
Abstract
Chronic obstructive pulmonary disease (COPD) is characterized by reduced lung function and is the third leading cause of death globally. Through genome-wide association discovery in 48,943 individuals, selected from extremes of the lung function distribution in UK Biobank, and follow-up in 95,375 individuals, we increased the yield of independent signals for lung function from 54 to 97. A genetic risk score was associated with COPD susceptibility (odds ratio per 1 s.d. of the risk score (∼6 alleles) (95% confidence interval) = 1.24 (1.20–1.27), P = 5.05 × 10−49), and we observed a 3.7-fold difference in COPD risk between individuals in the highest and lowest genetic risk score deciles in UK Biobank. The 97 signals show enrichment in genes for development, elastic fibers and epigenetic regulation pathways. We highlight targets for drugs and compounds in development for COPD and asthma (genes in the inositol phosphate metabolism pathway and CHRM3) and describe targets for potential drug repositioning from other clinical indications.
Similar content being viewed by others

Main
Maximally attained lung function and subsequent decline in lung function together determine the risk of developing COPD1,2. COPD, characterized by irreversible airflow obstruction and chronic airway inflammation, is the third leading cause of death globally3. Smoking is the primary risk factor for COPD, but not all smokers develop COPD and more than 25% of COPD cases occur in never smokers4. Patients with COPD exhibit variable presentation of symptoms and pathology, with or without exacerbations, with variable amounts of emphysema and with differing rates of progression. Although risk factors for COPD are known, including smoking and environmental exposures in early5,6 and later life, the causal mechanisms are not well understood7. Disease-modifying treatments for COPD are required7.
Understanding genetic factors associated with reduced lung function and COPD susceptibility could inform drug target identification, risk prediction, and stratified prevention or treatment. Previous genome-wide association studies (GWAS) of COPD identified several independent COPD-associated variants8,9,10, but the rate and scale of discovery have been limited by available sample sizes. We conducted a GWAS for lung function and followed up robustly associated variants in COPD case–control studies. Although previous GWAS have reported genome-wide significant associations with lung function11,12,13,14,15,16, there has not been a comprehensive study confirming the effect of these variants on COPD susceptibility. In this study, we hypothesized that (i) GWAS of lung function with high power and large scale would detect novel loci associated with quantitative measures of lung function; (ii) collectively, these variants would be associated with risk of developing COPD; and (iii) aggregate analyses of all new and previously reported signals of association, and the identification of genes through which their effects are mediated, would give further insight into biological mechanisms underlying the associations. Together, these findings could provide potential novel targets17 for therapeutic intervention and pinpoint existing drugs that could be candidates for repositioning18 for the treatment of COPD.
Results
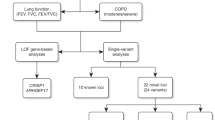
43 new signals for lung function
For stage 1, genome-wide association analyses of forced expired volume in 1 s (FEV1), forced vital capacity (FVC) and FEV1/FVC were undertaken in 48,943 individuals from the UK BiLEVE study16 who were selected from the extremes of the lung function distribution in UK Biobank (total n = 502,682). From analysis of 27,624,732 variants, 81 independent variants associated with one or more traits with P < 5 × 10−7 were selected for follow-up in stage 2, consisting of a further 95,375 independent individuals from UK Biobank, the SpiroMeta consortium and the UK Households Longitudinal Study (UKHLS) (Supplementary Table 1). No evidence of sample overlap between stage 1 and stage 2 studies or among stage 2 studies was found using LD score regression (Supplementary Table 2). Following meta-analysis of stage 1 and stage 2 results, 43 signals showed genome-wide significant (P < 5 × 10−8) association with one or more of FEV1, FVC or FEV1/FVC (Table 1, Supplementary Fig. 1 and Supplementary Table 3). We report these 43 signals as new independent signals (Fig. 1), almost doubling the number of confirmed independent genomic signals for lung function to 97 (Supplementary Table 4). Of the 43 newly identified signals, 33 represented new loci whereas 10 were statistically independent signals (conditional P < 5 × 10−7) within 500 kb of another association signal. On the basis of an assumed heritability of 40% (refs. 19,20) for each lung function trait, the new signals explained 4.3% of the heritability for FEV1, 3.2% of the heritability for FVC and 5.2% of the heritability for FEV1/FVC, bringing the total heritability explained by the 97 signals to 9.6%, 6.4% and 14.3%, respectively. The estimated effect sizes of lung-function-associated variants in children were correlated with those in adults (r = 0.65, 73 variants with high imputation quality; Supplementary Fig. 2). A genetic risk score based on these 73 variants was also significantly associated with FEV1 and FEV1/FVC in children (per-risk-allele β (standard error (s.e.)) = –0.0177 (0.0040), P = 1.03 × 10−5 and per-risk-allele β (s.e.) = –0.0213 (0.0037), P = 1.27 × 10−8, respectively), but not with FVC (per-risk-allele β (s.e.) = –0.0037 (0.0041), P = 0.366).

Plots are shown of genome-wide association results for FEV1 (forced expired volume in 1 s; top), FEV1/FVC (middle) and FVC (forced vital capacity; bottom). Previously reported signals are shown in dark blue (except signals with P > 5 × 10−4 in this study), and new signals are shown in red. Signals are shown only for the trait with which they exhibited the strongest association. The red and blue lines correspond to the genome-wide significance level (P = 5 × 10−8; –log10 P = 7.3) and the threshold used to select signals for follow-up in stage 2 (P = 5 × 10−7; –log10 P = 6.3), respectively. Labels are for the nearest gene to the new sentinel variants. There were two new independent signals near CDC7 and TGFBR3 on chromosome 1 (labeled as CDC7–TGFBR3). See Supplementary Table 3 for full results. The image was created using a modified version of the R package qqman.
Using the stage 1 results, a 95% 'credible set' of variants (the set of variants that were 95% likely to contain the underlying causal variant, based on Bayesian refinement) was defined for all (new and previously reported) association signals for which this was feasible (67 signals; Online Methods, Supplementary Figs. 3–5 and Supplementary Table 5); 13 of these signals were fine-mapped to ≤10 plausible causal variants and, for 63 of the 67 signals fine-mapped, the sentinel variant (with the lowest P value) was also the top ranked variant by posterior probability. In addition, by refining six chromosome 6 major histocompatibility complex (MHC) association signals using imputation of classical alleles and amino acid changes (Online Methods), we identified an amino acid change at position 57 of the MHC class II HLA-DQB1 gene product HLA-DQβ1 (alanine as compared to non-alanine) as the main driver of signals in the MHC region for both FEV1 (β (s.e.) = 0.048 (0.007), P = 5.71 × 10−13; Supplementary Fig. 6a) and FEV1/FVC (β (s.e.) = 0.062 (0.007), P = 1.17 × 10−20; Supplementary Fig. 6c), with secondary non-HLA (human leukocyte antigen) gene signals in the MHC region remaining after conditioning on the HLA-DQβ1 position 57 variant for rs34864796[G>A] (near ZKSCAN3, FEV1; conditional β (s.e.) = –0.058 (0.01), P = 1.26 × 10−9; Supplementary Fig. 6b) and rs2070600[C>T] (in AGER, FEV1/FVC; conditional β (s.e.) = 0.120 (0.013), P = 4.23 × 10−20; Supplementary Fig. 6d and Supplementary Table 6).
We found that 29 of the lung-function-associated signals had previously shown genome-wide significant associations in GWAS of traits other than lung function or COPD. These traits included inflammatory bowel disease (Crohn's disease and/or ulcerative colitis; three signals) and height (nine signals, three of which showed a consistent direction of effect on height and the lung function measure with which they were most strongly associated) (Supplementary Table 7). With the exception of KANSL1 (ref. 16), there was no significant (P < 5.15 × 10−4) association with smoking for any of the signals (Supplementary Table 8).
Ninety-five variants and COPD susceptibility
The disease relevance of lung-function-associated variants has been questioned21. Therefore, we tested association with COPD susceptibility for variants representing 95 of the 97 lung-function-associated signals in up to 20,086 COPD cases and 215,630 controls (data were unavailable for further study for the X-chromosome variant rs7050036[A>T] near AP1S2 and a rare variant, chr12:114743533[C>T]) (Supplementary Table 9). These cases and controls made up the COPD study at deCODE Genetics22 (COPD cases defined using spirometry data, population-based controls excluding known cases, up to 1,964 moderate-to-severe cases, up to 142,262 controls), three lung resection cohorts23,24,25 (COPD definition based on spirometry data, 310 moderate-to-severe cases, 332 controls), four case–control studies employing post-bronchodilator spirometry8,9,10,26,27,28,29 (5,778 moderate-to-severe cases, 3,950 controls), two studies within which COPD was determined from electronic medical records30 (eMR; total of 1,487 cases, 15,138 controls), additional UK Biobank samples (COPD definition based on spirometry data, 984 moderate-to-severe cases, 26,561 controls) and UK BiLEVE (COPD definition based on spirometry data, 9,563 moderate-to-severe cases, 27,387 controls). UK BiLEVE COPD cases and controls were only used for single-variant COPD association tests for the subset of 47 variants discovered independently of UK BiLEVE (that is, excluding the 43 variants discovered using the UK BiLEVE data described in this paper and 5 variants reported in our previous study in the UK BiLEVE population16). Across all 95 variants, 51 showed nominal COPD association (P < 0.05) and 30 showed association with COPD susceptibility reaching a Bonferroni-corrected threshold for 95 tests (P < 5.26 × 10−4; Supplementary Table 10). Of these 30 variants, 27 were variants discovered independently of UK BiLEVE and 3 were from the 48 lower-powered association tests not including UK BiLEVE cases and controls.
Using a risk score based on the available 95 sentinel variants or their best proxies, and using data from up to 9,791 COPD cases and 120,462 controls (Online Methods), for the meta-analysis the odds ratio (OR) (95% confidence interval (CI)) per 1 s.d. change in risk score (∼6 alleles) was 1.24 (1.20–1.27), P = 5.05 × 10−49 (Fig. 2a and Supplementary Table 11). We observed considerable heterogeneity in effect estimates among the different COPD studies (I2 = 92%), which had different approaches to ascertainment of COPD cases and variable disease severity. In UK Biobank (including UK BiLEVE), we found broadly similar effect size estimates for moderate-to-severe COPD to those found in COPD case–control studies employing post-bronchodilator spirometry (OR = 1.42 versus 1.36, respectively), and we therefore undertook further modeling showing a gradation in susceptibility to moderate-to-severe COPD across deciles of allelic risk score (Online Methods). The risk for moderate-to-severe COPD was more than three times higher in the top decile than it was in the bottom decile (OR = 3.71, 95% CI = 3.34–4.12; Fig. 2b). The estimated proportion of COPD cases attributable to allelic risk scores above the first decile (population attributable risk fraction) was 48.0% (95% CI = 43.6–52.2%).

(a) Forest plots of COPD results for the risk score analysis. Odds ratios per 1 s.d. of the risk score (∼6 alleles) are presented for each study (bars, 95% CI). Studies are grouped according to study design and phenotyping: “eMR”, electronic medical records study using ICD codes to define COPD (DiscovEHR also used spirometry data to refine the COPD definition); “case–control”, COPD case–control study using post-bronchodilator spirometry data to define COPD; “lung resection cohort”, study using a combination of pre- and post-bronchodilator spirometry data to define COPD; Icelandic Biobank (deCODE), cohort where cases were selected from a population-based study and a study of patients with COPD using a spirometric definition and controls were selected as individuals within the cohort who were not known cases (no spirometric definition was used for controls); and UK Biobank (excluding UK BiLEVE), cohort where spirometry data were used to define both COPD cases and controls. Further details are provided in the Supplementary Note. (b) Odds ratios (bars, 95% CI) for spirometrically defined COPD from weighted genetic risk score deciles in UK Biobank (10,547 cases (pre-bronchodilator percent predicted FEV1 <80% and FEV1/FVC <0.7) and 53,948 controls (FEV1/FVC >0.7 and percent predicted FEV1 >80%); weights were derived from non-discovery populations). For each decile, odds ratios were obtained using logistic regression adjusted for age, age2, sex, height, smoking status, pack-years and the first ten ancestry principal components. The odds ratio for comparison of the 10th and 1st deciles in ever smokers only was 3.35 (95% CI = 2.93–3.84) and in never smokers only was 4.27 (95% CI = 3.61–5.06).
We tested association of individual variants and the 95-variant risk score with COPD exacerbations in subsets of individuals from UK Biobank, deCODE, four COPD case–control studies and two eMR studies (total of 2,462 COPD exacerbation cases and 15,288 COPD non-exacerbation controls) and the Lung Health Study (100 exacerbation cases and 4,002 controls). There was no association of individual variants or genetic risk score with acute exacerbations of COPD (Supplementary Tables 12 and 13).
To evaluate whether these variants showed disease-relevant associations in a non-European population, we studied 71 variants for which data were available in 7,116 COPD cases (20,919 controls) and 5,292 exacerbation cases (1,824 controls) from the China Kadoorie Biobank (CKB) cohort (Supplementary Tables 10–13). The allelic risk score was associated with COPD susceptibility (OR per 1 s.d. change in risk score (95% CI) = 1.08 (1.04–1.11), P = 4.2 × 10−6), suggesting some shared genetic contributions to COPD in populations of European and East Asian descent. Thirty-nine of the variants showed a consistent direction of effect on COPD in European and Chinese samples, and seven of these were significant (P < 0.05). Two signals were significant after correction for multiple testing (Supplementary Table 10c).
To assess the impact of including individuals with asthma in a COPD case–control analysis, we tested for association with COPD in UK Biobank both before and after excluding individuals with self-reported doctor-diagnosed asthma and show that the effect size estimates were similar (Supplementary Fig. 7).
Implicated genes highlight pathways and druggable targets
Gene expression and genotype data from lung, blood and multi-tissue resources were queried to determine whether the top variant at each of the 97 signals, or a proxy, was significantly associated with changes in expression of any gene (was an expression quantitative trait locus (eQTL) for any gene). Using this approach and identification of deleterious variants within the association signal (Online Methods and Supplementary Table 14), we identified 234 genes with potentially causal effects on lung function (Supplementary Table 15). These 234 genes were enriched (false discovery rate (FDR) ≤5%) in elastic fiber pathways and in 'signaling events mediated by the Hedgehog family', with the latter including CDON, which was implicated by a new intergenic signal (rs567508; between CDON and RPUSD4) on chromosome 11. We narrowed this group of 234 genes to 68 'high-priority genes' that were implicated via a deleterious variant or on stricter criteria for colocalization with a gene expression signal (r2 ≥ 0.9 between the sentinel variant and top expression-associated variant; Table 2). We found that the 68 high-priority genes were over-represented (FDR ≤ 5%) among a number of gene ontology terms, including SH3 domain binding, GTPase binding, actin binding and fibroblast migration (Supplementary Table 16). Alternative approaches to pathway analyses, which instead use all genome-wide association results, supported previous reports of enrichment of histone and systemic lupus erythematosus pathways14,15,16 and additional autoimmune and inflammatory pathways (Supplementary Table 17). Tests for tissue-specific enrichment of lung function signals overlapping histone marks identified enrichment in fetal lung, fetal heart and fibroblasts (H3K4me1) and stomach smooth muscle (H3K4me1 and H3K4me3) (Supplementary Table 18).
Approved drugs, or drugs in development, target the protein products of 7 of the 234 genes (Supplementary Table 19a). This includes three high-priority genes—CHRM3, SLC6A4 and CRHR1. CHRM3 and SLC6A4 were both implicated by new signals (rs6688537[C>A] in an intron of CHRM3 and rs59835752[–/A] in an intron of EFCAB5, respectively) and encode targets for drugs approved for the treatment of asthma and COPD (CHRM3, muscarinic acetylcholine receptor M3) and anxiety and depression (SLC6A4, serotonin transporter). CRHR1 (implicated by rs35524223[T >A] in an intron of KANSL1) encodes corticotropin-releasing factor receptor 1, which is a target for compounds in development for the treatment of anxiety, depression and irritable bowel syndrome. The other four genes include NDUFA12 (implicated by rs113745635[C>T] in an intron of FGD6) encoding an NADH dehydrogenase that is a target for metformin hydrochloride, primarily used to treat type 2 diabetes, and ITK (implicated by rs10515750 in an intron of CYFIP2) encoding a tyrosine protein kinase, a target for the cancer drug pazopanib.
Using STRING31 to find proteins that interact with the proteins encoded by the high-priority genes, we highlighted further druggable targets (Supplementary Table 19b). These included the phosphoinositol 3-kinase p110-delta subunit (part of the inositol phosphate metabolism pathway with INPP5E, which was implicated as a high-priority gene by rs10870202 in an intron of DNLZ, and a target for compounds in development for the treatment of COPD and asthma) and matrix metalloproteinases 1, 7 and 8 (targets for doxycycline, which is an antibiotic and antimalarial).
Discussion
In this study, the power gained by sampling from the extremes of a large biobank while retaining the power of a quantitative trait analysis, coupled with strategies to improve coverage of the genome and extensive follow-up, enabled a near-doubling of the number of signals of association with lung function identified thus far. We further explored 95 variants, representing 43 new signals and 52 previously reported signals, and we showed that collectively these variants are strongly associated with COPD susceptibility.
Using functional evidence from eQTL studies and deleterious variants to link signals to genes, we found that 41 of the 97 lung function signals are also the strongest signals of association for expression of, or contain deleterious variants within, 68 genes (which we term 'high-priority genes'). Among these, new signals in or near FAM13A and ADAM19, both previously associated with lung function and COPD susceptibility9,32, along with evidence that these signals are themselves eQTLs for FAM13A and ADAM19, provide further evidence for FAM13A and ADAM19 themselves being the drivers of those signals. There was significant enrichment amongst the 68 genes for SH3 domain (including ADAM19), GTPase and actin binding, and fibroblast migration, highlighting the potential importance of pathways relating to the cytoskeleton.
The 68 genes identified as high priority included genes at new signals encoding targets for which there are approved drugs or drugs in development (Supplementary Table 19). Of note, the muscarinic acetylcholine receptor M3, encoded by CHRM3, is a well-characterized drug target for which many approved drugs exist, including for the treatment of asthma and obstructive lung disease. SLC6A4 encodes a serotonin transporter, a target for a number of drugs approved for treating depression and anxiety disorders, one of which (nortriptyline hydrochloride) has been trialed for use in inflammatory skin disorders (psoriasis and eczema); HTR4, which encodes a serotonin receptor, was identified in one of the earliest lung function GWAS13. INPP5E, identified as a high-priority gene for a new signal of association with FVC (and FEV1) on chromosome 9, encodes inositol polyphosphate-5-phosphatase E, a component of the inositol phosphate metabolism pathway. Another component of the same pathway, phosphoinositide 3-kinase (PI3K) delta is a target of drugs under development for the treatment of a range of indications, including COPD and asthma. Mutations in INPP5E cause ciliopathy (Joubert and MORM syndromes).
Protective genetic variants that reduce the function or expression of a target protein could be mimicked by drugs and so are of particular interest. The minor allele (minor allele frequency (MAF) = 17%) at the new signal in an intron of FAM13A was associated with decreased expression of FAM13A in lung tissue and reduced risk of COPD. This, together with recent evidence from a study of the Fam13a knockout mouse33, suggests that pharmacological inhibition of FAM13A may be protective.
Extending our pathway analyses to all 234 genes implicated by gene expression or deleterious variants, we observed enrichment of genes related to 'signaling events mediated by the Hedgehog family' pathway. Hedgehog signaling has a crucial role in early development. Three members of this pathway, PTCH1, TGFB2 and HHIP, have previously been reported as likely causal genes underlying lung function association signals34. In this study, we additionally report PTHLH, encoding a parathyroid-hormone-like hormone, and CDON¸ encoding a Hedgehog co-receptor, as likely causal genes (the latter at a newly associated signal). Of the 73 well-imputed variants available in children, we show correlation (r = 0.62) of variant effect size estimates with those in adults. Should this pattern of correlation apply across all 97 lung-function-associated variants, this would suggest that many of these variants may act, at least in part, via effects on lung development. Elastic fiber pathways were over-represented; products of elastin degradation have been shown to be elevated during acute exacerbations of COPD35,36. In addition, degradation of elastin by excess neutrophil-released elastase in the lung leads to emphysema in individuals with α-1 antitrypsin deficiency. CARD9, another high-priority gene at a new signal, encodes an adaptor protein involved in neutrophil recruitment in respiratory fungal infection37. Tissue-specific enrichment of lung function signals overlapping H3K4me1 was seen in stomach smooth muscle. Although comparable H3K4me1 data were not available for airway smooth muscle, similar findings have been reported previously for rectal smooth muscle38.
The 17q21.31 inversion has previously been associated with lung function. Custom imputation of additional structural variation at the locus, along with eQTL evidence and deleterious variants in the gene, suggested that KANSL1 might drive the association. Among the new signals reported in this study, SNPs in an intron of EEFSEC on chromosome 3 are correlated with expression of nearby gene RUVBL1. Both KANSL1 and RUVBL1 encode members of histone modification complexes.
A new signal on chromosome 20 (rs72448466, intronic in ZGPAT), which showed association with FVC almost as strong as its association with FEV1, is an eQTL for the telomere gene RTEL1. Although rs72448466[–>GT] was not the strongest eQTL for RTEL1 (r2 = 0.6 with the top eQTL variant), RTEL1 is of interest as it has recently been implicated in familial pulmonary fibrosis39. Variant rs72448466 has also been associated with inflammatory bowel disease, prostate cancer and atopic dermatitis.
Our implication of genes of potential functional relevance to the 97 signals was based on gene expression data (eQTL) and associated deleterious variants within a gene. Although eQTL evidence currently gives the best in silico indication of which gene (or genes) might be functionally relevant to a signal, conclusive evidence for a causal relationship between SNP genotype and gene expression can only be obtained through direct molecular experiments.
Six signals of association have previously been identified within the HLA region. Using a custom imputation approach, we identified the presence of alanine (compared to aspartic acid, valine or serine) at amino acid position 57 in HLA-DQβ1 as associated with decreased lung function and the main driver of signals in this region. The presence of alanine is also strongly associated with risk of type 1 diabetes40.
The three lung function traits we studied are correlated. The overall and genetic correlations were as follows: 0.88 and 0.87 between FEV1 and FVC; 0.46 versus 0.35 between FEV1 and FEV1/FVC; and 0.038 and –0.17 between FVC and FEV1/FVC (transformed traits, as studied in UK Biobank and SpiroMeta15, respectively). One might expect variants showing the strongest association with FEV1 and FEV1/FVC to be of the greatest relevance for COPD, and genetic correlations of –0.76 and –0.9 have been reported between COPD and FEV1 and between COPD and FEV1/FVC, respectively41. We show, however, that variants associated with one of these traits also tend to be associated with one of the other two lung function traits studied (for example, all but two signals for FVC are also associated (P < 0.05) with FEV1; Supplementary Table 4). Although classification of COPD in UK Biobank was based on pre-bronchodilator spirometry data, we have previously shown that this leads to minimal misclassification of moderate-to-severe (GOLD score 2–4) COPD42. The effect size estimates for COPD associations could be influenced by differences in case ascertainment between the follow-up studies. Motivated by avoidance of potential winner's curse bias for the 48 variants discovered using UK BiLEVE, we excluded UK BiLEVE from individual variant analyses. However, this excluded 9,563 moderate-to-severe COPD cases, and the significance of COPD association tests for these variants should therefore be interpreted with caution. Notably, we found effect size estimates only slightly smaller in deeply characterized COPD case–control studies than in UK Biobank (OR per 1 s.d. change in allelic risk score = 1.36 as compared to 1.42). While we show that an appreciable proportion of COPD cases could be attributable to allelic risk scores above the first decile, great caution must be exercised in interpretation of population attributable risk fraction estimates, given considerations of shared etiologic responsibility43. The lung-function-associated variants we report were not associated with acute exacerbations of COPD. Although more powerful studies of exacerbations will be required, this suggests that different genetic mechanisms could underlie risk of acute exacerbations.
A threshold of P < 5 × 10−8 is a valid threshold for genome-wide significance in GWAS analyses of common variants44. Our genotyping and imputation strategy resulted in testing of 27.6 million variants, of which 21.6 million had MAF <5% and 18.2 million had MAF <1%. Although all of our 43 signals were common, had we adopted a stricter threshold for genome-wide significance, for example, P < 1 × 10−8 (recommended in a recent report of significance thresholds in whole-genome sequencing44), only two of our signals (rs10246303[A>T] in the 3′ UTR of C1GALT1 on chromosome 7 and rs1698268[A>T] near LINC00911 on chromosome 14) would not have reached significance. Thirty-nine of the 43 signals were additionally supported by statistically significant independent replication in stage 2 (P < 0.05/43; Supplementary Table 3).
In summary, our study provides comprehensive evidence regarding genetic variants associated with lung function and their association with susceptibility to COPD, with a more than threefold difference in COPD risk between the highest and lowest allelic risk score deciles. While translation of GWAS findings can take some years and requires extensive additional work, selecting genetically supported targets could double the drug development success rate17. The future clinical relevance of our findings include contributions toward understanding of disease pathogenesis, identification of drug targets for targeting or repositioning of drugs18, and potentially improved prediction of COPD or its subtypes.
Methods
Study governance.
UK Biobank has ethical approval from the NHS National Research Ethics Service (11/NW/0382). Informed consent was obtained from all participants. All other studies were approved by an appropriate ethics committee or data protection authority (Supplementary Note).
Stage 1 study sample selection.
A genome-wide discovery study for variants associated with lung function measures was performed in 48,943 individuals from the UK BiLEVE16 subset of UK Biobank (UK BiLEVE, stage 1). In brief, UK Biobank comprised 502,682 individuals, of whom 275,939 were of self-reported European ancestry and had ≥2 FEV1 and FVC measures (Vitalograph Pneumotrac 6800) passing ATS/ERS criteria45. On the basis of the best (highest) available FEV1 measurement, 50,008 individuals from groups with extremely low (n = 10,002), near-average (n = 10,000) and extremely high (n = 5,002) percent predicted FEV1 were selected from among never smokers (total n = 105,272), and the same numbers were selected from among heavy smokers (mean of 35 pack-years of smoking; total n = 46,758). FEV1, FVC and FEV1/FVC distributions are summarized in Supplementary Figure 8. Genotyping was undertaken using the Affymetrix Axiom UK BiLEVE array16, and genotypes were imputed to a 1000 Genomes Project Phase 1 (ref. 46) and UK10K47,48 combined panel. A total of 27,624,732 imputed or directly genotyped autosomal variants with imputation quality (info) >0.5 and MAC ≥3 were included in the analysis. In total, 48,943 unrelated individuals passed all quality control steps and were used in this analysis.
Association testing and selection of signals from stage 1 for follow-up in stage 2.
Power calculations were undertaken using Quanto (see URLs) (Supplementary Fig. 9). For stage 1, GWAS of FEV1, FVC and FEV1/FVC were undertaken separately in heavy smokers and never smokers and meta-analysis was then performed for each trait. Linear regression of age, age2, sex, height, the first ten principal components of genetic ancestry and pack-years of smoking (in smokers) on each trait was undertaken, and residuals were ranked and transformed to inverse normally distributed z scores. For the first 26 lung function variants reported11,13,14,49, stage 2 effect size estimates14 were comparable to those from inverse normally distributed z scores in UK BiLEVE (Supplementary Fig. 10). Subsequently, these z scores were used for genome-wide association testing with an additive genetic model (SNPTEST v2.5). The full genome-wide stage 1 results are available via UK Biobank (see URLs).
From each of the three discovery GWAS, signals were selected for follow-up in stage 2 if they met an initial threshold of P < 5 × 10−7. Variants with low MAC (MAC between 3 and 20), were selected for follow-up only if the imputation quality (info) exceeded 0.8. The independence of signals was assessed as follows: the most strongly associated (P < 5 × 10−7) variant within a 1-Mb region was selected as a putative signal and the analysis was then repeated for that 1-Mb region conditioning on the most strongly associated variant. Any variant that then had conditional P < 5 × 10−7 was assigned as a secondary putative signal and also included in the conditional analysis. This process was repeated until no variants with P < 5 × 10−7 remained within the 1-Mb region. Results were confirmed using joint conditional analysis (GCTA50) and visual inspection of region plots. Previously reported signals were not included in the final list of putative signals to be taken for follow-up in stage 2. Where new signals for different traits were in LD (r2 > 0.2), the variant for the trait with the most significant association was followed up. Because of the extended LD structure in the MHC region, conditional analyses and GCTA were run over a 9-Mb region (chr. 6: 24,126,750–33,126,689). Two pairs of signals previously reported as being independent (rs16909859[G>A]11 and rs16909898[A>G]14 in PTCH1 and rs34712979[G>A]16 and rs6856422[T>G]15 in NPNT) were found to be correlated in our data.
Stage 2: follow-up in independent studies (quantitative lung function).
Putative new signals of association from stage 1 were followed up in three independent sets of samples (stage 2): (i) an independent subset of UK Biobank participants (UK Biobank; n = 49,727), (ii) a population-based consortium (SpiroMeta; n = 38,199)15 and (iii) the UK Households Longitudinal Study (UKHLS; n = 7,449). We did not include these studies in stage 1 as the SpiroMeta consortium was to be used for independent replication and the UKHLS and independent subset of UK Biobank participants were not yet available when stage 1 was undertaken. Each signal was followed up only for the trait with which it was most strongly associated in stage 1. The first tranche of genotype data and imputation output (merged 1000 Genomes Project Phase 3 and UK10K imputation panel) from UK Biobank was released in May 2015 (see URLs) and comprised the 49,979 individuals originally genotyped for UK BiLEVE (an unrelated subset of 48,943 of whom were used for discovery in this study) and an additional 102,757 individuals selected at random from the entire UK Biobank. From these 102,757 individuals, we initially selected 51,117 samples that had lung function measurements (FEV1 and FVC) meeting ATS/ERS criteria and had data for the covariates age, sex, height, principal components and smoking status recorded. Following further exclusion of individuals with sex mismatch (n = 41), individuals of non-European ancestry (on the basis of k-means clustering of principal components 1 and 2 with four clusters; n = 124) and one individual from each pair of related samples (KING relatedness > 0.088 (second-degree relatives); n = 1,225), a total of 49,727 individuals remained for analysis.
Details for the SpiroMeta consortium analysis (including contributing studies, spirometry details and methods) appear elsewhere15. In brief, this was an inverse-variance-weighted fixed-effects meta-analysis of 17 studies with imputation to the 1000 Genomes Project Phase 1 reference panel. Within each study, FEV1, FVC and FEV1/FVC measures were adjusted for age, age2, sex, height and population structure, separately for ever and never smokers. Inverse-normal-transformed residuals were then tested for association within each smoking stratum assuming an additive genetic effect, and meta-analysis was performed. Genomic control was applied to account for residual population structure. We only included SpiroMeta meta-analysis results in the meta-analysis in this study if neffective > 70% (>70% of 38,199), where neffective is the effective sample size after scaling for imputation quality15.
Summary statistics from a GWAS of FEV1, FVC and FEV1/FVC in 7,449 individuals were available from UKHLS (Supplementary Note). SNPs were genotyped using the Illumina Infinium HumanCoreExome BeadChip kit and imputed against the same 1000 Genomes Project + UK10K combined imputation panel as used in discovery in this study. Association testing was performed separately for ever and never smokers with covariates for age, age2, sex, height and ancestry principal components included, as for stage 1. We only included UKHLS results in the meta-analysis in this study if imputation info > 0.5 and MAC ≥3.
Meta-analysis of stage 1 and stage 2.
All meta-analyses were undertaken using fixed-effects inverse variance weighting, which takes the directionality of each association into account. Effect estimates for all variants followed up in stage 2 were subjected to meta-analysis across the three stage 2 studies, and the combined result was then subjected to meta-analysis with stage 1 results. When the discovery variant was not present in any stage 2 study, a proxy (r2 > 0.8) that was available in all stage 1 and stage 2 studies was used. We report signals with association P < 5 × 10−8 in the meta-analysis of stage 1 and stage 2 as new signals of association with lung function.
Assessment of stage 1 and stage 2 sample overlap by LD score regression.
LD score regression was used to assess the extent of confounding. Absence of significant confounding indicates that factors such as sample overlap and/or population stratification are not evident. Precomputed LD scores from a European population were used (see URLs), based on genotypes for 1,293,150 HapMap 3 SNPs in samples from the 1000 Genomes Project EUR population. Association results were filtered (info > 0.9 and MAF > 1%) before running LD score regression on (i) three pairwise meta-analyses of results from UK BiLEVE (stage 1) and UK Biobank (stage 2), UK BiLEVE and SpiroMeta (stage 2), and UK Biobank and SpiroMeta and (ii) bivariate analyses of the three pairs of cohorts.
Effect sizes in adults and children.
The effects of variants on lung function in children were also tested in 5,062 children from ALSPAC (mean age of 8.6 years) and 1,220 children from the Raine study (mean age of 8.1 years). Data were available for 81 of the 97 variants (a proxy variant with r2 >0.7 was used for 11 signals) with imputation quality >0.5, of which 73 had imputation quality >0.8 (71 variants in ALSPAC and 35 variants in the Raine study). Association results from the two cohorts were combined using inverse-variance-weighted meta-analysis. A weighted risk score was approximated using pooled single-SNP results, as described in Dastani et al.51, and weights were obtained using estimated effect sizes from either SpiroMeta15 summary data (for SNPs discovered in UK Biobank) or from UK Biobank (for SNPs discovered elsewhere). The risk score was tested for the three lung function traits: FEV1, FVC and FEV1/FVC.
Refinement of signals.
A Bayesian method52 was used to fine-map lung-function-associated signals to the set of variants that were 95% likely to contain the underlying causal variant (assuming that the causal variant has been analyzed). This was undertaken for new signals and for previously reported signals that reached P < 1 × 10−5 in the stage 1 results. Following van de Bunt et al.53, we set the value of the prior W as 0.4 in the approximate Bayes factor formula. Signals in the HLA region were not included.
We re-imputed our 48,943 discovery samples across the HLA region (chr. 6: 29,607,078–33,267,103 (Build 37)) using IMPUTE2 v2.3.1 with a reference panel incorporating classical HLA alleles and amino acid changes54. The reference panel contained haplotypes for 5,225 samples from the Type 1 Diabetes Genetics Consortium (T1DGC) across 8,961 biallelic variants comprising 5,863 directly genotyped biallelic SNPs and 3,098 surrogate biallelic variants encoding multiallelic SNPs, indels, classical HLA alleles and amino acid changes. Association testing was then undertaken as described for stage 1 for FEV1 and FEV1/FVC.
Effects of lung-function-associated variants on other traits.
To assess whether the new and previously reported lung-function-associated variants had been reported in previous GWAS as associated with traits other than lung function and COPD, we queried the GWAS Catalog55 (last updated on 13 March 2016, downloaded on 17 March 2016) and GRASP56 (v2.0; downloaded on 17 March 2016) for genome-wide significant (P < 5 × 10−8) signals using the 95% credible set (if calculated) or all proxy SNPs (r2 > 0.8) within 2 Mb of the top variant in our data.
Clinical relevance: COPD susceptibility and risk of COPD exacerbations in European and Chinese populations.
The effect on COPD susceptibility of up to 95 of the 97 lung-function-associated signals was tested in the COPD study at deCODE Genetics (deCODE COPD study; 1,964 COPD cases and 142,262 controls for single-variant analyses and 1,248 COPD cases and 74,700 controls for risk score analyses); in three lung resection studies—Groningen, Laval and UBC (310 COPD cases and 332 controls); in COPD case–control studies—the COPDGene Study (2,812 COPD cases and 2,534 controls), Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-Points (ECLIPSE; 1,736 COPD cases and 176 controls), the National Emphysema Treatment Trial (NETT) and the Normative Aging Study (NAS) (NETT/NAS; 376 COPD cases and 435 controls) and the Norway GenKOLS study (Genetics of Chronic Obstructive Lung Disease; 854 cases and 805 controls); in eMR studies—Mount Sinai BioMe Biobank (BioMe; 207 COPD cases and 1,817 controls) and the Geisinger–Regeneron DiscovEHR Study (DiscovEHR; 1,280 COPD cases and 13,321 controls for single-variant analyses and 1,264 COPD cases and 13,032 controls for risk score analyses); and in UK Biobank (not including UK BiLEVE samples; 984 cases and 26,561 controls in total) and UK BiLEVE (9,563 moderate-to-severe cases and 27,387 controls). rs7050036, located on chromosome X, and chr12:114743533, with MAF = 0.15%, were not present in most studies and were therefore excluded from these analyses, bringing the 97 signals to 95. Of the 95 signals, 47 were previously discovered independently of UK BiLEVE and were tested for association using all available COPD cases and controls (20,086 COPD cases and 215,630 controls). The remaining 48 signals were discovered using UK BiLEVE data and so were tested for association using 10,523 COPD cases and 188,243 controls (UK BiLEVE excluded). The effect on risk of COPD exacerbation was additionally tested in the Lung Health Study (LHS; 100 COPD exacerbation cases and 4,002 COPD controls) as well as subsets of UK Biobank (including UK BiLEVE; 647 cases and 9,900 controls), COPDGene (557 cases and 2,255 controls), ECLIPSE (278 cases and 1,458 controls), NETT/NAS (87 cases and 277 controls), GenKOLS (120 cases and 734 controls), BioMe (8 cases and 199 controls) and DiscovEHR (774 cases and 472 controls). Analyses of the effect of lung-function-associated variants on COPD susceptibility and on risk of COPD exacerbation in a Chinese-ancestry population were undertaken using the China Kadoorie Biobank (CKB) prospective cohort, in which data were available for 71 (single-variant analyses) or 70 (risk score analyses) of the 95 variants (or proxies) for analyses of COPD susceptibility (7,116 COPD cases and 20,919 controls) and risk of COPD exacerbation (5,292 cases and 1,824 controls). Further details on all studies, including case and control definitions, are given in the Supplementary Note and Supplementary Table 20.
To test single-variant associations with COPD susceptibility and risk of exacerbation, logistic regression was applied using age, age2, sex and height as covariates (unless otherwise indicated; Supplementary Note) and assuming an additive genetic effect. To test the joint effect of these variants, risk alleles for the subset of the 95 signals with data available in each study (from 86 to 95) were summed to create an unweighted genetic risk score and logistic regression was used to test the effect of the risk score, as a continuous variable, on COPD status and COPD exacerbation status (adjusted for age, age2, sex and height, unless otherwise indicated; Supplementary Note). Results, both from single-variant and risk score analyses, were subjected to meta-analysis separately for studies where similar study designs and phenotyping were used—eMR, case–control and lung resection studies—and results were also subjected to meta-analysis across studies. Inverse-variance-weighted meta-analysis was used. In CKB, analyses were adjusted for sex, age, age2, height, region (n = 10) and disease status (n = 5) and final results were genomic control corrected on the basis of genome-wide inflation estimates. Heterogeneity was evaluated using the I2 statistic57.
We calculated odds ratios for spirometrically defined COPD for weighted risk score deciles in UK Biobank (incorporating UK BiLEVE; 10,547 cases (pre-bronchodilator percent predicted FEV1 <80% and FEV1/FVC <0.7) and 53,948 controls (FEV1/FVC >0.7 and percent predicted FEV1 >80%)). Weighting of the risk score was undertaken using log-transformed odds ratios for COPD calculated in studies free of winner's curse bias (Supplementary Table 21). We scaled the log-transformed odds ratios so that the weights added up to 95.
Population attributable risk fraction calculation.
The population attributable risk fraction (PARF) was calculated using the formula

where P(E) is the probability of the exposure, in this case the probability of having more risk alleles than individuals in the lowest decile of the risk score distribution (P(E) = 0.9), and OR refers to the odds of having COPD for individuals in deciles 2–10 of the risk score distribution as compared to the odds of having COPD for individuals in the lowest decile (decile 1) of the risk score distribution. The odds ratios were calculated separately in ever and heavy smokers using logistic regression adjusted for age, age2, sex, height and the first ten ancestry principal components and an additional pack-years adjustment for heavy smokers, and meta-analysis was then performed using inverse variance weighting. Confidence intervals were estimated using the formula above with the lower and upper bounds of the meta-analysis odds ratios estimated by logistic regression. These analyses were performed using UK Biobank data and the COPD case definition described above: individuals with percent predicted FEV1 <80% and FEV1/FVC <0.7 were selected as COPD cases, and those with FEV1/FVC >0.7 and percent predicted FEV1 >80% were selected as controls.
Implication of causal genes.
To implicate the likely causal gene (or genes) for each of the new and previously reported signals (97 in total), we employed functional annotation and analysis of gene expression data. All variants within 25 kb of the top SNP at each locus, variants within 500 kb of the top SNP with r2 >0.5, and variants within 1 Mb of the top SNP with r2 >0.8 were annotated using the Ensembl Variant Effect Predictor (VEP). A variant was labeled as deleterious if it was a missense coding variant that was annotated as 'deleterious' by SIFT, was annotated as 'probably damaging' or 'potentially damaging' by PolyPhen-2, had a CADD scaled score ≥20 (CADD_PHRED ≥20) or had a GWAVA score >0.5. Deleterious variants were each, in turn, included as a covariate in the association analysis for the top SNP. If inclusion of the deleterious variant as a covariate reduced the association signal for the top SNP such that P > 0.01, that deleterious variant was deemed to explain part of the signal. If annotation (for example, as a coding variant) implicated a specific gene, then the gene was classified as a high-priority gene for the relevant signal.
At each signal, the sentinel SNP and top proxies with r2 >0.4 and within 2 Mb (no limit on the number of proxies) were used to query three eQTL resources: lung eQTLs23,24,25, blood eQTLs58 and GTEx59 (artery (aorta and tibia), adrenal gland, colon sigmoid, esophagus (gastroesophageal junction and mucosa), transformed fibroblasts, lung, spleen, skin (sun-exposed lower leg), stomach, testis, thyroid, whole blood). An FDR of 10% was used as a threshold for significance in the lung and blood eQTL data sets, and an FDR threshold of 5% was used in GTEx (because of the large number of different tissues and cell types and the small sample size). A gene was classified as a potential causal gene if the sentinel SNP or proxy (r2 >0.4) showed significant evidence of being an eQTL signal for that gene. Genes were further classified as high-priority genes if the variant most strongly associated with the lung function traits (or a proxy with r2 >0.9) was also the variant most strongly associated with expression of the gene in one or more of the eQTL data sets (that is, there was colocalization of the lung-function-associated SNP and the gene-expression-associated SNP). Because of the extended LD across the MHC region, only high-priority genes were identified for the signals in the MHC region.
Pathway analyses.
The genes implicated for each signal (high-priority genes only and all genes) were tested for enrichment of gene sets and pathways using ConsensusPathDB60. Pathways or gene sets represented entirely by genes implicated by the same association signal were excluded. Pathways or gene sets represented by two or more genes from the same association signal were flagged. Pathway enrichment using all genome-wide P values was undertaken using MAGENTA61 as previously described15. Gene sets and pathways with FDR < 5% either including or excluding the HLA region are reported.
Tissue-specific enrichment of overlap with histone marks.
Two methods were used to test for enrichment of the 97 signals of association with lung function for H3K4me1 and H3K4me3 histone marks in up to 127 different tissue and cell types from the Encyclopedia of DNA Elements (ENCODE) and Roadmap Epigenomics projects38.
First, enrichment was investigated with a hypergeometric test (as previously described38) using SNPs from the GWAS Catalog (hg19; downloaded 2 November 2015) as background. The GWAS Catalog was pruned within each contributing GWAS to retain only SNPs that were at least 1 Mb apart in that study, resulting in 18,202 SNPs for further analysis. BEDtools was used to calculate overlap with precomputed 'gapped peaks' for H3K4me1 and H3K4me3 histone marks, and a hypergeometric test was used to test the significance of enrichment for the 97 lung function variants as compared to the background of GWAS Catalog SNPs. Control for multiple testing was undertaken by selecting 97 random variants from the pruned GWAS Catalog and repeating the enrichment computation. FDR was calculated from 10,000 randomizations, and FDR = 10% was used as the significance threshold.
The second method used, GoShifter, calculates overlap enrichment against a null distribution generated by locally shifting annotations62. LD was calculated using the stage 1 population. Precomputed 'narrow peaks' for H3K4me1 and H3K4me3 histone marks from the Roadmap Epigenomics project were used. Tissues and cell types with overlap enrichment P < 0.05 are reported.
Druggability.
We searched the ChEMBL database (v21; last updated on 1 February 2016, downloaded on 11 February 2016) to assess whether any of the implicated genes encode proteins that are targets of approved drugs or drug compounds in development. We additionally searched for genes predicted to interact (parameters: STRING score ≥0.90, maximum of ten interactions per gene) with each of the high-priority genes31.
Data availability.
The stage 1 (UK BiLEVE) genome-wide association results for FEV1, FVC and FEV1/FVC are available from UK Biobank at http://www.ukbiobank.ac.uk/. The sources of all other data used in this study can be found in the Online Methods and Supplementary Note.
URLs.
UK Biobank genetic data release, http://www.ukbiobank.ac.uk/scientists-3/genetic-data/; LD score regression, Broad Institute, http://ldsc.broadinstitute.org/; Global Initiative for Chronic Obstructive Lung Disease (GOLD), http://goldcopd.org/.
References
Fletcher, C. & Peto, R. The natural history of chronic airflow obstruction. BMJ 1, 1645–1648 (1977).
Lange, P., Celli, B. & Agustí, A. Lung-function trajectories and chronic obstructive pulmonary disease. N. Engl. J. Med. 373, 1575 (2015).
Lozano, R. et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2095–2128 (2012).
Salvi, S.S. & Barnes, P.J. Chronic obstructive pulmonary disease in non-smokers. Lancet 374, 733–743 (2009).
Melén, E. Bridging genetics, epidemiology, and respiratory medicine. Am. J. Respir. Crit. Care Med. 190, 716–718 (2014).
Svanes, C. et al. Early life origins of chronic obstructive pulmonary disease. Thorax 65, 14–20 (2010).
Celli, B.R. et al. An official American Thoracic Society/European Respiratory Society statement: research questions in COPD. Eur. Respir. J. 45, 879–905 (2015).
Pillai, S.G. et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet. 5, e1000421 (2009).
Cho, M.H. et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat. Genet. 42, 200–202 (2010).
Cho, M.H. et al. Risk loci for chronic obstructive pulmonary disease: a genome-wide association study and meta-analysis. Lancet Respir. Med. 2, 214–225 (2014).
Hancock, D.B. et al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat. Genet. 42, 45–52 (2010).
Loth, D.W. et al. Genome-wide association analysis identifies six new loci associated with forced vital capacity. Nat. Genet. 46, 669–677 (2014).
Repapi, E. et al. Genome-wide association study identifies five loci associated with lung function. Nat. Genet. 42, 36–44 (2010).
Soler Artigas, M. et al. Genome-wide association and large-scale follow up identifies 16 new loci influencing lung function. Nat. Genet. 43, 1082–1090 (2011).
Soler Artigas, M. et al. Sixteen new lung function signals identified through 1000 Genomes Project reference panel imputation. Nat. Commun. 6, 8658 (2015).
Wain, L.V. et al. Novel insights into the genetics of smoking behaviour, lung function, and chronic obstructive pulmonary disease (UK BiLEVE): a genetic association study in UK Biobank. Lancet Respir. Med. 3, 769–781 (2015).
Nelson, M.R. et al. The support of human genetic evidence for approved drug indications. Nat. Genet. 47, 856–860 (2015).
Sanseau, P. et al. Use of genome-wide association studies for drug repositioning. Nat. Biotechnol. 30, 317–320 (2012).
Palmer, L.J. et al. Familial aggregation and heritability of adult lung function: results from the Busselton Health Study. Eur. Respir. J. 17, 696–702 (2001).
Wilk, J.B. et al. Evidence for major genes influencing pulmonary function in the NHLBI family heart study. Genet. Epidemiol. 19, 81–94 (2000).
Weiss, S.T. Lung function and airway diseases. Nat. Genet. 42, 14–16 (2010).
Gudbjartsson, D.F. et al. Large-scale whole-genome sequencing of the Icelandic population. Nat. Genet. 47, 435–444 (2015).
Hao, K. et al. Lung eQTLs to help reveal the molecular underpinnings of asthma. PLoS Genet. 8, e1003029 (2012).
Lamontagne, M. et al. Refining susceptibility loci of chronic obstructive pulmonary disease with lung eqtls. PLoS One 8, e70220 (2013).
Obeidat, M. et al. GSTCD and INTS12 regulation and expression in the human lung. PLoS One 8, e74630 (2013).
Regan, E.A. et al. Genetic epidemiology of COPD (COPDGene) study design. COPD 7, 32–43 (2010).
Vestbo, J. et al. Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points (ECLIPSE). Eur. Respir. J. 31, 869–873 (2008).
Fishman, A. et al. A randomized trial comparing lung-volume-reduction surgery with medical therapy for severe emphysema. N. Engl. J. Med. 348, 2059–2073 (2003).
Bell, B., Rose, C.L. & Damon, H. The Normative Aging Study: an interdisciplinary and longitudinal study of health and aging. Aging Hum. Dev. 3, 5–17 (1972).
Dewey, F.E. et al. Inactivating variants in ANGPTL4 and risk of coronary artery disease. N. Engl. J. Med. 374, 1123–1133 (2016).
Szklarczyk, D. et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–D452 (2015).
Wilk, J.B. et al. Genome-wide association studies identify CHRNA5/3 and HTR4 in the development of airflow obstruction. Am. J. Respir. Crit. Care Med. 186, 622–632 (2012).
Jiang, Z. et al. A chronic obstructive pulmonary disease susceptibility gene, FAM13A, regulates protein stability of β-catenin. Am. J. Respir. Crit. Care Med. 194, 185–197 (2016).
Obeidat, M. et al. Molecular mechanisms underlying variations in lung function: a systems genetics analysis. Lancet Respir. Med. 3, 782–795 (2015).
Carter, R.I., Ungurs, M.J., Mumford, R.A. & Stockley, R.A. Aα-Val360: a marker of neutrophil elastase and COPD disease activity. Eur. Respir. J. 41, 31–38 (2013).
Sand, J.M. et al. Accelerated extracellular matrix turnover during exacerbations of COPD. Respir. Res. 16, 69 (2015).
Jhingran, A. et al. Compartment-specific and sequential role of MyD88 and CARD9 in chemokine induction and innate defense during respiratory fungal infection. PLoS Pathog. 11, e1004589 (2015).
Roadmap Epigenomics Consortium. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Cogan, J.D. et al. Rare variants in RTEL1 are associated with familial interstitial pneumonia. Am. J. Respir. Crit. Care Med. 191, 646–655 (2015).
Todd, J.A., Bell, J.I. & McDevitt, H.O. HLA-DQβ gene contributes to susceptibility and resistance to insulin-dependent diabetes mellitus. Nature 329, 599–604 (1987).
Hobbs, B.D. et al. Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat. Genet. http://dx.doi.org/10.0138/ng.3752 (2017).
Soler Artigas, M. et al. Effect of five genetic variants associated with lung function on the risk of chronic obstructive lung disease, and their joint effects on lung function. Am. J. Respir. Crit. Care Med. 184, 786–795 (2011).
Poole, C. A history of the population attributable fraction and related measures. Ann. Epidemiol. 25, 147–154 (2015).
Fadista, J., Manning, A.K., Florez, J.C. & Groop, L. The (in)famous GWAS P-value threshold revisited and updated for low-frequency variants. Eur. J. Hum. Genet. 24, 1202–1205 (2016).
Miller, M.R. et al. Standardisation of spirometry. Eur. Respir. J. 26, 319–338 (2005).
Abecasis, G.R. et al. A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 (2010).
Huang, J. et al. Improved imputation of low-frequency and rare variants using the UK10K haplotype reference panel. Nat. Commun. 6, 8111 (2015).
Walter, K. et al. The UK10K project identifies rare variants in health and disease. Nature 526, 82–90 (2015).
Wilk, J.B. et al. A genome-wide association study of pulmonary function measures in the Framingham Heart Study. PLoS Genet. 5, e1000429 (2009).
Yang, J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, 369–375 (2012).
Dastani, Z. et al. Novel loci for adiponectin levels and their influence on type 2 diabetes and metabolic traits: a multi-ethnic meta-analysis of 45,891 individuals. PLoS Genet. 8, e1002607 (2012).
Wakefield, J. A Bayesian measure of the probability of false discovery in genetic epidemiology studies. Am. J. Hum. Genet. 81, 208–227 (2007).
van de Bunt, M., Cortes, A., Brown, M.A., Morris, A.P. & McCarthy, M.I. Evaluating the performance of fine-mapping strategies at common variant GWAS loci. PLoS Genet. 11, e1005535 (2015).
Jia, X. et al. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS One 8, e64683 (2013).
Welter, D. et al. The NHGRI GWAS Catalog, a curated resource of SNP–trait associations. Nucleic Acids Res. 42, D1001–D1006 (2014).
Eicher, J.D. et al. GRASP v2.0: an update on the Genome-Wide Repository of Associations between SNPs and phenotypes. Nucleic Acids Res. 43, D799–D804 (2015).
Higgins, J.P., Thompson, S.G., Deeks, J.J. & Altman, D.G. Measuring inconsistency in meta-analyses. Br. Med. J. 327, 557–560 (2003).
Westra, H.J. et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 45, 1238–1243 (2013).
GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015).
Kamburov, A., Stelzl, U., Lehrach, H. & Herwig, R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 41, D793–D800 (2013).
Segrè, A.V., Groop, L., Mootha, V.K., Daly, M.J. & Altshuler, D. Common inherited variation in mitochondrial genes is not enriched for associations with type 2 diabetes or related glycemic traits. PLoS Genet. 6, e1001058 (2010).
Trynka, G. et al. Disentangling the Effects of Colocalizing Genomic Annotations to Functionally Prioritize Non-coding Variants within Complex-Trait Loci. Am. J. Hum. Genet. 97, 139–152 (2015).
Acknowledgements
This work was funded by a Medical Research Council (MRC) strategic award to M.D.T., I.P.H., D.S. and L.V.W. (MC_PC_12010). This research has been conducted using the UK Biobank Resource under application 648. This article presents independent research funded partially by the National Institute for Health Research (NIHR). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the UK Department of Health. This research used the ALICE and SPECTRE High-Performance Computing Facilities at the University of Leicester. Additional acknowledgments and funding details can be found in the Supplementary Note.
Author information
Authors and Affiliations
Consortia
Contributions
L.V.W., D.J.P., M.-R.J., A.L.J., N.J.W., J.F.W., B.S., H.S., N.M.P.-H., S.K., C.G., I.J.D., I. Rudan, S.M.K., O.P., M.K., C.H., T.L., O.T.R., A.J.H., C.E.P., P.D.S., A.G., P.S.B., J.D.C., T.H.B., N.N.H., R.A.M., I. Ruczinski, K.C.B., Y.B., P.J., P.D.P., D.D.S., K.H., E.P.B., R.J.F.L., R.G.W., Z.C., I.Y.M., L.L., E.Z., I. Sayers, D.P.S., I.P.H., U.G. and M.D.T. contributed to the conception and study design. L.V.W., N.S., M.S., A.M.E., B.N., L.B.-C., M.O., A.P.H., M.A.P., R.J.H., C.K.B., T.L.R., A.G.F., C.J., T.B., V.E.J., R.J.A., B.P.P., A.C., M.W., J.H., J.Z., P.K.J., B.S., R.R., M.I., N.M.P.-H., S.E.H., J.M., S.E., I. Surakka, V.V., C.H., T.L., D.M.E., C.A.W., E.S.W., R.B., B.D.H., A.A.L., D.W.S., M.v.d.B., C.-A.B., D.C.N., O.G., F.E.D., S.E.B., D.J.C., H.L.K., S.J., G. Thorleifsson, I.J., T.G., K.S., C.S., G.N., R.G.W., J.V., O.P.K., M.H.C., E.K.S., G. Trynka, J.H.Z. and D.P.S. contributed to data analysis. L.V.W., N.S., M.S., A.M.E., B.N., M.O., A.P.H., M.A.P., R.J.H., C.K.B., T.L.R., A.G.F., C.J., V.E.J., A.C., M.-R.J., B.S., R.R., H.S., M.I., N.M.P.-H., S.K., C.G., C.H., A.G., C.S., G.N., R.J.F.L., A.L.H., C.B., I. Sayers, A.P.M., D.P.S., I.P.H. and M.D.T. contributed to data interpretation.
Corresponding authors
Ethics declarations
Competing interests
F.E.D. and S.E.B. are employed by Regeneron Pharmaceuticals. D.C.N. is employed by Merck. In the past three years, E.K.S. received honoraria and consulting fees from Merck, grant support and consulting fees from GlaxoSmithKline, and honoraria and travel support from Novartis. S.J., G. Thorleifsson, I.J. and K.S. are employed by deCODE Genetics/Amgen. M.H.C. receives grant funding from GlaxoSmithKline.
Additional information
A list of members and affiliations appears in the Supplementary Note.
A list of members and affiliations appears in the Supplementary Note.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–10, Supplementary Tables 1, 2, 5–9, 11 and 13–21, and Supplementary Note (PDF 8709 kb)
Supplementary Tables 3, 4, 10 and 12
Supplementary Tables 3, 4, 10 and 12 (XLSX 95 kb)
Rights and permissions
About this article

Cite this article
Wain, L., Shrine, N., Artigas, M. et al. Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat Genet 49, 416–425 (2017). https://doi.org/10.1038/ng.3787
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3787
This article is cited by
-
Lipid metabolism-related genes are involved in the occurrence of asthma and regulate the immune microenvironment
BMC Genomics (2024)
-
Human genetic associations of the airway microbiome in chronic obstructive pulmonary disease
Respiratory Research (2024)
-
Genetics of chronic respiratory disease
Nature Reviews Genetics (2024)
-
Prediction and stratification of longitudinal risk for chronic obstructive pulmonary disease across smoking behaviors
Nature Communications (2023)
-
A genome-wide association study identifies distinct variants associated with pulmonary function among European and African ancestries from the UK Biobank
Communications Biology (2023)
