
Torday J, Hua J, Slavin R 1995 Metabolism and fate of neutral lipids of fetal lung fibroblast origin. Biochim Biophys Acta 1254: 198–206
Torday JS, Sun H, Qin J 1998 Prostaglandin E2 integrates the effects of fluid distension and glucocorticoid on lung maturation. Am J Physiol 274: L106–L111
Faridy EE, Permutt S, Riley RL 1966 Effect of ventilation on surface forces in excised dogs' lungs. J Appl Physiol 21: 1453–1462
Wyszogrodski I, Kyei-Aboagye K, Taeusch HW Jr Avery ME 1975 Surfactant inactivation by hyperventilation: conservation by end-expiratory pressure. J Appl Physiol 38: 461–466
Oyarzun MJ, Clements JA, Baritussio A 1980 Ventilation enhances pulmonary alveolar clearance of radioactive dipalmitoyl phosphatidylcholine in liposomes. Am Rev Respir Dis 121: 709–721
Hastings RH, Duong H, Burton DW, Deftos LJ 1994 Alveolar epithelial cells express and secrete parathyroid hormone-related protein. Am J Respir Cell Mol Biol 11: 701–706
Lee K, Deeds JD, Segre GV 1995 Expression of parathyroid hormone-related peptide and its receptor messenger ribonucleic acids during fetal development of rats. Endocrinology 136: 453–463
Yamamoto M, Harm SC, Grasser WA, Thiede MA 1992 Parathyroid hormone-related protein in the rat urinary bladder: a smooth muscle relaxant produced locally in response to mechanical stretch. Proc Natl Acad Sci U S A 89: 5326–5330
Daifotis AG, Weir EC, Dreyer BE, Broadus AE 1992 Stretch-induced parathyroid hormone-related peptide gene expression in the rat uterus. J Biol Chem 267: 23455–23458
Alcorn D, Adamson TM, Lambert TF, Maloney JE, Ritchie BC, Robinson PM 1977 Morphological effects of chronic tracheal ligation and drainage in the fetal lamb lung. J Anat 123: 649–660
Rubin LP, Kifor O, Hua J, Brown EM, Torday JS 1994 Parathyroid hormone (PTH) and PTH-related protein stimulate surfactant phospholipid synthesis in rat fetal lung, apparently by a mesenchymal-epithelial mechanism. Biochim Biophys Acta 1223: 91–100
Torday JS, Sanchez-Esteban J, Rubin LP 1998 Paracrine mediators of mechanotransduction in lung development. Am J Med Sci 316: 205–208
McGowan SE, Torday JS 1997 The pulmonary lipofibroblast (lipid interstitial cell) and its contributions to alveolar development. Annu Rev Physiol 59: 43–62
Gao J, Serrero G 1999 Adipose differentiation related protein (ADRP) expressed in transfected COS-7 cells selectively stimulates long chain fatty acid uptake. J Biol Chem 274: 16825–16830
Schultz CJ, Torres E, Londos C, Torday JS 2002 Role of adipocyte differentiation-related protein in surfactant phospholipid synthesis by type II cells. Am J Physiol Lung Cell Mol Physiol 283: L288–L296
Torday JS, Sun H, Wang L, Torres E, Sunday ME, Rubin LP 2002 Leptin mediates the parathyroid hormone-related protein paracrine stimulation of fetal lung maturation. Am J Physiol Lung Cell Mol Physiol 282: L405–L410
Torday JS, Rehan VK 2002 Stretch-stimulated surfactant synthesis is coordinated by the paracrine actions of PTHrP and leptin. Am J Physiol Lung Cell Mol Physiol 283: L130–L135
Coalson JJ 1997 Experimental models of bronchopulmonary dysplasia. Biol Neonate 71: 35–38
Davis JM 2002 Role of oxidant injury in the pathogenesis of neonatal lung disease. Acta Paediatr Suppl 91: 23–25
Jobe AH 2005 Antenatal associations with lung maturation and infection. J Perinatol 25: S31–S35
Torday JS, Torres E, Rehan VK 2003 The role of fibroblast transdifferentiation in lung epithelial cell proliferation, differentiation, and repair in vitro. Pediatr Pathol Mol Med 22: 189–207
Scarpelli EM 1984 Perinatal lung mechanics and the first breath. Lung 162: 61–71
Ikegami M, Kallapur S, Michna J, Jobe AH 2000 Lung injury and surfactant metabolism after hyperventilation of premature lambs. Pediatr Res 47: 398–404
Wirtz HR, Dobbs LG 1990 Calcium mobilization and exocytosis after one mechanical stretch of lung epithelial cells. Science 250: 1266–1269
Sanchez-Esteban J, Tsai SW, Sang J, Qin J, Torday JS, Rubin LP 1998 Effects of mechanical forces on lung-specific gene expression. Am J Med Sci 316: 200–204
Torday JS 2003 Parathyroid hormone-related protein is a gravisensor in lung and bone cell biology. Adv Space Res 32: 1569–1576
Rehan V, Torday J 2003 Hyperoxia augments pulmonary lipofibroblast-to-myofibroblast transdifferentiation. Cell Biochem Biophys 38: 239–250
Boros LG, Torday JS, Paul Lee WN, Rehan VK 2002 Oxygen-induced metabolic changes and transdifferentiation in immature fetal rat lung lipofibroblasts. Mol Genet Metab 77: 230–236
Dasgupta C et al. 2006 Western Society for Pediatric Research, Carmel, CA. J Invest Med 54: S168
Rehan VK, Wang Y, Patel S, Santos J, Torday JS 2006 Rosiglitazone, a peroxisome proliferator-activated receptor-gamma agonist, prevents hyperoxia-induced neonatal rat lung injury in vivo. Pediatr Pulmonol 41: 558–569
Rehan VK, Dargan-Batra SK, Wang Y, Cerny L, Sakurai R, Santos J, Beloosesky R, Gayle D, Torday JS 2007 A paradoxical temporal response of PTHrP/PPARγ signaling pathway to lipopolysaccharide in an in vitro model of the developing rat lung. Am J Physiol Lung Cell Mol Physiol Apr 13; [Epub ahead of print]
Torday JS, Torday DP, Gutnick J, Qin J, Rehan V 2001 Biologic role of fetal lung fibroblast triglycerides as antioxidants. Pediatr Res 49: 843–849
Jobe AJ 1999 The new BPD: an arrest of lung development. Pediatr Res 46: 641–643
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported by grants from the American Heart Association (grant no. 0265127), the National Institutes of Health (grant nos. HL55268 and HL75405), the March of Dimes Foundation, Philip Morris USA Inc. and Philip Morris International, and the Tobacco-Related Disease Research Program (grant nos. 14RT-0073 and 15IT-0250).
Rights and permissions
About this article
Cite this article
Torday, J., Rehan, V. Developmental Cell/Molecular Biologic Approach to the Etiology and Treatment of Bronchopulmonary Dysplasia. Pediatr Res 62, 2–7 (2007). https://doi.org/10.1203/PDR.0b013e31806772a1
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e31806772a1
This article is cited by
-
Adult onset lung disease following transient disruption of fetal stretch-induced differentiation
Respiratory Research (2009)
-
CFTR and Wnt/beta-catenin signaling in lung development
BMC Developmental Biology (2008)
