
Abstract
The discovery of somatic mutations in the isocitrate dehydrogenase (IDH) enzymes through a genome-wide mutational analysis in glioblastoma represents a milestone event in cancer biology. The nature of the heterozygous, point mutations mapping to arginine residues involved in the substrate binding inspired several research teams to investigate their impact on the biochemical activity of these enzymes. Soon, it became clear that the mutations identified impaired the ability of IDH1 and IDH2 to catalyze the conversion of isocitrate to α-ketoglutarate (αKG), whereas conferring a gain of a novel enzymatic activity leading to the reduction of αKG to the metabolite D2-hydroxyglutarate (D-2HG). Across glioma as well as several hematologic malignancies, mutations in IDH1 and IDH2 have shown prognostic value. Several hypotheses implicating the elevated levels of D-2HG and tumorigenesis, and the therapeutic potential of targeting mutant IDH enzymes will be discussed.
Similar content being viewed by others


Introduction
Malignant transformation is most often a multistep process in which a series of genetic, epigenetic and post-transcriptional events ultimately confer emerging neoplastic cells with a growth and survival advantage (Vogelstein and Kinzler, 1993; Hanahan and Weinberg, 2000; Jones and Baylin, 2002). It is becoming increasingly clear that metabolic adaptation is an integral aspect of this process (Gillies and Gatenby, 2007; Kroemer and Pouyssegur, 2008). Fast proliferating cancer cells meet their elevated energetic and anabolic demands by preferentially ‘feasting’ on certain carbohydrates, lipids and amino acids and resorting to unique metabolic strategies. In fact, the elevated glucose avidity of hematologic malignancies and solid tumors has been widely exploited for diagnostic purposes. But only more recently researchers have begun to appreciate the far reaching implications of metabolic alterations in malignant cells, and their potential contribution to initiation and development of cancer. In addition to their intermediary role in energy and biomass producing reactions, certain privileged metabolites have key roles as allosteric regulators and cofactors of enzymes involved in fundamental processes including chromatin remodeling, mitochondrial respiration, angiogenesis and migration. Furthermore, the same molecular mechanisms leading to uncoupling of cell proliferation and survival from growth factor signaling and other extracellular cues, also contribute to metabolic rewiring.
It has long been recognized that the aberrant activity of proto-oncogenes and tumor suppressors affect the expression and activity of nutrient transporters and metabolic enzymes (Dang and Semenza, 1999; Kroemer and Pouyssegur, 2008; Dang et al., 2009; Vander Heiden et al., 2009). However, the first examples of mutations in genes coding for mitochondrial enzymes involved in metabolism have only come to light in the past few years. Those include mutations in fumarate hydratase (FH) and succinate dehydrogenase (SDH) in hereditary leiomyomatosis and renal cell carcinoma as well as paragangliomas (Baysal et al., 2000; Tomlinson et al., 2002; Brandon et al., 2006; King et al., 2006; Kaelin, 2009). A mutation in the isocitrate dehydrogenase 1 (IDH1) gene was first discovered during a sequencing effort across colorectal cancers (Sjöblom et al., 2006). More recently, somatic mutations in IDH1 were identified through a genome wide mutation analysis in glioblastoma (Parsons et al., 2008). Following this pivotal study, high throughput deep sequencing revealed the presence of mutations in either IDH1 or its mitochondrial counterpart IDH2 in >70% of grade II–III gliomas and secondary glioblastomas (GBMs; Yan et al., 2009). Since these initial reports, mutations in IDH1 and IDH2 have been identified in ∼16–17% of patients with acute myeloid leukemia, and spotted in a variety of other malignancies at lower frequencies (Abbas et al., 2010; Paschka et al., 2010). Unlike the loss-of-function mutations in FH and SDH genes, the IDH1 and IDH2 mutations described to date confer a neoactivity to their enzyme products (Dang et al., 2009; Mardis et al., 2009; Gross et al., 2010; Ward et al., 2010). This review will attempt to summarize our current understanding of the recently identified mutations in IDH1 and IDH2 and provide several potential molecular mechanisms linking them to malignant transformation.
Biochemistry of IDHs
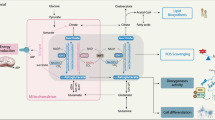
The family of IDHs consists of three members: IDH1, IDH2 and IDH3. All three enzymes catalyze the oxidative decarboxylation of isocitrate to produce CO2 and α-ketoglutarate (αKG). These reactions require the presence of a divalent ion (Mg2+ or Mn2+) and the cofactors NADP+ (IDH1 and IDH2) or NAD+ (IDH3) as electron acceptors, generating nicotinamide adenine dinucleotide phosphate (NADPH) or NADH respectively. The NADP+-dependent family members, IDH1 and IDH2, are homodimeric enzymes that share significant structural similarity. The human proteins are 69% identical and crystallography studies have shown that the overall quaternary structure is nearly identical between the two proteins (Ceccarelli et al., 2002; Xu et al., 2004). One fundamental difference between IDH1 and IDH2 is the subcellular localization of the two proteins. IDH1 contains a C-terminal peroxisomal localization sequence and is localized in both peroxisomes and cytosol (Geisbrecht and Gould, 1999; Yoshihara et al., 2001). IDH2 and IDH3 are mitochondrial isoforms of IDH (Figure 1a). Unlike the NADP+-dependent enzymes, the IDH3 holoenzyme is a heterotetrameric protein consisting of two subunits of IDH3A and one subunit each of IDH3B and IDH3G. To date, there have been no reports of cancer-associated mutations in any of the IDH3 subunits. In yeast, peroxisomal IDH is important for maintaining NADPH levels that are essential for the metabolism of branched chain fatty acids (Henke et al., 1998; van Roermund et al., 1998). Although NAD+-dependent IDH3 is believed to be the principal IDH enzyme in the tricarboxylic acid cycle, the exact roles of IDH1 and IDH2 in cellular metabolism have been less well defined.

Metabolic reactions catalyzed by the IDH family. (a) IDH1 catalyzes the NADP+-dependent decarboxylation of isocitrate to αKG in the peroxisome and cytoplasm, whereas IDH2 mediates the same reaction in the mitochondria. Also, within the mitochondrial compartment, heterotetrameric IDH3 catalyzes the conversion of isocitrate to αKG in a NADP+-dependent manner. (b) Mutations in IDH1 and IDH2 impart these enzymes the ability to catalyze the reduction of αKG to 2HG. On the basis of the chemical similarity between these two metabolites, it is possible that the activity of αKG-dependent enzymes is affected by the elevated level of 2HG in tumors harboring IDH1 and IDH2 mutations. In this model, these events may trigger alterations in angiogenesis, epigenetic state, extracellular matrix dynamics, and in turn may affect proliferation, survival and invasive properties of these tumors. Cit, citrate; Fum, fumarate; Mal, malate; OAA, oxaloacetate; Suc, succinate.
The oxidation of isocitrate to αKG by IDH is widely viewed as one of the irreversible enzymatic reactions in the central metabolism. This conversion involves a large free energy change associated with the production of CO2. On binding of isocitrate and NADP+, IDH1 shifts from an open conformation to a catalytically active closed conformation (Xu et al., 2004). The first step in the enzymatic reaction is the oxidation of isocitrate to oxalosuccinate with the concomitant reduction of NADP+ to NADPH. Enzyme-bound oxalosuccinate then undergoes decarboxylation to form αKG that is then released as the enzyme transitions back to the open conformation in preparation for another round of catalysis.
Neoactivity of IDH1/IDH2 mutant enzymes
As mentioned earlier, somatic mutations in IDH1 and IDH2 were initially identified in low-grade gliomas and secondary GBMs. Those reports established the presence of a heterozygous mutation affecting Arg132 in IDH1as well as the equivalent residue, Arg172, in IDH2, albeit at lower frequency. Since Arg132 together with Arg100 and Arg109 are part of a catalytic arginine triad involved in isocitrate binding, it was immediately hypothesized that these mutations would affect IDH1 enzymatic activity (Yan et al., 2009; Figure 2). Initial biochemical studies established that mutant IDH1/IDH2 were unable to efficiently catalyze the oxidative decarboxylation of isocitrate, consistent with the mutations resulting in a loss of enzymatic activity. A subsequent report suggested that a single mutant subunit within the enzyme dimer could exert a dominant negative effect (Zhao et al., 2009). In this case, purified heterodimers of wild-type and mutant IDH1 were unable to efficiently catalyze the conversion of NADP+ to NADPH when isocitrate was provided as a substrate.

Localization of mutated amino acid residues in IDH1. Active site of wild-type IDH1, showing in green amino acid residues Arg 132, Gly97, Val71 and Gly123 reported to be mutated in cancer. Amino acids Arg 100 and Arg 109 (in blue) together with Arg 132 (corresponding to Arg172 on IDH2) form the catalytic triad that coordinates isocitrate (red). The cofactor NADP+ is depicted in cyan. In AML, the frequently mutated Arg140 on IDH2 corresponds to Arg100 on IDH1.
Recently, we demonstrated that mutant IDH1 and IDH2 are not catalytically inactive enzymes, but rather possess novel enzymatic activities, reconciling the proposed gain-of-function model based on the heterozygous nature of the point mutations (Dang et al., 2009). Untargeted metabolic profiling by liquid chromatography-mass spectrometry identified one metabolite, 2-hydroxyglutarate (2HG), that was elevated over 100-fold in U87-MG cells expressing mutant IDH1 relative to the same cells expressing wild-type IDH1. The chemical similarity of 2-HG to αKG, in combination with observations from isotope labeling experiments in whole cells showing carbon flow from αKG to 2HG, led our group to immediately hypothesize that mutant IDH1 could catalyze a novel enzymatic reaction in which the reduction of αKG to 2HG is coupled to the oxidation of NADPH to NADP+. Indeed, our studies established that purified mutant protein efficiently catalyzed the proposed reduction of αKG to 2HG, while being unable to synthesize isocitrate (Dang et al., 2009). There are two possible enantiomers of 2HG because of the presence of a chiral center in this molecule, and mutant IDH1 produces exclusively R(–)-2HG (also commonly called D-2HG) as established by chemical derivatization of the product. Supporting X-ray crystallography studies with IDH1R132H showed that within the active site, αKG and NADPH are positioned as expected for the reduction of the substrate to the expected enantiomer. Overall, the enzymatic characterization of mutant enzyme, suggests that the enzymatic gain-of-function is likely driven by both the loss in affinity for isocitrate as well as significant increases in affinity for αKG and NADPH (Dang et al., 2009).
Supporting the in vitro biochemical data and the observations in engineered cell lines, D-2HG levels were dramatically elevated in malignant gliomas harboring IDH1 and IDH2 mutations. In a panel of human secondary GBMs samples, tumors with IDH1 mutations contained up to ∼100-fold higher levels of D-2HG than tumors harboring wild-type IDH1 (Dang et al., 2009). Two subsequent studies in acute myeloid leukemia (AML) also found elevated levels of D-2HG in cells with mutant IDH1 and IDH2 as compared with cells with wild-type alleles (Gross et al., 2010; Ward et al., 2010). To date IDH1R132, IDH2172 and IDH2R140 mutant enzymes of IDH1 and IDH2 have shown the ability to synthesize D-2HG (Dang et al., 2009; Gross et al., 2010; Ward et al., 2010). The fact that IDH1 and IDH2 mutant enzymes possess the ability to catalyze the partial reverse reaction, suggests that this gain-of-function activity is the relevant consequence of IDH mutations in cancer.
Loss-of-heterozygosity at the IDH locus is very rarely observed in tumors with IDH1 mutations (Zhao et al., 2009). It is likely that wild-type enzymatic activity is essential for tumor initiation or progression, possibly by potentiating 2HG production by forming heterodimers with mutant subunits. Although heterodimers of mutant and wild-type IDH subunits have been purified and initially characterized (Zhao et al., 2009), the enzymatic activity was only indirectly monitored by measuring steady state NADP/NADPH ratios. It will be important to evaluate the ability of these heterodimers to produce D-2HG. If both subunits of the heterodimers retain their respective enzymatic activities, it is possible that the heterodimeric enzyme is more efficient than the mutant homodimeric enzyme in catalyzing the partial reverse reaction. The close proximity of the two active sites could lead to substrate channeling or local concentration effects that would drive the production of D-2HG from αKG. Alternatively, if cells heterozygous for IDH mutations produce sufficient levels of D-2HG to drive tumorigenesis, irrespective of the presence of heterodimeric enzyme, there may simply be no selective pressure to drive loss-of-heterozygosity. It is equally possible that cancer cells require wild-type IDH enzymatic activity in order to remain viable. As future studies define the role of mutant enzyme and D-2HG in tumorigenesis, the exact role of wild-type IDH in these tumors will likely become apparent.
Incidence of IDH1/IDH2 mutations and correlations to other genetic alterations
Following the initial discovery of IDH1 and IDH2 mutations in low-grade gliomas and secondary GBMs, subsequent sequencing efforts revealed alterations in these two genes across additional cancers including acute myeloid leukemia, prostate, B-acute lymphoblastic leukemia and others (Yan et al., 2009). In gliomas and AML, mutations in IDH1 and IDH2 were mutually exclusive of one another and occur at very early stages of tumor development suggesting that the generation of D-2HG may somehow function as an oncometabolite and promote formation and progression of tumors (Watanabe et al., 2009; Pardanani et al., 2010b).
I. Cancers of the central nervous system
As introduced in previous sections, in low-grade gliomas and secondary GBMs, heterozygous IDH1 and IDH2 mutations occur at a single arginine residue, Arg132 of IDH1, and the analogous Arg172 residue of IDH2. Mutations in IDH1 and IDH2 are mutually exclusive, suggesting that they independently confer a growth advantage to mutant cells (Hartmann et al., 2009). Proliferation of poorly differentiated gliomas with mixed astrocytic and oligodendroglial features is key to the progression of the disease and their pathogenesis may be related to a block in differentiation rather than merely just an increase in cell proliferation (Ward et al., 2010). This observation raises the possibility that D-2HG may interfere with processes involved in cellular differentiation, and ongoing studies will shed light on this question. Patients that harbor these mutations tend to be of younger age and have a better clinical prognosis.
Understanding the link between IDH1/IDH2 mutations and other common genetic events in gliomas may provide further insights into their contribution to pathogenesis (Yan et al., 2009; Verhaak et al., 2010). In general, TP53 mutations are associated more frequently with low-grade astrocytomas and secondary GBMs compared with primary GBMs. Within these lower grade tumors, TP53 mutations are more frequent across diffuse astrocytomas (74%), anaplastic astrocytomas (65%) and secondary GBMs (62%), whereas they are less frequent in oligodendrogliomas and anaplastic oligodendrogliomas, with an overall prevalence of 16–9%, respectively. Conversely, deletions in 1p and 19q are found more often in oligodendroglial tumors than in astrocytic tumors. Interestingly, in these two tumor types IDH1/IDH2 mutations are found at relatively similar frequency (60–100%; Yan et al., 2009). Unlike the clear association between TP53 loss, 1p19q loss and tumor lineage, IDH1/IDH2 mutations may contribute to the pathogenesis of both astrocytic and oligodendroglial tumors. Noteworthy, sequential biopsies from multiple patients showed no cases in which an IDH1 mutation occurred after the acquisition of a TP53 mutation or loss of 1p/19q suggesting that acquisition of IDH1 mutations are early events in gliomagenesis and may affect a common glial precursor cell (Hartmann et al., 2009; Watanabe et al., 2009).
High-grade primary GBMs are often associated with activating mutations or amplification of epidermal growth factor receptor and PTEN (phosphatase and tensin homolog) deletion (Voelzke et al., 2008; Huang et al., 2009). Analysis of the relationship between genetic alterations in epidermal growth factor receptor and IDH1/IDH2 in GBMs revealed that amplification of EGFR and IDH1/IDH2 mutations are mutually exclusive events (Sanson et al., 2009). Similarly, genomic profiling of 200 GBMs and two normal brain samples has defined four subtypes of tumors with a common morphologic diagnosis of GBM. Consistent with earlier reports, IDH1 mutations are most commonly found in the proneural class along with TP53 mutations, whereas PTEN, neurofibromin 1 (NFI) and epidermal growth factor receptor alterations are more commonly associated with classical and mesenchymal subclasses (Verhaak et al., 2010). Finally, hypermethylation of a large number of loci has revealed the existence of a glioma–CpG island methylator phenotype that belongs to the proneural subgroup of gliomas (Noushmehr et al., 2010). In particular, methylation of the 06-Methylguanine-DNA Methyltransferase (MGMT) gene promoter confers a better prognosis for patients and is often associated with IDH1 and IDH2 mutations (Sanson et al., 2009; Noushmehr et al., 2010). Whether this is a direct consequence of the activity of mutant IDH or a surrogate marker for epigenetic changes in tumors harboring IDH1/IDH2 mutation remains to be determined.
IDH1 mutations (R132H) have also been found in up to 33% of rare, aggressive adult supratentorial primitive neuroectodermal tumors of the cerebral hemispheres (Balss et al., 2008; Hayden et al., 2009). The fact that none of the supratentorial primitive neuroectodermal tumors arising in the infratentorial compartment (that is, medulloblastomas) contain IDH1 mutations further supports the idea that the supratentorial and infratentorial compartments have distinct mechanisms of disease pathogenesis despite having nearly identical histopathology. These findings may help to stratify patients with supratentorial primitive neuroectodermal tumors most likely to respond to targeted therapy directed toward IDH1 mutations once available. Noteworthy, these tumors primarily affect children and interestingly, the presence of the IDH1 mutation was found exclusively in the adult patient tumors (⩾18 years of age) in two independent studies suggesting that this mutation does not have a significant role in childhood tumors of this type (Hayden et al., 2009).
II. Acute myeloid leukemia
The first study on IDH mutations in AML, reported the identification on IDH1R132 alterations in ∼8% of the 188 cases analyzed (Mardis et al., 2009). An association with normal cytogenetics was noted. Subsequent sequencing efforts revealed the existence of mitochondrial IDH2 mutations (Lalisang et al., 1997; Gross et al., 2010; Ward et al., 2010). Interestingly IDH2 mutations are significantly more common in AML than gliomas. Two groups reported the identification of a novel leukemia-associated mutation in IDH2, resulting in the substitution of Arg140 for glutamate in the catalytic arginine triad (Green and Beer, 2010; Ward et al., 2010; Figure 2). In a small cohort of western patients, IDH1 mutations (R132 H, R132C and R132G) were observed in 6/78 samples (7.7%), whereas IDH2 mutations were seen at a higher frequency of 12/78 (15.4%; Ward et al., 2010). Subsequently, within a more homogenous group comprised of adult de novo cytogenetically normal AML patients, mutations in IDH1 and IDH2 were found in 14 and 19% of the cases respectively. More recently, unlike gliomas, two patients were found to harbor both an IDH1 and IDH2 mutation (Marcucci et al., 2010; Paschka et al., 2010). Unbiased sequencing has also revealed the presence of 12 non-synonymous recurring mutations (including IDH1) in more than one AML genome. The data suggest that these alterations may be important for the pathogenesis of the tumor and indeed all these mutations were retained in the dominant clone (Mardis et al., 2009). Similar to that in glioma, samples from AML patients harboring IDH1 and IDH2 mutations showed elevated levels of D-2HG (Gross et al., 2010; Ward et al., 2010).
To gain further insights into potential cooperating events in AML, several groups have examined the correlation between IDH1/IDH2 mutations and other common genetic alterations in AML (Lalisang et al., 1997; Marcucci et al., 2010). Mutations in the phosphoprotein NPM1 are found in 25–35% of adult de novo AML and are associated with a wide spectrum of morphological subtypes. Recent studies across large patient cohorts revealed that IDH1/IDH2 mutations are strongly associated with NPM1 mutation. Activating FLT3/ITD mutations are also common in AML (30–35%) and result in increased proliferation and reduced susceptibility to apoptosis in hematopoietic cells. Both IDH1 and IDH2 mutations are seen in AMLs and subgroup analysis of AML patients have shown an adverse effect of IDH mutations on overall survival among patients with normal karyotype AML (Paschka et al., 2010). Retrospective clinical trial data has revealed that the presence of IDH1 mutations is associated with a worse prognosis in younger cytogenetically normal AML patients with mutated NPM1 without FLT3-ITD and patients with IDH2 mutations had a significantly lower complete response rate (Marcucci et al., 2010; Paschka et al., 2010). Interestingly a unique microRNA signature previously found to block myeloid cell and stem cell differentiation was also observed among these patients (Marcucci et al., 2010). Additional studies will help understand whether they may contribute to block differentiation of these cell populations and impact disease progression.
III. Myelodysplastic syndrome (MDS) and myeloproliferative neoplasm (MPN)
MDS and MPN are clonal disorders of hematopoiesis characterized by the proliferation of myeloid cells. Patients with MDS and MPN have a significantly higher risk of developing secondary AML. Importantly, mutations in IDH enzymes have been linked to this leukemic transformation, an event that is associated with a poor clinical prognosis (Abdel-Wahab et al., 2010). Sequence analysis has shown that IDH mutations occur in ∼5% MDS, 8.8% of MPN and 9.7% of secondary AML patients and were always heterozygous and mutually exclusive of one another (Kosmider et al., 2010). Of note, mutations in IDH2 were restricted to MPN, whereas the IDH1R132H mutation was detected in MDS and secondary AML patient samples (Kosmider et al., 2010). Previous studies have shown that mutations in JAK2 (V617F), TET2 and ASXL1 are also commonly found in these diseases however, were typically wild type in patient samples that contained mutations in either IDH1 or IDH2 as noted above (Abdel-Wahab et al., 2010; Kosmider et al., 2010). Finally, recent data indicates that IDH1 mutations in MDS are associated with a shorter overall survival and a higher rate of transformation into AML (Thol et al., 2010). In blast phase MPN, the presence of IDH mutations predicted worse survival (Pardanani et al., 2010a). In addition, studies both in MPN as well as in high-risk MDS with del (5q) suggest that acquisitions of IDH1 and IDH2 mutations are early events and have a role in disease progression (Pardanani et al., 2010a, 2010b).
IV. IDH mutations in other malignancies
In addition to cancers of the central nervous system and AML, IDH1 mutations have been identified at low frequency in prostate cancer (2.7%) and B-acute lymphoblastic leukemias (1.7%), suggesting that IDH1 mutations may have a role in the pathogenesis of both tumors types (Kang et al., 2009; Table 1). Follow-up studies across many more patients will clarify the prevalence of mutations in IDH enzymes in these cancers. In addition, analysis of 365 pheochromocytomas and paragangliomas for alterations in codons 132 and 172 of IDH1 and IDH2, respectively, identified one case of a heterozygous IDH1R132C mutation (Gaal et al., 2010).
A series of less characterized mutations in IDH1 have been reported in thyroid and colorectal cancers. The two most aggressive subtypes of thyroid cancer, anaplastic thyroid cancer and follicular thyroid cancer, both harbor novel mutations in IDH1 corresponding to the predicted G123R and V71I amino acid substitutions (Murugan et al., 2010; Figure 2). Unlike Arg132 involved in substrate coordination, both residues are located in the vicinity of the catalytic domain region and their impact on IDH1 enzyme activity remains unclear. Interestingly, follicular thyroid cancer is a precursor to anaplastic thyroid cancer suggesting that IDH1 mutations could be an early event in tumorigenesis and have a role in the progression of the disease. In addition, IDH1 mutations have been observed in a few human cancer cell lines. For example, a V71I mutation has been found in a plasma cell myeloma line RPMI-8226, and G97D mutations have been found in the colorectal cancer cell lines DLD-1 and HCT15 both derived from the same tumor, as well as in a cell line derived from glioma, HGG153 (Catalogue of Somatic Mutations in Cancer; Bleeker et al., 2009). One cell line MZ7-mel derived from a malignant melanoma, also contains a V294M mutation in IDH2 (Catalogue of Somatic Mutations in Cancer). Future studies will elucidate whether these mutations are present in primary tumors and whether they affect the biochemical activity of IDH1 and IDH2.
D-2HG as a potential biomarker of cancers harboring IDH1 and IDH2 mutations
Elevated levels of D-2HG are a common feature of human tumors with IDH1 and IDH2 mutations. Because this metabolite is a direct product of mutant IDH1 and IDH2, it provides a means to track the activity of mutant IDH enzymes. Our initial studies suggest that D-2HG may represent a biomarker with potential clinical utility. Across a panel of glioma samples, we observed an absolute correlation between elevated intracellular D-2HG levels and the presence of IDH mutations (Dang et al., 2009). Similarly, in AML intracellular D-2HG levels correlated with IDH1/IDH2 mutational status (Gross et al., 2010). Because D-2HG is excreted from cells, the presence of this metabolite in serum from patients with IDH mutant AML was examined. In this case, high levels of D-2HG in serum also correlated with the presence of IDH mutations (Gross et al., 2010). These results underscore the potential utility of 2HG as serum biomarker. In parallel, Ward et al. (2010) determined relative levels of intracellular 2HG in a panel of 18 AML clinical samples, and based on metabolite levels, the presence of IDH mutations was identified with 100% accuracy. Additionally, the novel IDH2(R140Q) mutation in AML was identified through this screening approach, further demonstrating the utility of D-2HG as a biomarker of IDH1/2 mutations.
Although these initial results hold great promise, clearly, a number of important questions await investigation. In particular, the correlation of serum D-2HG levels and disease burden as well as the potential of D-2HG levels as a predictive marker of therapeutic response to treatment with standard of care deserve careful evaluation. It has been reported that D-2HG may be tractable via magnetic resonance spectroscopy (Aydin et al., 2003; Sener, 2003). If that were the case, non-invasive detection of D-2HG by magnetic resonance spectroscopy (MRS) could help to diagnose and guide therapy before the surgery in patients with less accessible tumors such as gliomas. As additional clinical data emerges on efficacy of specific therapeutic strategies in gliomas with IDH1 or IDH2 mutations, early diagnosis before surgery may have significant therapeutic impact. Additionally, it will be important to establish whether this metabolite constitutes a pharmacodynamic biomarker to monitor the activity of inhibitors targeting mutant IDH enzymes once available.
The metabolic product of somatic mutant IDH enzymes as a modulator of cancer-relevant processes
The five carbon dicarboxylic acid D-2HG is normally found in human urine, and is an intermediate in the synthesis of 5-aminolaevulinate and porphyrin for heme synthesis, as well as metabolism of hydroxy-lysine (Lindahl et al., 1967; Chalmers et al., 1980). Our current understanding of the metabolic origin and fate of D-2HG in mammalian cells is rather limited. This metabolite is produced by the action of hydroxyacid oxoacid transhydrogenase enzyme (HOT) in the liver, kidney and brain of mammals. In this reaction, the formation of D-2HG from α-KG is coupled to oxidation of γ-hydroxybutyrate to succinic semialdehyde (Kaufman et al., 1988). In addition, the promiscuous activity of metabolic enzymes is an alternative source of 2HG (Rzem et al., 2007). Under physiologic conditions the cellular levels of D-2HG rate are maintained in check by the activity of the enantiomer selective D-2-hydroxyglutaric dehydrogenase (D-2HGDH). Alterations in the balance between production and metabolism of both 2HG enantiomers are associated with pathological acidurias. Indeed, elevated D-2HG and L-2HG in urine, plasma and CSF characterize this group of rare neurometabolic disorders with inherited mutations in D-2HGDH as well as L-2HGDH.
Characterization of the molecular events triggered by elevated D-2HG has revealed that this metabolite has an excitotoxic effect on neurons. Activation of N-methyl-D-aspartic acid receptors by D-2HG results in disruption of Ca2+ homeostasis and an increase in reactive oxygen species levels of primary neuronal cultures (Kolker et al., 2002). In addition, several observations provide the framework to begin to link D-2HG and tumorigenesis (Figure 1b). D-2HG exerts a direct inhibitory effect on adenosine-5′-triphosphate synthase contributing to mitochondrial dysfunction. Whether these observations in primary neurons are relevant to the malignant state remains to be determined. It is tempting to speculate that 2HG-driven mitochondrial dysfunction may constitute selective pressure that promotes metabolic adaptation and a shift towards aerobic glycolysis, which could ultimately confer a growth advantage in a hyperproliferative cellular state.
The dicarboxylic acids αKG and D-2HG are very similar whereby a ketone group and hydroxyl group on the second carbon functionally distinguish these two molecules. On the basis of their chemical similarity, several groups have proposed that D-2HG could modulate the activity of αKG-dependent enzymes or interfere with αKG-dependent processes linked to oncogenesis (Frezza et al., 2010; Gross et al., 2010). This hypothesis is also in part rooted in our current understanding of the impact of alterations in the tricarboxylic acid cycle enzymes and tumor suppressors SDH and FH (Pollard et al., 2005; Selak et al., 2005; Kaelin, 2009). In this case, loss of SDH and FH results in elevated levels of succinate and fumarate, respectively, which in turn interfere with the activity of the superfamily of αKG-dependent dioxygenases that catalyze a wide range of biochemical reactions (Loenarz and Schofield, 2008). Potential targets of D-2HG include histone and DNA demethylases that regulate chromatin remodeling, prolyl-hydroxylases involved in HIF1α degradation as well as prolyl and lysyl hydroxylases that mediate collagen biosynthesis. Thus, the intriguing possibility that the elevated levels of D-2HG, the metabolic product of mutant IDH1 and IDH2 enzymes, could impact epigenetic state and gene transcription, angiogenesis as well as extracellular matrix dynamics is actively being explored by many groups. Interestingly, a recent correlation between CpG island methylation phenotype and IDH1 mutational status has been noted (Noushmehr et al., 2010). This observation adds to the hypothesis linking D-2HG and epigenetic rewiring.
Finally, because D-2HG is excreted by malignant cells it is possible that this metabolite may mediate non-cell autonomous effects, impacting the tumor stroma, normal surrounding tissue and even distal metastasis. Availability of selective inhibitors of mutant IDH enzymes and of relevant genetically engineered mouse cancer models will enable the study of cell autonomous and paracrine effects of D-2HG.
Concluding remarks
The discovery of cancer-associated mutations in IDH1 and IDH2 constitutes the first example of gain-of-function somatic genetic alterations in metabolic enzymes. The discovery that all mutants characterized to date possess the ability to catalyze the reduction of αKG to D-2HG has profound implications. From a pragmatic stand point, the metabolite D-2HG provides a biomarker of mutant enzyme activity with potential clinical applicability. In addition, based on the similarities between αKG and D-2HG, a series of hypotheses have been proposed to molecularly link IDH mutations and tumorigenesis. Ongoing studies will help determine whether elevated D-2HG levels interfere with mitochondrial activity and with αKG-dependent enzymes, and whether mutations in IDH, SDH and FH enzymes may represent another example of evolutionary convergence.
On the basis of genetic and prognostic correlations, mutant IDH1 and IDH2 have emerged as potential therapeutic targets. In addition, replacement of Arg 132 by smaller side chains or hydrophobic amino acids suggests that the identification of selective inhibitors of mutant IDH1 may be feasible. Clearly, the availability of a genetic biomarker for patient stratification and of a metabolic biomarker to track target activity makes mutant IDH1 and IDH2 unique opportunities for therapeutic intervention and drug development. Multiple research teams are actively working to define whether interfering with D-2HG production results in a therapeutic benefit. Furthermore, understanding the prevalence of specific mutations in particular disease types (Table 1) will provide valuable insight into the mechanism of disease progression associated with mutant IDH1/IDH2. Finally, the utility of D-2HG as a biomarker may offer a unique opportunity for clinicians and patients. For example, in AML, the ability to detect D-2HG in serum and the potential to image D-2HG in gliomas via MRS may provide a means to track the progression and/or relapse of disease without painful invasive procedures. Although the ultimate diagnosis will come via genetic confirmation of the presence or absence of IDH1 and IDH2 mutations, plasma D-2HG may serve as a pharmacodynamic biomarker and potentially as a response biomarker, once inhibitors become available.
References
Abbas S, Lugthart S, Kavelaars FG, Schelen A, Koenders J, Zeilemaker A et al. (2010). Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia (AML): prevalence and prognostic value. Blood 116: 2122–2126.
Abdel-Wahab O, Manshouri T, Patel J, Harris K, Yao J, Hedvat C et al. (2010). Genetic analysis of transforming events that convert chronic myeloproliferative neoplasms to leukemias. Cancer Res 70: 447–452.
Aydin K, Ozmen M, Tatli B, Sencer S . (2003). Single-voxel MR spectroscopy and diffusion-weighted MRI in two patients with l-2-hydroxyglutaric aciduria. Pediatr Radiol 33: 872–876.
Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A . (2008). Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 116: 597–602.
Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A et al. (2000). Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287: 848–851.
Bleeker FE, Lamba S, Leenstra S, Troost D, Hulsebos T, Vandertop WP et al. (2009). IDH1 mutations at residue p. R132 (IDH1(R132)) occur frequently in high-grade gliomas but not in other solid tumors. Hum Mutat 30: 7–11.
Brandon M, Baldi P, Wallace DC . (2006). Mitochondrial mutations in cancer. Oncogene 25: 4647–4662.
Ceccarelli C, Grodsky NB, Ariyaratne N, Colman RF, Bahnson BJ . (2002). Crystal structure of porcine mitochondrial NADP+-dependent isocitrate dehydrogenase complexed with Mn2+ and isocitrate. Insights into the enzyme mechanism. J Biol Chem 277: 43454–43462.
Chalmers RA, Lawson AM, Watts RW, Tavill AS, Kamerling JP, Hey E et al. (1980). D-2-hydroxyglutaric aciduria: case report and biochemical studies. J Inherit Metab Dis 3: 11–15.
Chou WC, Hou HA, Chen CY, Tang JL, Yao M, Tsay W et al. (2010). Distinct clinical and biological characteristics in adult acute myeloid leukemia bearing isocitrate dehydrogenase 1 (IDH1) mutation. Blood 115: 2749–2754.
Dang CV, Semenza GL . (1999). Oncogenic alterations of metabolism. Trends Biochem Sci 24: 68–72.
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM et al. (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462: 739–744.
Frezza C, Tennant DA, Gottlieb E . (2010). IDH1 mutations in gliomas: when an enzyme loses its grip. Cancer Cell 17: 7–9.
Gaal J, Burnichon N, Korpershoek E, Roncelin I, Bertherat J, Plouin PF et al. (2010). Isocitrate dehydrogenase mutations are rare in pheochromocytomas and paragangliomas. J Clin Endocrinol Metab 95: 1274–1278.
Geisbrecht BV, Gould SJ . (1999). The human PICD gene encodes a cytoplasmic and peroxisomal NADP(+)-dependent isocitrate dehydrogenase. J Biol Chem 274: 30527–30533.
Gillies RJ, Gatenby RA . (2007). Hypoxia and adaptive landscapes in the evolution of carcinogenesis. Cancer Metastasis Rev 26: 311–317.
Green A, Beer P . (2010). Somatic mutations of IDH1 and IDH2 in the leukemic transformation of myeloproliferative neoplasms. N Engl J Med 362: 369–370.
Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG et al. (2010). Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med 207: 339–344.
Hanahan D, Weinberg RA . (2000). The hallmarks of cancer. Cell 100: 57–70.
Hartmann C, Meyer J, Balss J, Capper D, Mueller W, Christians A et al. (2009). Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1010 diffuse gliomas. Acta Neuropathol 118: 469–474.
Hayden JT, Fruhwald MC, Hasselblatt M, Ellison DW, Bailey S, Clifford SC . (2009). Frequent IDH1 mutations in supratentorial primitive neuroectodermal tumors (sPNET) of adults but not children. Cell Cycle 8: 1806–1807.
Henke B, Girzalsky W, Berteaux-Lecellier V, Erdmann R . (1998). IDP3 encodes a peroxisomal NADP-dependent isocitrate dehydrogenase required for the beta-oxidation of unsaturated fatty acids. J Biol Chem 273: 3702–3711.
Ho PA, Alonzo TA, Kopecky KJ, Miller KL, Kuhn J, Zeng R et al. (2010). Molecular alterations of the IDH1 gene in AML: a Children's Oncology Group and Southwest Oncology Group study. Leukemia 24: 909–913.
Huang PH, Xu AM, White FM . (2009). Oncogenic EGFR signaling networks in glioma. Sci Signal 2: re6.
Jones PA, Baylin SB . (2002). The fundamental role of epigenetic events in cancer. Nat Rev Genet 3: 415–428.
Kaelin Jr WG . (2009). SDH5 mutations and familial paraganglioma: somewhere Warburg is smiling. Cancer Cell 16: 180–182.
Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Seo SI et al. (2009). Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int J Cancer 125: 353–355.
Kaufman EE, Nelson T, Miller D, Stadlan N . (1988). Oxidation of gamma-hydroxybutyrate to succinic semialdehyde by a mitochondrial pyridine nucleotide-independent enzyme. J Neurochem 51: 1079–1084.
King A, Selak MA, Gottlieb E . (2006). Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene 25: 4675–4682.
Kolker S, Pawlak V, Ahlemeyer B, Okun JG, Horster F, Mayatepek E et al. (2002). NMDA receptor activation and respiratory chain complex V inhibition contribute to neurodegeneration in d-2-hydroxyglutaric aciduria. Eur J Neurosci 16: 21–28.
Kosmider O, Gelsi-Boyer V, Slama L, Dreyfus F, Beyne-Rauzy O, Quesnel B et al. (2010). Mutations of IDH1 and IDH2 genes in early and accelerated phases of myelodysplastic syndromes and MDS/myeloproliferative neoplasms. Leukemia 24: 1094–1096.
Kroemer G, Pouyssegur J . (2008). Tumor cell metabolism: cancer′s Achilles’ heel. Cancer Cell 13: 472–482.
Lalisang RI, Wils JA, Nortier HW, Burghouts JT, Hupperets PS, Erdkamp FL et al. (1997). Comparative study of dose escalation versus interval reduction to obtain dose-intensification of epirubicin and cyclophosphamide with granulocyte colony-stimulating factor in advanced breast cancer. J Clin Oncol 15: 1367–1376.
Lindahl G, Lindstedt G, Lindstedt S . (1967). Metabolism of 2-amino-5-hydroxyadipic acid in the rat. Arch Biochem Biophys 119: 347–352.
Loenarz C, Schofield CJ . (2008). Expanding chemical biology of 2-oxoglutarate oxygenases. Nat Chem Biol 4: 152–156.
Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrozek K, Margeson D et al. (2010). IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol 28: 2348–2355.
Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K et al. (2009). Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 361: 1058–1066.
Murugan AK, Bojdani E, Xing M . (2010). Identification and functional characterization of isocitrate dehydrogenase 1 (IDH1) mutations in thyroid cancer. Biochem Biophys Res Commun 393: 555–559.
Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP et al. (2010). Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17: 510–522.
Pardanani A, Lasho TL, Finke CM, Mai M, McClure RF, Tefferi A . (2010a). IDH1 and IDH2 mutation analysis in chronic- and blast-phase myeloproliferative neoplasms. Leukemia 24: 1146–1151.
Pardanani A, Patnaik MM, Lasho TL, Mai M, Knudson RA, Finke C et al. (2010b). Recurrent IDH mutations in high-risk myelodysplastic syndrome or acute myeloid leukemia with isolated del(5q). Leukemia 24: 1370–1372.
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P et al. (2008). An integrated genomic analysis of human glioblastoma multiforme. Science 321: 1807–1812.
Paschka P, Schlenk RF, Gaidzik VI, Habdank M, Kronke J, Bullinger L et al. (2010). IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J Clin Oncol 28: 3636–3643.
Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC et al. (2005). Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet 14: 2231–2239.
Rzem R, Vincent MF, Van Schaftingen E, Veiga-da-Cunha M . (2007). L-2-hydroxyglutaric aciduria, a defect of metabolite repair. J Inherit Metab Dis 30: 681–689.
Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F et al. (2009). Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 27: 4150–4154.
Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD et al. (2005). Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7: 77–85.
Sener RN . (2003). L-2 hydroxyglutaric aciduria: proton magnetic resonance spectroscopy and diffusion magnetic resonance imaging findings. J Comput Assist Tomogr 27: 38–43.
Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD et al. (2006). The consensus coding sequences of human breast and colorectal cancers. Science 314: 268–274.
Thol F, Weissinger EM, Krauter J, Wagner K, Damm F, Wichmann M et al. (2010). IDH1 mutations in patients with myelodysplastic syndromes are associated with an unfavorable prognosis. Haematologica (e-pub ahead of print 21 May 2010; doi:10.3324/haematol.2010.025494).
Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D et al. (2002). Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 30: 406–410.
van Roermund CW, Hettema EH, Kal AJ, van den Berg M, Tabak HF, Wanders RJ . (1998). Peroxisomal beta-oxidation of polyunsaturated fatty acids in Saccharomyces cerevisiae: isocitrate dehydrogenase provides NADPH for reduction of double bonds at even positions. EMBO J 17: 677–687.
Vander Heiden MG, Cantley LC, Thompson CB . (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029–1033.
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD et al. (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17: 98–110.
Voelzke WR, Petty WJ, Lesser GJ . (2008). Targeting the epidermal growth factor receptor in high-grade astrocytomas. Curr Treat Options Oncol 9: 23–31.
Vogelstein B, Kinzler KW . (1993). The multistep nature of cancer. Trends Genet 9: 138–141.
Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA et al. (2010). The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17: 225–234.
Watanabe T, Nobusawa S, Kleihues P, Ohgaki H . (2009). IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 174: 1149–1153.
Xu X, Zhao J, Xu Z, Peng B, Huang Q, Arnold E et al. (2004). Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J Biol Chem 279: 33946–33957.
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W et al. (2009). IDH1 and IDH2 mutations in gliomas. N Engl J Med 360: 765–773.
Yoshihara T, Hamamoto T, Munakata R, Tajiri R, Ohsumi M, Yokota S . (2001). Localization of cytosolic NADP-dependent isocitrate dehydrogenase in the peroxisomes of rat liver cells: biochemical and immunocytochemical studies. J Histochem Cytochem 49: 1123–1131.
Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P et al. (2009). Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 324: 261–265.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors are employees of Agios Pharmaceuticals.
Rights and permissions
About this article
Cite this article
Yen, K., Bittinger, M., Su, S. et al. Cancer-associated IDH mutations: biomarker and therapeutic opportunities. Oncogene 29, 6409–6417 (2010). https://doi.org/10.1038/onc.2010.444
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2010.444
Keywords
This article is cited by
-
Therapies for IDH-Mutant Gliomas
Current Neurology and Neuroscience Reports (2023)
-
Liquid biopsies to occult brain metastasis
Molecular Cancer (2022)
-
Atypical cartilage in type II germ cell tumors of the mediastinum show significantly different patterns of IDH1/2 mutations from conventional chondrosarcoma
Modern Pathology (2022)
-
The Clinical Frailty Scale as predictor of overall survival after resection of high-grade glioma
Journal of Neuro-Oncology (2022)
-
The oncometabolite R-2-hydroxyglutarate dysregulates the differentiation of human mesenchymal stromal cells via inducing DNA hypermethylation
BMC Cancer (2021)
