
Abstract
A submicroscopic deletion containing SOX2 was identified at the 3q breakpoint in a child with t(3;11)(q26.3;p11.2) associated with bilateral anophthalmia. Subsequent SOX2 mutation analysis identified de novo truncating mutations of SOX2 in 4 of 35 (11%) individuals with anophthalmia. Both eyes were affected in all cases with an identified mutation.
Similar content being viewed by others


Main
Bilateral anophthalmia is the rarest and most severe form of structural eye malformation. Constitutional chromosome deletions involving 3q27 have been identified in three unrelated individuals with anophthalmia1,2. De novo apparently balanced reciprocal translocations involving 3q27 were reported in two cases3,4 of severe bilateral microphthalmia and microphthalmia/anophthalmia.
We obtained a sample of fixed cell suspension from a female infant4 with isolated bilateral anophthalmia and a de novo t(3;11)(q27;p11.2), with the aim of mapping the 3q27 breakpoint using fluorescence in situ hybridization (FISH). Because we could identify only a few cells in metaphase in this sample and could not obtain a fresh sample, conventional FISH analysis was not possible. But by co-hybridizing the sample with a 3q paint5, we were able to unambiguously locate locus-specific BAC probes relative to the breakpoint. BAC signals that co-localized with the two larger domains (normal 3q & derivative 3) mapped centromeric to the breakpoint; those sited in a large and a small 3q domain (normal 3q & derivative 11) were telomeric to the breakpoint; BACs crossing the breakpoint give signals in all three domains (Fig. 1a). Using this strategy, we found that BAC clone RP11-225N10 spanned the breakpoint. Using FISH analysis with BACs flanking the breakpoint, we identified a deletion of roughly 740 kb between RP11-4B14 and RP11-431C22 on the der(3) chromosome (Fig. 1b). This deletion was centromeric to the translocation breakpoint and separated from it by roughly 600 kb of non-deleted DNA (Fig. 1b and Supplementary Fig. 1 online).

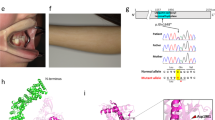
See Supplementary Note 1 online. a, Interphase nuclei (some multilobed granulocytes) from the individual with t(3;11) and anophthalmia hybridized with a 3q chromosome paint (green signals) and the named BAC clones (red signals). The two large green domains represent the normal chromosome 3 and the der(3) chromosome; the small green domain is the der(11). The left-to-right order of the BAC clones is centromeric to telomeric. RP11-262M14 and RP11-646E18 map centromeric to the breakpoint. RP11-223B4 and RP11-463A6 are deleted. RP11-225N10 crosses the translocation breakpoint. RP11-29D4 maps telomeric to the translocation breakpoint. b, An ideogram of the complex genomic rearrangement surrounding the translocation breakpoint in this individual. c, Cartoon representation of the genomic region surrounding the single-exon SOX2 gene, which lies within an intron of a conserved, seemingly non-coding gene, SOX2OT. d, Electropherograms showing the three de novo, heterozygous nonsense mutations that were detected in four unrelated children. The position of the protein truncation with respect to the known functional domains is indicated on the cartoon of the SOX2 protein of 317 amino acids.
The BAC spanning the translocation breakpoint contained no obvious candidate genes for association with anophthalmia. The available sequence from the deleted region was incomplete, showing only one annotated gene with homology to the 60S ribosomal protein L7a. The gene SOX2 (encoding the sex determining region Y–box 2) had been previously mapped to 3q26.3–q27 by FISH6 but could not be identified in the Ensembl assembly. SOX2 is a transcription factor with site- and stage-specific expression in the developing eye and nervous system7,8,9,10 and with a regulatory role in lens development9, making it a good candidate gene for association with severe structural eye defects.
BLAST searches using the 3′ region of the sheep ortholog of SOX2 identified a series of human BAC clones that contained the single-exon gene SOX2 by both PCR and Southern-blot analysis. We found that four of these BACs (RP11-335I3, RP11-349O18, RP11-203N24 and RP11-223B4) were deleted or partially deleted in the individual with t(3;11). During the course of this study, 217 kb of complete sequence from overlapping BAC clones RP11-43F17 and RP11-379M20 became available. This sequence contained the open reading frame (ORF) of SOX2, which codes for 317 amino acids.
Genomic analysis indicated that SOX2 lies in an intron of another gene, which we called SOX2OT (SOX2 overlapping transcript (non-coding RNA)). SOX2OT has at least five exons and produces a mRNA of roughly 3.4 kb from the same strand as SOX2 (Fig. 1c). It seems to be a non-coding gene with no ORF that encodes a peptide of more than 70 amino acids. SOX2OT cDNA is evolutionarily conserved, with 88% nucleotide identity between the human cDNA and available mouse expressed-sequence tags. The sequence over the whole genomic region (roughly 40 kb) encompassing the SOX2OT transcription unit, within which SOX2 is embedded, is highly conserved (see Supplementary Fig. 2 online). Non-coding RNAs often have complex regulatory roles in the cell11, suggesting that SOX2OT may be involved in the transcriptional regulation of SOX2 in some tissues.
To test whether haploinsufficiency for SOX2 might have a specific and crucial role in human eye development, we carried out mutation analysis using denaturing high-performance liquid chromatography (DHPLC). We tested 102 individuals with microphthalmia (n = 46), anophthalmia (n = 35) and coloboma (n = 21) without visible cytogenetic abnormalities after the study was approved by the Multicentre Research Ethics Committee for Scotland and London. In total, we detected ten DHPLC shifts in 10 of 102 (9.8%) of these cases. Five of these shifts were due to a heterozygous change (976G→A) in SOX2 3′ UTR that we also observed in 5 of 88 (5.7%) unrelated control individuals. The other five unrelated individuals each had a mutation in the SOX2 coding region.
One mutation involved a neutral change in the third position of codon 303 (V303V) and was inherited from the individual's phenotypically normal mother; this mutation is assumed to be non-pathogenic. We identified two nonsense amino-acid changes (S83X and E93X) in the HMG box (high-mobility group DNA-binding domain) at CpG dinucleotides; these are predicted to ablate DNA binding. We observed a recurrent de novo nonsense mutation (529C→T resulting in the amino-acid change Q177X) in two unrelated individuals. Q177X occurs C-terminal to the HMG box but removes 141 amino acids, including much of the highly conserved SOX B1 subgroup activation domain12. Each of these nonsense mutations probably results in loss of function (Fig. 1d).
The parents of each individual with a mutation in SOX2 had normal SOX2 sequence (see Supplementary Fig. 3 online). We confirmed family structure using a panel of 12 autosomal microsatellite markers. No mutations were identified in 176 unrelated control chromosomes from unaffected adults living in Scotland. We excluded large genomic rearrangements around SOX2 using FISH analysis in 23 cases for which cell lines were available.
Clinically, the individuals with nonsense mutations in SOX2 presented with anophthalmia (two with bilateral anophthalmia and two with unilateral anopthalmia and contralateral microphthalmia). The associated extraocular abnormalities are variable and include male genital tract anomalies, myopathy and spastic diplegia (Table 1). It is not clear how these phenotypes are related to expression and function of SOX2. No missense mutations were detected in individuals with the many milder eye malformations that we screened, suggesting that missense changes may give rise to different phenotypes. Mice heterozygous with respect to a targeted Sox2-null mutation show no eye phenotype whereas homozygotes die early in gestation13. Such human–mouse differences in phenotypes associated with haploinsufficiency are not uncommon.
Accession numbers. Sheep SOX2 ortholog, AQ473552; BAC clone RP11-43F17, AC117415; BAC clone RP11-379M20, AC125613; SOX2OT (SOX2 overlapping transcript), AL157425.
Note: Supplementary information is available on the Nature Genetics website.
References
Male, A. et al. Eur. J. Hum. Genet. 10, 807–812 (2002).
Chitayat, D. et al. Am. J. Med. Genet. 61, 45–48 (1996).
Kurbasic, M., Jones, F.V. & Cook, L.N. Ophthalmic. Genet. 21, 239–242 (2000).
Driggers, R.W. et al. Am. J. Med. Genet. 87, 201–202 (1999).
Guan, X.Y. et al. Nat. Genet. 12, 10–11 (1996).
Stevanovic, M., Zuffardi, O., Collignon, J., Lovell-Badge, R. & Goodfellow, P. Mamm. Genome. 5, 640–642 (1994).
Uwanogho, D. et al. Mech. Dev. 49, 23–36 (1995).
Le, R.D., Rayner, K., Rex, M., Wigmore, P.M. & Scotting, P.J. J. Anat. 200, 51–56 (2002).
Kamachi, Y., Uchikawa, M., Collignon, J., Lovell-Badge, R. & Kondoh, H. Development 125, 2521–2532 (1998).
Zappone, M.V. et al. Development 127, 2367–2382 (2000).
Szymanski, M. & Barciszewski, J. Genome. Biol. 3, Reviews 0005.1–0005.8 (2002).
Kamachi, Y., Uchikawa, M. & Kondoh, H. Trends. Genet. 16, 182–1827 (2000).
Avilion, A.A. et al. Genes Dev. 17, 126–140 (2003).
Acknowledgements
We thank all the children and their parents for participating in this study, J. Trent for providing the 3q chromosome paint and W. Bickmore, N. Gilbert and N. Hastie for advice and discussion.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Fantes, J., Ragge, N., Lynch, SA. et al. Mutations in SOX2 cause anophthalmia. Nat Genet 33, 462–463 (2003). https://doi.org/10.1038/ng1120
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng1120
This article is cited by
-
Phase separation as a possible mechanism for dosage sensitivity
Genome Biology (2024)
-
Distinct gene expression patterns of SOX2 and SOX2OT variants in different types of brain tumours
Journal of Genetics (2023)
-
Novel SOX2 mutation in autosomal dominant cataract-microcornea syndrome
BMC Ophthalmology (2022)
-
LncRNA SOX2OT alleviates mesangial cell proliferation and fibrosis in diabetic nephropathy via Akt/mTOR-mediated autophagy
Molecular Medicine (2021)
-
Multiocular organoids from human induced pluripotent stem cells displayed retinal, corneal, and retinal pigment epithelium lineages
Stem Cell Research & Therapy (2021)
