
Abstract
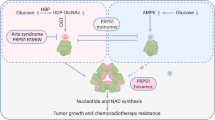
The oxidative pentose phosphate pathway (PPP) contributes to tumour growth, but the precise contribution of 6-phosphogluconate dehydrogenase (6PGD), the third enzyme in this pathway, to tumorigenesis remains unclear. We found that suppression of 6PGD decreased lipogenesis and RNA biosynthesis and elevated ROS levels in cancer cells, attenuating cell proliferation and tumour growth. 6PGD-mediated production of ribulose-5-phosphate (Ru-5-P) inhibits AMPK activation by disrupting the active LKB1 complex, thereby activating acetyl-CoA carboxylase 1 and lipogenesis. Ru-5-P and NADPH are thought to be precursors in RNA biosynthesis and lipogenesis, respectively; thus, our findings provide an additional link between the oxidative PPP and lipogenesis through Ru-5-P-dependent inhibition of LKB1–AMPK signalling. Moreover, we identified and developed 6PGD inhibitors, physcion and its derivative S3, that effectively inhibited 6PGD, cancer cell proliferation and tumour growth in nude mice xenografts without obvious toxicity, suggesting that 6PGD could be an anticancer target.
Similar content being viewed by others


Main
The Warburg effect in cancer cells describes increased aerobic glycolysis, producing not only ATP but also precursors for anabolic biosynthesis of macromolecules that are necessary for cell proliferation and rapid tumour growth1,2,3. The glycolytic intermediate glucose-6-phosphate is diverted into the oxidative pentose phosphate pathway (PPP), which produces ribose-5-phosphate (R-5-P), a precursor for nucleotide synthesis3,4,5,6,7,8. The oxidative PPP also produces nicotinamide adenine dinucleotide phosphate (NADPH), which is not only required for biosynthesis of lipids but is also a crucial antioxidant that quenches the reactive oxygen species (ROS) produced during rapid proliferation of cancer cells and maintains redox homeostasis2. Therefore, the oxidative PPP plays a crucial role in metabolic coordination of glycolysis, biosynthesis and appropriate redox status to provide an overall metabolic advantage to tumour cell proliferation and disease development. Indeed, inhibition of glucose-6-phosphate dehydrogenase (G6PD), the first enzyme of the oxidative PPP that produces NADPH, results in attenuated cell growth with potentiated H2O2-mediated cell death, probably due to lack of NADPH (refs 9,10,11,12). Moreover, matrix-detachment upregulates G6PD, which confers anoikis resistance to detached ErbB2-transformed MCF-10A breast cancer cells13. Furthermore, 6-amino-nicotinamide (6-AN), an inhibitor of G6PD, has demonstrated anti-tumorigenic effects in leukaemia, glioblastoma and lung cancer cells14.
6PGD is the third enzyme in the oxidative PPP, which converts 6-phosphogluconate (6-PG) to Ru-5-P and produces NADPH. We recently reported that glycolytic enzyme phosphoglycerate mutase 1 (PGAM1) signals through 6PGD to coordinate glycolysis and the oxidative PPP in cancer cells, suggesting an important role for 6PGD in cancer cell metabolism and tumour growth5. However, although 6PGD has been reported to be upregulated in many cancers, including colorectal cancers15, cervical intraepithelial neoplasia16,17, thyroid tumours18 and lung cancers19, it remains unclear whether and how 6PGD contributes to oxidative PPP flux and subsequent biosynthesis and redox homeostasis in cancer cells, as well as cancer cell proliferation and tumour growth. It was recently reported that knockdown of 6PGD in lung cancer H1975 cells resulted in attenuated cell proliferation and tumour size in xenograft mice. However, suppression of 6PGD in these cells did not cause defects in the oxidative PPP, nor affected intracellular levels of NADPH. Instead, 6PGD knockdown inhibited H1975 cell proliferation through induction of senescence19,20. Thus, it remains important to determine whether 6PGD is commonly important for the oxidative PPP flux and related metabolic and proliferative properties in cancer cells.
The tumour suppressor liver kinase B1 (LKB1) is a crucial upstream kinase of adenine monophosphate-activated protein kinase (AMPK), and LKB1–AMPK signalling plays a central role in the regulation of cell metabolism, survival and proliferation in response to nutrient and energy levels21,22,23. In particular, AMPK governs lipid metabolism by inhibiting fatty-acid and cholesterol synthesis through direct phosphorylation of the metabolic enzymes acetyl-CoA carboxylase (ACC) 1 and 2 (refs 23,24,25). In addition, AMPK-dependent inhibition of ACC1 and ACC2 contributes to regulation of NADPH homeostasis by decreasing NADPH consumption in fatty-acid synthesis, which promotes tumour cell survival during energy stress26,27. Here, we report that 6PGD activation is important for the oxidative PPP flux and tumour growth in diverse cancer cells, and thus represents a promising anticancer target. We present a molecular mechanism that explains how 6PGD regulates lipogenesis by controlling intracellular concentrations of its product Ru-5-P to inhibit LKB1–AMPK signalling, providing additional crosstalk between metabolic pathways and cell signalling networks.
RESULTS
6PGD is important for the oxidative PPP and lipogenesis, as well as proliferative and tumour growth potential of cancer cells
We found that stable knockdown of 6PGD resulted in decreased cell proliferation with reduced 6PGD activity in a group of human tumour and leukaemia cells, including human lung cancer H1299, H157 and H322, leukaemia K562 and head and neck cancer 212LN cells, but not the control normal proliferating keratinocyte HaCaT cells (Fig. 1a, b). Moreover, in a xenograft experiment in which nude mice were injected with control H1299 cells and 6PGD knockdown cells on the left and right flanks, respectively, the growth rate (Fig. 1c) and masses (Fig. 1d) of tumours derived from 6PGD knockdown cells were significantly reduced with decreased expression of the cell proliferation marker Ki-67 assessed by immunohistochemical (IHC) staining (Fig. 1e) compared with those of tumours formed by control cells over a ∼6-week time period. In addition, H1299 cells harbouring an inducible 6PGD short hairpin shRNA (shRNA) construct showed decreased 6PGD expression and activity (Fig. 1f), reduced cell proliferation (Fig. 1g) and attenuated tumour growth in xenograft mice (Fig. 1h–j) in the presence of doxycycline, compared with cells without treatment. Similar results were obtained in K562 leukaemia cells with stable 6PGD knockdown cells (Supplementary Fig. 1a, b), or K562 cells harbouring an inducible 6PGD shRNA construct in the presence and absence of doxycycline (Supplementary Fig. 1c–g).

(a,b) Cell proliferation rates determined by cell counting (a) and 6PGD activity (b; upper) in diverse human cancer cells with 6PGD stable knockdown (b; lower), which were normalized to the corresponding control vector cells. Keratinocyte HaCaT cells were included as controls. (c) Tumour growth was compared between xenograft nude mice injected with 6PGD KD H1299 cells and control vector cells. (d) Left: Dissected tumours in a representative nude mouse. Right: Tumour mass in xenograft nude mice injected with 6PGD KD H1299 cells compared with mice injected with the control vector cells. (e) Representative images of IHC staining of Ki-67 from xenograft mice injected with control vector or 6PGD KD H1299 cells. (f) 6PGD activity (upper) and protein expression (lower) in H1299 cells with inducible knockdown of 6PGD in the presence and absence of doxycycline (Dox). (g) Cell proliferation rates determined by cell counting in H1299 cells with inducible 6PGD knockdown and control cells in the presence and absence of Dox. (h) Tumour growth was compared between xenograft mice injected with H1299 cells with inducible 6PGD knockdown fed with drinking water in the presence or absence of Dox. (i) Dissected tumours in two representative nude mice (i; left) and tumour mass of xenograft mice injected with H1299 cells with inducible 6PGD knockdown (i; right) fed with drinking water in the presence or absence of Dox. (j) 6PGD activity (upper) and protein expression (lower) in tumour lysates. In a–g,j, data are from a single experiment that is representative of 2 independent experiments for a and 3 independent experiments for b–g,j. Source data for independently repeated experiments are provided in Supplementary Table 1. (c) Mean ± s.e.m.; n = 10 tumours from 10 mice, (d) n = 10 tumours from 10 mice; centre lines represent means, (h) mean ± s.e.m.; n = 9 tumours from 9 mice, (i) n = 9 tumours from 9 mice; centre lines represent means. The P values were determined by two-sided unpaired Student’s t-test for c,h,i and paired two-sided Student’s t-test for d (∗, 0.01 < P < 0.05; ∗∗, 0.001 < P < 0.01; ∗∗∗, P < 0.001). (a–g,j) Source data for independent replicates and experiments with sample size <5 are available in Supplementary Table 1. Unprocessed original scans of western blots are shown in Supplementary Fig. 9.
6PGD knockdown in H1299 cells also resulted in reduced oxidative PPP flux, NADPH/NADP+ ratio and intracellular levels of Ru-5-P (6PGD product) but increased 6-PG (6PGD substrate; Fig. 2a), as well as subsequently decreased intracellular levels of ribose-5-phosphate (R-5-P) and RNA biosynthesis (Fig. 2b). Similar results were obtained using additional cancer cell lines (Supplementary Fig. 1h–k). The decreased R-5-P levels may be predominantly due to defective oxidative PPP in 6PGD KD cells, because the protein expression and enzyme activity levels of transketolase (TKT) in non-oxidative PPP (Fig. 2c, left) and sensitivity to the TKT inhibitor oxythiamine in terms of R-5-P levels (Fig. 2c, right) were not altered in these cells. We also observed increased glycolytic rate (Fig. 2d) and lactate production (Fig. 2e) with elevated glucose uptake rate (Fig. 2f) and intracellular ATP levels (Fig. 2g) in distinct human cancer cells with 6PGD knockdown, whereas the O2 consumption rate in either the presence or absence of ATP synthase inhibitor oligomycin was not altered (Fig. 2i). Such increased glycolysis was probably due to 6-PG-dependent activation of the glycolytic enzyme phosphofructokinase28 (PFK), as 6-PG activated PFK in a dose-dependent manner (Fig. 2j) and 6PGD knockdown increased the activity of PFK, but not other glycolytic enzymes in cells (Fig. 2k). These results demonstrate an important role for 6PGD in regulation and coordination between the oxidative PPP and glycolysis in cancer cells and subsequent tumour growth, suggesting 6PGD as a promising anticancer target.

(a) 6PGD KD and control cells harbouring an empty vector were tested for oxidative PPP flux and NADPH/NADP+ ratio (left panels), and intracellular levels of Ru-5-P and 6-PG (right panels). (b) 6PGD KD H1299 cells and control vector cells were tested for intracellular R-5-P levels (left) and RNA biosynthesis (right). (c) Left: H1299 and K562 cells with stable knockdown of 6PGD were tested for transketolase (TKT) protein expression (lower) and enzyme activity (upper) levels. Right: H1299 and K562 cells with stable knockdown of 6PGD were tested for intracellular R-5-P levels in the presence and absence of TKT inhibitor oxythiamine (OT). (d–g) 6PGD KD and vector control cells were tested for glycolytic rate (d), lactate production (e), glucose uptake rate (f) and intracellular ATP level (g). (h) 6PGD KD H1299 cells and control vector cells were tested for oxygen consumption rate in the presence or absence of 100 nM oligomycin (ATP synthase inhibitor). (i) H1299 cell lysates were incubated with increasing concentrations of 6-PG, followed by in vitro PFK enzyme activity assay. PFK activities were normalized to the control sample without 6-PG treatment. (j) 6PGD KD and control vector H1299 cells were tested for activity of glycolytic enzymes including PFK, PGI, PGAM1 and LDHA. In a–j, data are from a single experiment that is representative of 2 independent experiments for a, g; middle, and h–j, 3 independent experiments for b–d, and f, and 4 independent experiments for e and g; left and right. Source data for independent replications and experiments with sample size <5 are available in Supplementary Table 1. Unprocessed original scans of western blots are shown in Supplementary Fig. 9.
6PGD promotes lipogenesis by controlling intracellular Ru-5-P levels to inhibit AMPK
Interestingly, we observed decreased lipogenesis in cells with stable 6PGD knockdown (Fig. 3a) or induced 6PGD shRNA in the presence of doxycycline compared with control cells (Fig. 3b and Supplementary Fig. 2a). To test whether 6PGD regulates lipogenesis through its products Ru-5-P and NADPH, cell contents were obtained from parental or 6PGD knockdown H1299 cells after sonication, followed by incubation with increasing concentrations of Ru-5-P or NADPH to rescue the levels of Ru-5-P (∼320 μM) or NADPH (7.2 μM) in 6PGD knockdown cells to be close to and exceed the physiological levels of Ru-5-P (∼800 μM) or NADPH (9.0 μM) in control cells, respectively, before lipid biosynthesis assay. We found that restoration of Ru-5-P to physiological levels rescued not only the decreased intracellular R-5-P levels (Supplementary Fig. 2b) but also the reduced lipid biosynthesis (Fig. 3c, upper) in 6PGD knockdown cells, whereas rescue of NADPH to physiological levels did not affect the decreased lipogenesis in these cells, although amounts of NADPH exceeding physiological levels eventually resulted in increased lipogenesis (Fig. 3c, lower).

(a) 6PGD KD and control cells harbouring an empty vector were tested for lipogenesis. (b) H1299 cells with inducible knockdown of 6PGD were tested for oxidative PPP flux (left) and lipogenesis (right) in the presence and absence of Dox. (c) Cell lysates from 6PGD KD H1299 and K562 cells were treated with increasing concentrations of Ru-5-P (upper panels) or NADPH (lower panels) for 12 h, followed by lipid biosynthesis assay. Final levels (fold) of Ru-5-P and NADPH were normalized to the control vector cells without treatment with Ru-5-P or NADPH, respectively. Lipid biosynthesis rates (%) were normalized to the control vector cells without treatment with Ru-5-P (upper) or NADPH (lower). (d) 6PGD KD cells and control vector H1299 cells were tested for enzyme activity of ACLY (left), FASN (middle) and ACC1 (right). Enzyme activities were normalized to the control vector cells. (e) Cell lysates from 6PGD KD H1299 cells were treated with or without AMPK inhibitor Compound C (10 μM) for 12 h (left) and 6PGD stable KD cells were infected with lentivirus harbouring AMPK shRNA (right). The samples were applied to lipid biosynthesis assay. In a–e, data are from a single experiment that is representative of 3 independent experiments for a,c and 2 independent experiments for b,d,e. Source data for independent replications and experiments with sample size <5 are available in Supplementary Table 1.
These data suggest that although NADPH serves as a precursor for lipid biosynthesis, the defect of lipogenesis in 6PGD knockdown cells is predominantly due to a Ru-5-P-dependent mechanism, which may affect a crucial step in lipogenesis. Indeed, we found that among the three key enzymes in the lipogenesis, acetyl-CoA carboxylase 1 (ACC1), but not ATP citrate lyase (ACLY) and fatty-acid synthase (FASN), has decreased enzyme activity in 6PGD knockdown cells (Fig. 3d). AMPK inhibits ACC1 by phosphorylating Ser 79, Ser 1200 and Ser 1215 (refs 29,30,31,32,33). Consistently, inhibition of AMPK by Compound C or shRNA (Supplementary Fig. 2c) also led to increased lipogenesis in 6PGD KD cells (Fig. 3e), suggesting that 6PGD may promote lipogenesis by controlling Ru-5-P to inhibit AMPK and subsequently activate ACC1.
Ru-5-P disrupts active LKB1 complex
Consistently, 6PGD knockdown resulted in increased activating Thr 172 phosphorylation of AMPK (ref. 21) and inhibitory Ser 79 phosphorylation of ACC1, and incubation with increasing concentrations of Ru-5-P (Fig. 4a, left), but not R-5-P (Fig. 4a, right) or NADPH (Supplementary Fig. 2d), reduced AMPK and ACC1 phosphorylation levels. Similar results were obtained using K562 cells with 6PGD knockdown (Supplementary Fig. 2e). However, incubation with Ru-5-P did not affect AMPK activity in an in vitro kinase assay using purified AMPK (Supplementary Fig. 2f), suggesting that Ru-5-P may inhibit an upstream activating kinase of AMPK, probably LKB1. Indeed, Ru-5-P inhibited the kinase activity of LKB1 wild type (WT) but not a kinase-dead form (K78M) in a dose-dependent manner in an in vitro LKB1 kinase assay using purified LKB1 protein incubated with purified AMPK as an exogenous substrate (Fig. 4b, left). Similar results were obtained using purified active LKB1 incubated with purified AMPK (Fig. 4b, upper right and Supplementary Fig. 2g) or myelin basic protein (MBP; Fig. 4b, lower right) as exogenous substrates. In contrast, incubation with increasing concentrations of R-5-P or NADPH did not affect LKB1 activity (Fig. 4b, right and Supplementary Fig. 2h, respectively).

(a) Cell lysates from 6PGD KD H1299 cells were treated with increasing concentrations of Ru-5-P (left) or R-5-P (right) for 4 h, followed by western blot for phosphorylation levels of AMPK (pT172) and ACC1 (pS79). Final levels (fold) of Ru-5-P or R-5-P were normalized to the control vector cells without treatment. (b) In vitro LKB1 kinase assays were performed using LKB1 wild type (WT) or a kinase-dead form (K78M) purified from A549 cells (left panel) or LKB1 WT purified from H1299 cells (right panel) incubated with recombinant AMPK (left and upper right) or MBP (lower right) as substrates in the presence of increasing concentrations of Ru-5-P (left), or Ru-5-P or R-5-P (right) at 37 °C for 20 min. Samples were applied for western blot. (c) Cell lysates of 6PGD KD H1299 cells were incubated with increasing concentrations of Ru-5-P (left) or R-5-P (right), followed by immunoprecipitation of MO25 and western blot to detect co-immunoprecipitated LKB1 and STRAD. (d) Purified active LKB1 complex purchased from Upstate (Millipore) was incubated with Ru-5-P (100 or 200 μM) for the indicated times, followed by immunoprecipitation of LKB1 and western blot to detect co-immunoprecipiated MO25. (e) Cell lysates of 6PGD KD H1299 cells were incubated with increasing concentrations of Ru-5-P, followed by immunoprecipitation of LKB1 and western blot to detect co-immunoprecipiated AMPK. a–e show results of 1 representative experiment from 2 independent experiments (a,b) and 3 independent experiments (c–e). Unprocessed original scans of western blots are shown in Supplementary Fig. 9.
LKB1 forms a complex with pseudokinase Ste20-related adaptor (STRAD) and scaffolding-like adaptor protein mouse protein 25 (MO25) to achieve full activation33,34,35,36. We found that treatment with increasing concentrations of Ru-5-P led to decreased association between LKB1, STRAD and MO25 in H1299 cell lysates (Supplementary Fig. 2i) without affecting the protein levels of LKB1, MO25 and STRAD (Supplementary Fig. 2j). Moreover, 6PGD knockdown resulted in increased binding among LKB1, STRAD and MO25, whereas rescue of decreased levels of Ru-5-P (Fig. 4c, left), but not R-5-P (Fig. 4c, right) or NADPH (Supplementary Fig. 2k), in 6PGD KD cell lysates reversed the increased formation of the LKB1–STRAD–MO25 complex. Similar results were obtained using K562 cells (Supplementary Fig. 2l). Furthermore, treatment with Ru-5-P was sufficient to trigger the disruption of purified active LKB1 complex within 5 min (Fig. 4d), which eventually attenuated AMPK binding to LKB1 (Fig. 4e), whereas incubation with R-5-P or NADPH for 20 min had no effect (Supplementary Fig. 2m).
6PGD is important for coordination between biosynthesis and redox homeostasis to promote cancer cell proliferation
6PGD knockdown also resulted in increased ROS levels, which was consistent with the decreased NADPH/NADP+ ratio, and treatment with an antioxidant agent N-acetylcysteine (NAC) did not rescue reduced lipogenesis (Supplementary Fig. 3a) but significantly reduced ROS levels (Fig. 5a and Supplementary Fig. 3b). Although treatments with either Compound C or NAC or both (Supplementary Fig. 3c–e), or knockdown of AMPK (Supplementary Fig. 3f) did not affect H1299 cell proliferation, such treatments in 6PGD knockdown cells significantly rescued the reduced cell proliferation (Supplementary Fig. 3g), and combined treatment with NAC and Compound C or AMPK shRNA demonstrated a further rescue effect (Fig. 5b). Moreover, we found that knockdown of LKB1 abolished AMPK phosphorylation enhanced by 6PGD knockdown (Supplementary Fig. 3h), which, although not affecting the decreased Ru-5-P levels, reversed the reduced lipogenesis in 6PGD knockdown cells (Supplementary Fig. 3i).

(a) 6PGD KD H1299 (left) and K562 (right) cells were assayed for general ROS levels in the absence and presence of NAC (1 and 3 mM) by measuring intracellular ROS-mediated DCFDA oxidation to fluorescent DCF by flow cytometry. The relative general ROS levels were normalized to the control vector cells without NAC treatment. (b) Cell proliferation rates were determined by cell counting in 6PGD KD H1299 cells treated with either or both NAC and Compound C (left) or 6PGD KD cells treated with either or both AMPK shRNA and NAC (right). (c–f) LKB1-deficient A549 cells with 6PGD knockdown (c) and control vector cells were assayed for intracellular Ru-5-P levels (d), phosphorylation levels of AMPK (e), and lipogenesis (f). (g,h) Cell lysates from 6PGD KD A549 cells were treated with increasing concentrations of Ru-5-P, followed by western blot for phosphorylation levels of AMPK (g) or lipogenesis assay (h). Final levels (fold) of Ru-5-P were normalized to the control vector cells without treatment. (i,j) A549 vector and 6PGD knockdown cells were tested for cell proliferation rate by cell counting (i) and ROS level (j) in the presence or absence of NAC (3 mM). (k–n) Normal proliferating HaCaT vector and 6PGD knockdown cells were assayed for phosphorylation levels of AMPK (k), oxidative PPP flux rate (l), intracellular Ru-5-P levels (m), and lipogenesis (n). a–n show data from a single experiment that is representative of 3 independent experiments for a,c,e,g and 2 independent experiments for b,d,f,h–n. Source data for independent replications and experiments with sample size <5 are available in Supplementary Table 1. Unprocessed original scans of western blots are shown in Supplementary Fig. 9.
Consistent with these findings, although stable knockdown of 6PGD in LKB1-deficient A549 cells resulted in decreased 6PGD activity and subsequent Ru-5-P levels (Fig. 5c and d, respectively), 6PGD knockdown did not affect phosphorylation levels of AMPK and lipogenesis (Fig. 5e and f, respectively). Additionally, rescue of the decreased Ru-5-P levels in cell lysates from A549 cells with 6PGD knockdown did not affect AMPK phosphorylation levels or lipogenesis (Fig. 5g and h, respectively), whereas NAC treatment completely rescued decreased cell proliferation (Fig. 5i) by reversing increased ROS level (Fig. 5j) in A549 cells with stable 6PGD knockdown. Furthermore, expression of LKB1 WT but not the kinase-dead form K78M in LKB1-deficient A549 cells with 6PGD knockdown resulted in increased AMPK activation, decreased lipogenesis that was rescued by Ru-5-P treatment, and further decreased cell proliferation that was only partially rescued by NAC treatment, whereas expression of either LKB1 WT or K78M did not affect the decreased Ru-5-P levels in these cells (Supplementary Fig. 3j–n, respectively).
Moreover, although knockdown of 6PGD in normal proliferating HaCaT cells also resulted in decreased 6PGD activity (Fig. 1b), Ru-5-P level and oxidative PPP flux along with increased AMPK phosphorylation and reduced lipogenesis (Fig. 5k–n, respectively), 6PGD knockdown did not significantly affect cell proliferation of HaCaT cells (Fig. 1a), suggesting that the 6PGD-dependent regulation of the LKB1–AMPK cascade and subsequent lipogenesis through Ru-5-P also exists in normal proliferating cells, but that these cells rely less on this regulatory mechanism for cell proliferation compared with cancer cells.
Discovery and development of physcion and its derivative S3 as 6PGD inhibitors
We next designed a screening strategy using an in vitro 6PGD assay (Fig. 6a). We identified physcion (C16H12O5; 1,8-dihydroxy-3-methoxy-6-methyl-anthraquinone; emodin-3-methyl ether) as a 6PGD inhibitor from a library of 2,000 FDA-approved small-molecule compounds (Fig. 6a), which effectively inhibits 6PGD but not G6PD (Fig. 6b). A physcion derivative S3 (C15H10O4; 1-hydroxy-8-methoxy-anthraquinone; Fig. 6a) was identified with improved solubility from a group of 10 commercially available physcion derivatives. Physcion and S3 inhibited 6PGD with absolute IC50 values of approximately 38.5 μM and 17.8 μM, respectively (Fig. 6c) and the Kd values of the physcion–6PGD and S3–6PGD interaction were determined to be 26.0 μM and 17.1 μM, respectively, using a tryptophan fluorescence-based binding assay (Fig. 6d). Physcion did not effectively inhibit other NADP+-dependent metabolic enzymes including G6PD, glutamate dehydrogenase 1 (GLUD1) and isocitrate dehydrogenase 1 (IDH1), nor glycolytic enzymes LDHA and PGAM1 (Fig. 6c). Moreover, physcion inhibits 6PGD but not G6PD (Fig. 6e and Supplementary Fig. 4a), leading to reduced proliferation of H1299 and K562 cells, whereas 6PGD knockdown cells were resistant to physcion treatment (Fig. 6f and Supplementary Fig. 4b).

(a) Upper: Screening strategy for lead compounds as 6PGD inhibitors. Lower: Structure of physcion and its derivative S3. (b) Purified 6PGD (left) and G6PD (right) were assayed for 6PGD and G6PD activity, respectively, in the presence of physcion. (c) Absolute IC50 values of physcion and S3 were determined in activity assays using purified enzymes. (d) Kd values were determined for physcion or S3 binding to purified human 6PGD proteins. The fluorescence intensity (Ex: 280 nm, Em: 350 nm) was measured37. (e,f) Physcion-treated H1299 cells were assayed for 6PGD (e; left) and G6PD (e; right) activity, and cell viability (f). (g) Schematic representation of the molecular docking study of physcion based on the crystal structure of 6PGD (PDB code: 3FWN) in complex with its substrate 6-PG. Physcion (green) is docked in a pocket near the binding site of 6-PG (yellow) that is surrounded by residues including Met 15, Lys 76, Lys 261 and His 452. (h) Purified 6PGD WT (left) and M15A mutant (right) were treated with physcion and assayed for 6PGD activity. Absolute IC50 values are shown; NR, not reached. (i) H1299 6PGD knockdown cells were transfected with 6PGD WT (left) and M15A mutant (right), followed by cell proliferation assay based on cell numbers in the presence of physcion. (j) Cell viability of physcion-treated human cancer cells was determined by MTT assay. Normal proliferating human dermal fibroblasts (HDF) and melanocyte PIG1 cells were included as controls. (k) Cell proliferation rates of H1299 cells treated with physcion were determined. (l) Apoptotic cell death (48 h) of H1299 cells harbouring AMPK shRNA or an empty vector in the presence of physcion was determined by annexin V staining. Data are from a single experiment that is representative of 3 independent experiments for b,e,h, 2 independent experiments for f,i,l, and 4 independent experiments for j,k. Source data for independent replications and experiments with sample size <5 are available in Supplementary Table 1.
The molecular docking study based on the crystal structure of 6PGD (PDB code: 3FWN) in complex with its substrate 6-PG suggests that physcion fits in a pocket near the binding site of 6-PG surrounded by residues including Met 15, Lys 76, Lys 261 and His 452 of 6PGD (Fig. 6g). Physcion forms hydrophobic interactions with these residues and a hydrogen bond with Asn 103 through its 10-keto group (Fig. 6g). In support of this model, the 6PGD M15A mutant showed resistance to physcion or S3 treatment in an in vitro 6PGD enzyme activity assay (Fig. 6h and Supplementary Fig. 4c, respectively), and expression of M15A (Supplementary Fig. 4d) conferred physcion or S3 resistance to H1299 cells (Fig. 6i and Supplementary Fig. 4e, respectively), suggesting that 6PGD is a predominant target of physcion and S3 in cancer cells.
Physcion and S3 inhibit cancer cell proliferation in vitro and tumour growth in vivo
Physcion treatment resulted in decreased cell viability of H1299, A549, 212LN and K562 cells in a dose-dependent manner, but did not significantly affect the control proliferating cells including human dermal fibroblasts (HDFs) and immortalized human melanocyte PIG1 cells (Fig. 6j). Physcion also inhibited cell proliferation of diverse human cancer cells including H1299, K562, MDA-MB-231 and 212LN cells (Fig. 6k and Supplementary Fig. 4f), and induced apoptosis in H1299 after 48 h treatment, whereas knockdown of AMPK abolished induction of apoptosis on physcion treatment (Fig. 6l).
We found that physcion treatment for 12 h resulted in increased intracellular 6-PG levels but decreased Ru-5-P levels and NADPH/NADP+ ratio, as well as reduced oxidative PPP flux and biosynthesis of RNA and lipids (Fig. 7a–d, respectively), without induction of apoptosis (Supplementary Fig. 4g). Similar results were obtained using K562 cells treated with physcion (Supplementary Fig. 4h–l) and H1299 cells treated with S3 (Supplementary Fig. 5a and b, respectively). Physcion or S3 also resulted in increased PFK activity in H1299 cells with increased lactate production and intracellular ATP levels (Supplementary Fig. 5c–e, respectively), as well as increased phosphorylation levels of AMPK and ACC1 (Fig. 7e) and disruption of active LKB1 complex (Fig. 7f), and NAC treatment reversed the increased ROS levels in physcion-treated H1299 cells (Fig. 7g). Furthermore, NAC treatment (Fig. 7h and Supplementary Fig. 5f) or inhibition of AMPK by Compound C or shRNA (Fig. 7i) partially rescued the reduced cell proliferation of H1299 cells treated with 6PGD inhibitors. Similar results were obtained using K562 cells (Supplementary Fig. 5g–j). Together, these results were consistent with the phenomenon observed in 6PGD knockdown cells (Figs 2–5).

(a–d) H1299 cells were assayed for intracellular concentration of 6-PG (a) and Ru-5-P (b), NADPH/NADP+ ratio (c), and oxidative PPP flux and biosynthesis of RNA and lipids (d) in the presence and absence of physcion. (e) H1299 cells were treated with increasing concentrations of physcion, followed by western blot to detect phosphorylation levels of ACC1 (pS79; upper) and AMPK (pT172; lower). 6PGD KD cells were included as a control. (f) Cell lysates of S3-treated H1299 cells were used for immunoprecipitation of MO25 and western blot to detect co-immunoprecipiated LKB1 and STRAD. (g,h) H1299 cells treated with or without physcion were assayed for general ROS levels (g) and cell proliferation rates by cell counting (h) in the presence and absence of NAC. (i) H1299 cells treated with or without physcion were assayed for cell proliferation rates by cell counting in the presence and absence of Compound C (left) or lentivirus harbouring AMPK shRNA (right). (j–o) Effects of treatment with physcion on LKB1-deficient A549 cells were assayed for 6PGD activity (j), intracellular Ru-5-P levels (k), phosphorylation levels of AMPK (l) and lipid biosynthesis (m), as well as general ROS levels (n) and cell proliferation rates by cell counting (o) in the presence and absence of NAC. (p–s) Effects of physcion treatment on normal proliferating HaCaT cells assayed for 6PGD activity (p), intracellular Ru-5-P levels (q), phosphorylation levels of AMPK and lipid biosynthesis (r), as well as cell proliferation rates by cell counting (s). In a–s, data are from a single experiment that is representative of 3 independent experiments for b,c,e and 2 independent experiments for a,d,f–s. Source data for independent replications and experiments with sample size <5 are available in Supplementary Table 1. Unprocessed original scans of western blots are shown in Supplementary Fig. 9.
Consistently, physcion treatment decreased 6PGD activity and Ru-5-P levels in LKB1-deficient A549 cells (Fig. 7j and k, respectively) but did not affect AMPK phosphorylation levels or lipogenesis (Fig. 7l and m, respectively). Moreover, NAC treatment completely reversed the increased ROS level and rescued the decreased cell proliferation in physcion-treated A549 cells (Fig. 7n and o, respectively). Furthermore, normal proliferating HaCaT cells treated with physcion showed decreased 6PGD activity and Ru-5-P level along with increased AMPK phosphorylation and reduced lipogenesis (Fig. 7p–r respectively) but unaltered cell proliferation (Fig. 7s). Similar results were obtained using normal proliferating HFF cells (Supplementary Fig. 5k–o).
S3 similarly inhibited cell viability of H1299 and K562 cells but not control proliferating PIG1 and HDF cells (Supplementary Fig. 6a). Chronic injection of S3 to nude mice for ∼4 weeks revealed that 20 mg kg−1 d−1 administered intraperitoneally is a well-tolerated dose. In xenograft nude mice subcutaneously injected with H1299 cells, S3 treatment resulted in significantly decreased tumour growth and masses compared with mice receiving dimethylsulphoxide (DMSO; Fig. 8a–c) with decreased 6PGD activity (Fig. 8c, right) and reduced Ki-67 expression (Fig. 8d), suggesting that S3 inhibits 6PGD in vivo to confer a specific inhibitory effect on tumour cell proliferation. Similar results were obtained using K562 leukaemia cell xenograft nude mice (Supplementary Fig. 6b–e), and an orthotopic xenograft model of head and neck cancer, where human head and neck cancer Tu212 cells were orthotopically injected into the primary tumour site by submandibular injection to mylohyoid muscles of nude mice (Fig. 8e, f and Supplementary Fig. 6f, g). Additionally, chronic treatment with S3 to nude mice did not affect body weight, serum chemistry, or complete blood counts and haematopoietic properties, nor cause notable differences in histopathological analyses (Supplementary Fig. 6h–k, respectively) compared with the DMSO-treated group, suggesting that S3 treatment does not cause obvious toxicity in vivo.

(a,b) Tumour growth curve (a) and tumour mass (b) in H1299 xenograft mice treated with S3 or DMSO. (c) Left: Dissected tumours in representative mice treated with DMSO or S3. Right: 6PGD enzyme activity in tumour lysates of H1299 xenograft mice treated with DMSO or S3. (d) Representative images of IHC staining of Ki-67 from H1299 xenograft mice treated with DMSO or S3. (e) Tumour growth curve (left) and tumour mass (right) in orthotopic xenograft nude mice injected with Tu212 cells treated with S3 or DMSO. (f) 6PGD activity (upper) and protein (lower) levels in tumour lysates. (g) 6PGD activity in physcion- or S3-treated human primary leukaemia cells isolated from PB samples from a representative B-ALL patient. (h) 6-PG levels (left) and NADPH/NADP+ ratio (right) in physcion-treated human primary leukaemia cells from a representative CML patient. (i,j) AMPK (pT172; i) and ACC1 (pS79; j) phosphorylation levels were examined by immunoblotting using physcion-treated human primary leukaemia cells isolated from PB samples from representative AML patients. (k) Cell proliferation (left) and viability (right) in physcion-treated human primary leukaemia cells isolated from PB samples from a representative B-ALL patient. (l) Physcion shows no toxicity in treatment (72 h) of peripheral blood cells (top) and CD34+ cells isolated from bone marrow samples (bottom) from representative healthy human donors. PB, peripheral blood; BM, bone marrow; B-ALL, acute B lymphoblastic leukaemia; AML, acute myeloid leukaemia. (a) Mean ± s.e.m.; n = 7 tumours from 7 mice, (b) n = 7 tumours from 7 mice; centre lines represent means, (e) mean ± s.e.m.; n = 8 tumours from 8 mice; centre lines represent means. The P values were determined by two-sided unpaired Student’s t-test for b,e (∗, 0.01 < P < 0.05; ∗∗, 0.001 < P < 0.01). In a–c,f, data are from a single experiment that is representative of 2 independent experiments. Source data for independent replications and experiments with sample size <5 are available in Supplementary Table 1. Unprocessed original scans of western blots are shown in Supplementary Fig. 9.
6PGD represents a promising anti-leukaemia target
Moreover, physcion inhibited 6PGD in human primary leukaemia cells (Fig. 8g, left), leading to decreased cell viability (Supplementary Fig. 7a), 6-PG accumulation and NADPH/NADP+ ratio (Fig. 8h), increased phosphorylation levels of AMPK (Fig. 8i) and ACC1 (Fig. 8j), and increased lactate production and intracellular ATP levels (Supplementary Fig. 7b and c, respectively). Figure 8k shows results using samples from a representative B-ALL patient with decreased cell viability and proliferation on physcion treatment. Similar results were obtained using S3 (Fig. 8g, right and Supplementary Fig. 7d–f). In contrast, physcion or S3 treatment did not affect cell viability of mononucleocytes in peripheral blood samples or CD34+ progenitors isolated from bone marrow samples from healthy donors (Fig. 8l and Supplementary Fig. 7g, h), suggesting promising anticancer potential of physcion and S3 without obvious toxicity to human blood cells.
DISCUSSION
Our findings suggest that 6PGD is commonly important for cell proliferation and tumour growth; 6PGD coordinates anabolic biosynthesis and redox homeostasis, at least in part by controlling the intracellular levels of its products Ru-5-P and NADPH (Supplementary Fig. 8a). In a related study, we demonstrated that upregulation of 6PGD by lysine acetylation in cancer cells is common and important for cell proliferation and tumour growth37. Thus, in cancer cells, lysine acetylation enhances 6PGD activity37 to promote the oxidative PPP and nucleotide or RNA biosynthesis and keep intracellular Ru-5-P at a physiological level that is sufficient to inhibit LKB1–AMPK signalling, which in turn relieves AMPK-dependent inhibition of ACC1, permitting high levels of lipid biosynthesis to fulfil the request of rapidly growing tumours (Supplementary Fig. 8a, left). Attenuation of 6PGD results in decreased Ru-5-P to levels below the physiological concentrations, which in turn not only attenuate nucleotide biosynthesis but also relieve the inhibition of LKB1, leading to activation of AMPK and subsequent inhibition of ACC1 and lipogenesis (Supplementary Fig. 8a, right). Our results suggest that 6PGD provides an additional link between the oxidative PPP and lipogenesis through Ru-5-P-dependent inhibition of LKB1–AMPK signalling by disrupting the active LKB1–MO25–STRAD complex. Although the intracellular level of Ru-5-P decreased in 6PGD knockdown cells, which suggests an important role for 6PGD in maintaining the intracellular Ru-5-P level that cannot be compensated by other pathways, this does not exclude the potential contribution of non-oxidative PPP in maintaining physiological Ru-5-P levels to regulate lipogenesis. In addition, our findings regarding a commonly important role of 6PGD in the oxidative PPP in cancer cells are different from the previous report using H1975 lung cancer cells19,20, suggesting that 6PGD functions may vary in cancer cells owing to different cell types or oncogenic background.
A remaining question is whether the key enzymes along the oxidative PPP including G6PD, gluconolactonase (6-phosphogluconolactonase; also known as H6PD) and 6PGD participate in LKB1 regulation using a similar mechanism. Interestingly, we found that knockdown of H6PD essentially reconstitutes the phenotypes in cells with 6PGD knockdown, including decreased Ru-5-P levels, increased AMPK activation and reduced cell proliferation (Supplementary Fig. 8b). In contrast, although knockdown of G6PD similarly resulted in decreased Ru-5-P levels, loss of G6PD did not affect AMPK activation nor attenuate cell proliferation (Supplementary Fig. 8c). These results suggest that H6PD and 6PGD probably function in a linear fashion to regulate Ru-5-P levels and consequently AMPK activation and cell proliferation, whereas the role of G6PD in cells is probably much more complicated. G6PD seems dispensable for cell proliferation, suggesting that G6PD knockdown cells may signal through alternative networks via unknown mechanisms to diminish AMPK activation and maintain their proliferative potential. Further studies are warranted to decipher the role of distinct enzymes in the oxidative PPP in cancer cell metabolism and cell proliferation.
We observed that 6PGD inhibitors were more potent to inhibit cell proliferation than 6PGD enzyme activity. This may suggest that 6PGD activity is probably not linearly correlated with cell proliferative potential. Cancer cells might require a certain level of cellular 6PGD activity to maintain cell metabolism and subsequently proliferation. Once 6PGD activity drops below such a threshold (for example, treatment with 6PGD inhibitor), cancer cells may shut down all of the related functions, leading to decreased cell proliferation. Moreover, suppression of 6PGD in A549 cells resulted in reduced cell proliferation with unaltered AMPK phosphorylation and lipogenesis but decreased Ru-5-P and increased ROS, suggesting that such LKB1-deficient cells may still rely on 6PGD-dependent regulation of oxidative PPP flux and subsequent nucleotide biosynthesis and redox homeostasis. The extent of this reliance would determine the responsiveness and sensitivity of LKB1-deficient cells to treatment with 6PGD inhibitors. Additionally, although 6PGD-dependent regulation of the LKB1–AMPK cascade and subsequent lipogenesis through Ru-5-P exists in normal proliferating HaCaT and HFF cells, suppression of 6PGD in these cells does not significantly affect their proliferative potential. These findings provide mechanistic insight into the selective toxicity of 6PGD inhibitors to cancer cells. These translational studies together provide a ‘proof of principle’ to suggest anti-6PGD as a promising therapy in clinical treatment of human cancers with commonly elevated 6PGD activity and lysine acetylation37. □
Methods
Reagents.
The antibody information including antibody name, company, catalogue/clone number and dilutions used for every assay is provided in Supplementary Table 2. For the doxycycline-inducible shRNA construct, h6PGD shRNA was cloned into a Tet-pLKO-puro vector. N-acetyl-L-cysteine (NAC) and 1-hydroxy-8-methoxy-anthraquinone (S3) were from Sigma; physcion from Santa Cruz Biotech; Compound C from EMD Millipore. Purified LKB1 protein complex was from Upstate (Millipore) and recombinant human AMPK protein was from Abnova.
Cell culture.
H1299, A549, H322, H157, HEL, KG1a, Molm14, MO91, EOL1 and K562 cells were cultured in RPMI 1640 medium with 10% bovine serum (FBS). 293T, MDA-MB231, MCF7, HFF and HaCaT cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% FBS. 212LN and Tu212 cells were cultured in DMEM/F12 medium with 10% FBS. HDF cells were cultured in FibroLife medium. PIG1 cells were cultured in Medium 254. NAM+TSA treatment was performed by incubating cells with 10 mM NAM and 5 μM TSA for 16 h. None of the cell lines used in this study is listed in the database of commonly cross-contaminated or misidentified cell lines maintained by ICLAC and NCBI Biosample except Tu212 and 212LN. Tu212 and 212LN are genetically identical, because Tu212 is a primary tumour from a head and neck squamous cell carcinoma patient and 212LN is a tumour derived from a metastatic lymph node of the same patient. The Tu212-derived 212LN cell line was used for cell viability assay on physcion treatment (Fig. 6j). The primary tumour cell line Tu212 was used for in vivo S3 treatment (Supplementary Fig. 6f, g), because the primary tumour cell line, but not the metastatic cell line from the lymph node, should be used for orthotopic xenograft injection to monitor the tumour growth. However, the genetic identity of Tu212 and 212LN does not in any way affect the finding of the manuscript and data show that pharmacological inhibition of 6PGD attenuates cancer cell viability and tumour growth. Cell lines were authenticated by STR profiling and were tested for mycoplasma contamination.
Screening stable knockdown cell lines.
Stable knockdown of endogenous h6PGD, hAMPKα1, hH6PD and hG6PD was achieved using lentiviral vectors harbouring shRNA constructs targeting each respective gene. Detailed RNAi reagent information including gene names, shRNA sequence, company name and catalogue number is provided in Supplementary Table 3. To produce lentivirus, each construct was co-transfected with psPAX2 packaging plasmid and pMD2.G envelope plasmid into 293T human embryonic kidney-derived cells using TransIT-LT1 Transfection Reagent (Mirus Bio) according to the manufacturer’s instructions. Forty-eight hours later, lentivirus-containing supernatant medium was collected, filtered, and concentrated by ultracentrifugation. Target cells were transduced with lentivirus-containing supernatant and subjected to puromycin selection. Knockdown of endogenous proteins was confirmed by western blot.
Transient transfection.
FLAG-6PGD WT, FLAG-6PGD M15A, pcDNA-LKB1 WT and pcDNA-LKB1 K78M were transfected using TransIT-LT1 Transfection Reagent (Mirus Bio) according to the manufacturer’s instructions. Cells were collected 24 h after transfection.
Metabolic assays.
Detailed methodology for the metabolic assays is described in refs 5,37,38,39,40,41,42,43. In brief, for the 14C-RNA biosynthesis assay, subconfluent cells were spiked with 4 μCi ml−1 of D-[U-14C]glucose (Perkin Elmer) for 2 h. Total RNA was extracted using RNeasy columns (Qiagen). The amount of 14C-RNA was determined by liquid scintillation counting and normalized by the total amount of RNA. For the 14C-lipid biosynthesis assay, lipids were extracted by the addition of 500 μl of hexane/isopropanol (3:2 v/v), dried, resuspended in 50 μl of chloroform, and subjected to scintillation counting.
The glucose utilization assay was performed by culturing 1 × 106 cells per 6-well plate in phenol-red-free RPMI with 1% FBS for 3 days. Glucose concentrations in media were measured using a colorimetric glucose assay kit (Biovision) and normalized with cell numbers each day.
The glycolytic rate was measured by monitoring the conversion of 5-3H-glucose to 3H2O. Cells of 1 × 106 in number were incubated in 1 ml of Krebs buffer without glucose for 30 min at 37 °C. The Krebs buffer was then replaced with Krebs buffer containing 10 mM glucose spiked with 10 μCi of 5-3H-glucose. Following incubation for 1 h at 37 °C, 50 μl aliquots were transferred to uncapped PCR tubes containing 50 μl of 0.2 N HCl, and a tube was transferred into a tube containing 0.5 ml of H2O for diffusion. The tubes were sealed, and diffusion was allowed to proceed for 24 h at 34 °C. The amounts of diffused 3H2O were determined by scintillation counting.
Lactate production was measured with a fluorescence-based lactate assay kit (MBL). Plain phenol-red-free RPMI medium was added to subconfluent cells for 1 h and the medium was assessed for lactate level. The values were normalized with cell number.
Oxygen consumption rates were measured using a Clark-type micro cathode oxygen electrode attached to a 782 oxygen meter (Strathkelvin Instruments).
Intracellular ATP level was measured by using an ATP bioluminescent somatic cell assay kit (Sigma Aldrich). Cells of 1 × 105 in number were suspended in ultrapure water. The reaction was initiated by adding ATP enzyme mix to the cell suspension and the luminescence was recorded by spectrofluorometer.
Intracellular reactive oxygen species (ROS) was measured by staining the cells with carboxy-H2DCFDA (Invitrogen).
Oxidative PPP flux was determined by measuring 14CO2 release. Cells were seeded on 6-cm dishes that were placed in a 10-cm dish with 2 sealed pinholes. 14CO2 released from cells was collected by sealing the 10-cm dish, in which the cells on the 6-cm dish were incubated in 2 ml of medium containing 14C-glucose (10 μCi ml−1) at 37 °C for 3 h. The PPP flux was stopped by injecting 0.3 ml of 2N HCl through one of the holes and released 14CO2 was trapped by injecting 0.3 ml of hyamine hydroxide into a cup placed on the 10-cm dish through the second hole. Each dish was completely re-sealed and incubated for 12 h. Hyamine hydroxide was dissolved in 60% methanol and directly subjected to scintillation counting.
6PGD, G6PD, PGAM1, LDHA, PFK, PGI, IDH1, ACLY, ACC1, FASN, transketolase (TKT) enzyme assays.
6PGD activity was determined on the basis of the rate of NADPH production in assay buffer containing 0.1 mM NADP+, 1 mM MgCl2 and 50 mM Tris (pH 8.1) with 0.2 mM 6-phosphogluconate as a substrate. The increase of absorbance at 340 nm was measured by a spectrophotometer.
G6PD activity was determined by the NADPH production rate from G6PD and 6PGD, and then subtracting that of 6PGD, because a product of G6PD, 6-phosphogluconolactone, is rapidly hydrolysed to a substrate of 6PGD, 6-phosphogluconate, in cells.
PGAM1 activity was measured by multiple enzymes coupled assay. PGAM1 enzyme mix containing 100 mM Tris-HCl, 100 mM KCl, 5 mM MgCl2, 1 mM ADP, 0.2 mM NADH, 5 mg ml−1 PGAM1, 0.5 units ml−1 enolase, 0.5 units ml−1 recombinant pyruvate kinase M1, and 0.1 units ml−1 recombinant LDH was prepared. 3-PG was added last at a final concentration of 2 mM to initiate the reaction. The decrease in fluorescence (ex: 340 nm, em: 460 nm) from oxidation of NADH was measured.
LDHA activity was determined by measuring the decreased fluorescence intensity at 340 nm from the NADH oxidation in 20 mM HEPES-K+ (pH 7.2), 0.05% bovine serum albumin, 20 μM NADH and 2 mM pyruvate using a spectrofluorometer (ex: 340 nm, em: 460 nm).
PFK activity was measured as described previously44 by adding 80 μg cell lysate to a reaction mixture containing 50 mM Tris-HCl pH 7.5, 100 mM KCl, 5 mM MgCl2, 1 mM ATP, 0.2 mM NADH, 5 mM Na2HPO4, 0.1 mM AMP, 1 mM NH4Cl, 5 mM fructose-6-phosphate, 5 units triose phosphate isomerase (Sigma), 1 unit aldolase (Sigma) and 1 unit α-glycerophosphate dehydrogenase (Sigma). Decrease in 340 nm absorbance (OD340) as a measure of NADH oxidation was measured every 15 s for 10 min on a DU800 Spectrophotometer (Beckman Coulter).
PGI activity was measured as described previously45 by adding D-fructose-6-phosphate to a reaction mixture containing 42 mM Gly-Gly, 3.3 mM D-fructose-6-phosphate, 0.67 mM NADP+, 3.3 mM MgCl2, 5 units glucose-6-phosphate dehydrogenase and 10 μg cell lysate. Increase in 340 nm absorbance (OD340) as a measure of NADPH production was measured every 20 s for 10 min on a DU800 Spectrophotometer (Beckman Coulter).
IDH1 oxidative activity was measured as described previously46 by adding 0.5 μg of rIDH1 protein to a reaction mixture containing 20 mM Gly-Gly (pH 7.5), 0.6 mM MnCl2, 1 mM NADP+ and 0.44 mM D-(+)-threo-isocitrate. Increase in 340 nm absorbance as a measure of NADPH production was detected every 20 s for 10 min on a DU800 Spectrophotometer (Beckman Coulter).
ACLY enzyme activity was measured as described previously47 by adding 100 mM ATP to a reaction mixture containing 100 mM Tris, pH 7.5, 10 mM MgCl2, 10 mM dithiothreitol (DTT), 0.3 mM acetyl-CoA, 0.14 mM NADH and 100 μg cell lysate. Decrease in autofluorescence (ex: 340 nm, em: 460 nm) from oxidation of NADH was measured as ACLY activity.
ACC1 and FASN enzyme activities were measured with slight modifications to the described procedure48. In brief, ACC activity was determined as the incorporation of radiolabelled acetyl-CoA into fatty acids in a reaction coupled with that catalysed by FASN. FASN was purified from 293T cells transfected with GST-tagged FASN construct by GST pulldown followed by elution with reduced glutathione. Fatty acids were extracted three times with 3:2 (hexane/isopropyl).
TKT activity was measured as previously described49,50. In brief, 20 μg of cell lysates containing TKT were added to a reaction mixture containing 216 mM glycylglycine, 3.3 mM xylulose 5-phosphate, 1.7 mM ribose-5-phosphate, 0.002% (w/v) cocarboxylase, 0.14 mM β-NAD reduced form, 15 mM MgCl2 and 20 units of α-glycerophosphate dehydrogenase/triosephosphate isomerase. Decrease in absorbance at 340 nm from NADH oxidation was measured by a DU800 spectrophotometer for 15 min (Beckman Coulter).
Cellular metabolite extraction and measurement.
Cellular metabolites were extracted and spectrophotometrically measured as described previously by measuring the intracellular concentrations of 6-PG (ref. 5), Ru-5-P and R-5-P (ref. 46), with some modifications. In brief, 200 μl of packed cell pellets were homogenized in 0.6 ml of hypotonic lysis buffer, centrifuged at 4 °C for 10 min at maximum speed, and the supernatants were applied to Amicon Ultra tubes with 10 KDa cutoff filter (Millipore). The flowthrough containing the metabolites was used for measurements. For 6-PG, Tris-HCl (pH 8.1) and MgCl2 were added to the flowthrough to final concentrations of 50 mM and 1 mM, respectively. Recombinant 6PGD protein was added to a final concentration of 10 μg ml−1, and the reaction was initiated by adding NADP+ (final concentration of 0.1 mM). Increase in absorbance at 340 nm from NADPH production was measured by a DU800 spectrophotometer (Beckman Coulter). Two hundred microlitres of 6-PG (Sigma) diluted with 0.6 ml of hypotonic lysis buffer was used as the standard. For Ru-5-P and R-5-P, in a 3 ml reaction mix, the final concentrations are 58 mM Gly-Gly, 1.7 mM ribose-5-phosphate or 1.7 mM ribulose-5-phosphate, 0.002% (w/v) cocarboxylase, 15 mM MgCl2, 0.13 mM β-nicotinamide adenine dinucleotide (NADH), 0.5 units α-glycerophosphate dehydrogenase, 5 units triosephosphate isomerase, 0.5 units transketolase and 0.025–0.05 units D-ribulose-5-phosphate 3-epimerase. Once the reaction was initiated by D-ribulose-5-phosphate 3-epimerase, a decrease in absorbance at 340 nm from NADH oxidation was measured by a DU800 spectrophotometer (Beckman Coulter). Two hundred microlitres of Ru-5-P (Sigma) and R-5-P (Sigma) diluted with 0.6 ml of hypotonic lysis buffer were used as the standards.
Cell proliferation and viability assays.
The experiments were performed by culturing 5 × 104 cells in 6-well plates at 37 °C in normoxia (5% CO2 and 95% air). Twenty-four hours later cells used for hypoxic culture were cultured at 37 °C in a sealed hypoxia chamber filled with 1% O2, 5% CO2 and 94% N2. Cell proliferation was determined by recording cell numbers 48 and 96 h post-seeding, and normalizing to cell numbers at 0 h. For cell viability assays, 5 × 106 cells were seeded in 6-well plates and incubated with increasing concentrations of physcion or S3 at 37 °C for the indicated times. Relative cell viability at each experimental time point up to 72 h was determined using either CellTiter 96 Aqueous One solution proliferation kit (Promega), or by trypan blue exclusion using a TC10 Automated Cell Counter (Bio Rad).
Apoptosis assay.
Cells of 1 × 105 in number were treated with increasing concentrations of physcion or S3 for 12 or 48 h, then collected and stained using FITC-conjugated annexin V labelling reagent (BD Pharmingen) and propidium iodide (Sigma) followed by FACS analysis for apoptotic cell population.
Kinase assays.
The LKB1 kinase assay was performed as described previously21. In brief, active LKB1 was immunoprecipitated from cell extracts using LKB1 antibody (Cell Signaling Technologies) followed by incubation with Protein G Sepharose 4 Fast Flow beads (GE Healthcare). Beads were washed three times in lysis buffer and twice in kinase buffer (50 mM Tris, pH 7.5/10 mM MgCl2/1 mM DTT/100 μM ATP), and then resuspended in kinase buffer containing 5 μg MBP or AMPK per reaction and increasing concentrations of Ru-5-P. Kinase assay was carried out for 20 min at 30 °C. Samples were electrophoresed on 10% SDS–acrylamide gel, transferred onto a nitrocellulose membrane, and then detected with antibodies against phospho-serine/threonine (Abcam) and phospho-AMPK T172 (Cell Signaling Technologies).
The AMPK kinase assay was performed as described previously51. In brief, Flag–AMPKα1 was transfected into 293T cells with Lipofectamine 2000 (Invitrogen). Twenty-four hours after transfection, Flag–AMPK was purified from cells using Anti-Flag M2 affinity gel (Sigma). Beads were washed three times in TBS and twice in kinase buffer (50 mM MOPS, pH 7.2/25 mM β-glycerophosphate/10 mM EGTA/4 mM EDTA/50 mM MgCl2/0.5 mM DTT) and then resuspended in kinase buffer containing 100 μM AMP, 50 μM ATP, 1 μCi γ-32P-ATP, and 100 μg SAMS peptide per reaction and increasing concentrations of Ru-5-P. Kinase assay was carried out for 15 min at 30 °C. Samples were spotted onto P81 paper. The P81 paper was dried and washed in 1% phosphoric acid three times, and then subjected to scintillation counting.
Cell-free system assays.
H1299 cells were cultured in RPMI 1640 medium with 10% fetal bovine serum (FBS). Once confluent, cells were collected, pelleted and resuspended in PBS (final volume 100 μl), followed by sonication. Cell lysates were diluted 4 times. As intracellular Ru-5-P levels in control vector and 6PGD knockdown H1299 cells were determined as 800 μM and 320 μM, respectively, the diluted samples of control and 6PGD knockdown cell lysates contained 200 μM (1.0 fold) and 80 μM (0.4 fold), respectively. To perform Ru-5-P rescue experiments, Ru-5-P (Sigma) was added to lysates from 6PGD knockdown cells to achieve final concentrations of Ru-5-P at 0.8-, 1.0- and 1.3-fold. As for R-5-P levels, because intracellular R-5-P levels in control vector and 6PGD knockdown H1299 cells were determined as 200 μM and 160 μM, respectively, the diluted samples of control and 6PGD knockdown cell lysates contained 50 μM (1.0 fold) and 40 μM (0.8 fold), respectively. To perform R-5-P rescue experiments, R-5-P (Sigma) was added to lysates from 6PGD knockdown cells to achieve final concentrations of Ru-5-P at 1.0-, 1.3- and 1.5-fold. As for NADPH levels, because intracellular NADPH levels in control vector and 6PGD knockdown H1299 cells were determined as 9 μM and 7.2 μM, respectively, the diluted samples of control and 6PGD knockdown cell lysates contained 2.25 μM (1.0 fold) and 1.80 μM (0.8 fold), respectively. To perform NADPH rescue experiments, NADPH (Sigma) was added to lysates from 6PGD knockdown cells to achieve final concentrations of NADPH at 1.0-, 1.2-, 1.6- and 5.2-fold. To examine the effect of Ru-5-P, and R-5-P and NADPH on LKB1 complex stability, Ru-5-P, and R-5-P and NADPH treatment was carried out for 20 min at 30 °C. To examine the effect of Ru-5-P, and R-5-P and NADPH on AMPK and ACC1 phosphorylation, Ru-5-P, and R-5-P and NADPH treatment was carried out for 4 h at 30 °C. For lipid synthesis and NADPH/NADP+ ratio in response to Ru-5-P treatment, D-(U-14C)glucose and Ru-5-P were added to the cell lysates and incubated for 1, 2, 4, 6 or 12 h. Following centrifugation of the lysates, the supernatant was retained for determination of NADPH/NADP+ ratio as previously described. The pellet was retained for lipid extraction as previously described.
In vitro 6PGD assays for drug screening.
We performed an in vitro 6PGD enzyme assay to screen 2,000 biologically active compounds (The Spectrum Collection; MicroSource). In brief, 0.2 mM 6-phosphogluconate was added to assay buffer containing 50 mM Tris (pH 8.1), 1 mM MgCl2, 0.1 mM NADP+, 0.5 μg r6PGD and 10 μM drug. The increase in absorbance at 341 nm was measured using a SpectraMax Plus spectrophotometer (Molecular Devices).
Drug binding assay.
Drug binding assay was performed as previously described5; 1 μg r6PGD was incubated with different concentrations of physcion or S3 (0–500 μM), and tryptophan fluorescence (ex: 280 nm, em: 350 nm) from r6PGD was measured in 100 mM Tris-HCl buffer. Relative fluorescence intensity without any treatment is presented as 100%.
Molecular docking study.
We performed the molecular docking study on the basis of the crystal structure of 6PGD (PDB code: 3FWN) in complex with its substrate 6-PG. Physcion was initially positioned into the 6-PG-binding site and then docked by FlexiDock in SYBYL program (Tripos). The docking result was optimized by local energy minimization. The final docking model was drawn by PyMOL.
Xenograft studies.
Approval of the use of mice and the designed experiments was given by the Institutional Animal Care and Use Committee of Emory University. In brief, nude mice (nu/nu, female 4–6-week-old, Harlan Laboratories) were subcutaneously injected with 20 × 106 H1299 cells or 5 × 106 K562 cells harbouring empty vector on the left flank, and cells with stable knockdown of endogenous h6PGD on the right flank, respectively. Ten mice were included in each experiment. Tumour growth was recorded by measuring two perpendicular diameters using the formula 4π/3 × (width/2)2 × (length/2). For xenograft experiments using 6PGD inducible knockdown cells, nude mice were subcutaneously injected with 20 × 106 H1299 cells or 5 × 106 K562 expressing doxycycline-inducible shRNA constructs. Once tumours reached ∼65 mm3 volume, mice were distributed randomly into two groups and provided drinking water with or without doxycycline (Dox; 1 mg ml−1) (Sigma) that was additionally supplemented with 1% sucrose. Tumours were collected and weighed at the experimental endpoint, and tumour masses (g) derived from cells with and without stable knockdown of endogenous h6PGD were compared.
For drug evaluation of S3 using xenograft mice, the drug was administered by daily i.p. injection at a dose of 20 mg kg−1 from 20 days after subcutaneous injection of 20 × 106 H1299 cells on the right flank of each mouse (6 mice were randomized to each group), or 3 days after subcutaneous injection of 5 × 106 K562 cells on the right flank of each mouse (7 mice were randomized to each group). Tumour growth was recorded by measurement of two perpendicular diameters using the formula 4π/3 × (width/2)2 × (length/2). The tumours were collected and weighed at the experimental endpoint, and the masses of tumours (g) treated with vehicle control (DMSO) and S3 were compared.
Histopathology and complete blood counts.
For histopathological analysis, sections of mouse tissue were stained with haematoxylin and eosin and slides were digitally scanned at ×20 magnification using a ScanScope XT from Aperio Technologies. Images were analysed and captured using ImageScope software (Aperio Technologies) without any additional or subsequent image processing. For complete blood counts, blood was collected retro-orbitally and immediately applied to a HemaVet 950FS (Drew Scientific) for generation of a complete haematology profile.
Serum chemistry assays.
Blood was collected from mice retro-orbitally. Samples were incubated on ice for 2 h and centrifuged at 735g for 10 min (4 °C), and then immediately applied to the following serum chemistry assays. The serum concentrations of uric acid, urea and creatinine were measured according to the protocol of the Colorimetric Assay Kit (BioVision). Enzyme activity of alanine aminotransferase (ALT) in mouse serum was determined by measuring its product pyruvate in a reaction that concomitantly converts a nearly colourless probe to fluorescence (Ex/Em = 535/587) according to the instructions of the Alanine Aminotransferase Activity Assay Kit (BioVision). Enzyme activity of aspartate aminotransferase (AST) in mouse serum was determined by measuring its product glutamate in a reaction that concomitantly converts a nearly colourless probe to colour at 450 nm according to the instructions of the Aspartate Aminotransferase Activity Assay Kit (BioVision).
Immunohistochemical staining.
Assay was performed as previously described5. In brief, resected tumours from xenograft mice were fixed in 10% buffered formalin, embedded in paraffin and mounted on slides. After deparaffinization and rehydration, mouse tumour sections were incubated in 3% hydrogen peroxide to suppress endogenous peroxidase activity. Antigen retrieval was achieved by microwaving the sections in 10 mM sodium citrate (pH 6.0). Sections were then blocked by incubation in 2.5% horse serum containing avidin and biotin. Human prediluted Ki-67 antibody (Invitrogen) was applied overnight at 4 °C. Detection was achieved with the Avidin–Biotin Complex System (Vector Laboratories). Slides were stained with 3,3′-diaminobenzidine, washed, counterstained with haematoxylin, dehydrated, treated with xylene, and mounted.
Primary tissue samples from patients with leukaemia and healthy donors.
Approval of the use of human specimens was given by the Institutional Review Board of Emory University School of Medicine. All clinical samples were obtained with informed consent with approval by the Emory University Institutional Review Board. Clinical information for patients was obtained from the pathologic files at Emory University Hospital under the guidelines and with approval from the Institutional Review Board of Emory University School of Medicine and according to the Health Insurance Portability and Accountability Act. Only samples from patients that were not previously treated with chemotherapy or radiation therapy were used. Mononuclear cells were isolated from peripheral blood and bone marrow samples from patients with leukaemia or from healthy donors using lymphocyte separation medium (Cellgro). Cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum and penicillin/streptomycin and incubated with increasing concentrations of physcion or S3 for up to 72 h.
Statistical analysis.
Data are presented as means ± s.d. for Supplementary Fig. 6i, j and means ± s.e.m. for all the rest. Experimental groups were compared using paired two-sided Student’s t-test for Fig. 1d and Supplementary Fig. 1b and unpaired two-sided Student’s t-test for all the rest.
Reproducibility of experiments.
The results of one representative experiment from at least two independent experiments are shown except data obtained from primary patient samples and animal experiments shown in Figs 1h, i and 8e, and Supplementary Figs 1e, f and 6h–j. Precise numbers of independent repeats are stated in each figure legend. There is no estimate of variation in each group of data and the variance is similar between the groups. No statistical method was used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment. All data are expected to have normal distribution.
Accession numbers.
The screen data were deposited to the PubChem Bioassay (https://www.ncbi.nlm.nih.gov/pcassay; accession #1159550).
Accession codes
References
Warburg, O. On the origin of cancer cells. Science 123, 309–314 (1956).
Cairns, R. A., Harris, I. S. & Mak, T. W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 11, 85–95 (2011).
Kroemer, G. & Pouyssegur, J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell 13, 472–482 (2008).
Christofk, H. R. et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452, 230–233 (2008).
Hitosugi, T. et al. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell 22, 585–600 (2012).
Locasale, J. W. et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 43, 869–874 (2011).
Possemato, R. et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350 (2011).
Vander Heiden, M. G. et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 329, 1492–1499 (2010).
Tian, W. N. et al. Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J. Biol. Chem. 273, 10609–10617 (1998).
Farquharson, C., Milne, J. & Loveridge, N. Mitogenic action of insulin-like growth factor-I on human osteosarcoma MG-63 cells and rat osteoblasts maintained in situ: the role of glucose-6-phosphate dehydrogenase. Bone Miner. 22, 105–115 (1993).
Tian, W. N. et al. Importance of glucose-6-phosphate dehydrogenase activity in cell death. Am. J. Physiol. 276, C1121–C1131 (1999).
Li, D. et al. A new G6PD knockdown tumor-cell line with reduced proliferation and increased susceptibility to oxidative stress. Cancer Biother. Radiopharm. 24, 81–90 (2009).
Schafer, Z. T. et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–113 (2009).
Budihardjo, I. I. et al. 6-Aminonicotinamide sensitizes human tumor cell lines to cisplatin. Clin. Cancer Res. 4, 117–130 (1998).
Bravard, A., Luccioni, C., Muleris, M., Lefrancois, D. & Dutrillaux, B. Relationships between UMPK and PGD activities and deletions of chromosome 1p in colorectal cancers. Cancer Genet. Cytogenet. 56, 45–56 (1991).
Jonas, S. K. et al. Increased activity of 6-phosphogluconate dehydrogenase and glucose-6-phosphate dehydrogenase in purified cell suspensions and single cells from the uterine cervix in cervical intraepithelial neoplasia. Br. J. Cancer 66, 185–191 (1992).
Basu, J. et al. Alterations in erythrocyte glutathione metabolism associated with cervical dysplasias and carcinoma in situ. Cancer Invest. 11, 652–659 (1993).
Giusti, L. et al. Fine-needle aspiration of thyroid nodules: proteomic analysis to identify cancer biomarkers. J. Proteome Res. 7, 4079–4088 (2008).
Sukhatme, V. P. & Chan, B. Glycolytic cancer cells lacking 6-phosphogluconate dehydrogenase metabolize glucose to induce senescence. FEBS Lett. 586, 2389–2395 (2012).
Chan, B., VanderLaan, P. A. & Sukhatme, V. P. 6-Phosphogluconate dehydrogenase regulates tumor cell migration in vitro by regulating receptor tyrosine kinase c-Met. Biochem. Biophys. Res. Commun. 439, 247–251 (2013).
Shaw, R. J. et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl Acad. Sci. USA 101, 3329–3335 (2004).
Woods, A. et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 13, 2004–2008 (2003).
Shackelford, D. B. & Shaw, R. J. The LKB1–AMPK pathway: metabolism and growth control in tumour suppression. Nat. Rev. Cancer 9, 563–575 (2009).
Park, S. H. et al. Phosphorylation-activity relationships of AMPK and acetyl-CoA carboxylase in muscle. J. Appl. Physiol. 92, 2475–2482 (2002).
Hardie, D. G. Regulation of fatty-acid and cholesterol-metabolism by the AMP-activated protein-kinase. Biochim. Biophys. Acta 1123, 231–238 (1992).
Jeon, S. M., Chandel, N. S. & Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485, 661–665 (2012).
Fullerton, M. D. et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 19, 1649–1654 (2013).
Sommercorn, J. & Freedland, R. A. Regulation of hepatic phosphofructokinase by 6-phosphogluconate. J. Biol. Chem. 257, 9424–9428 (1982).
Davies, S. P., Sim, A. T. & Hardie, D. G. Location and function of three sites phosphorylated on rat acetyl-CoA carboxylase by the AMP-activated protein kinase. Eur. J. Biochem. 187, 183–190 (1990).
Ha, J., Daniel, S., Broyles, S. S. & Kim, K. H. Critical phosphorylation sites for acetyl-CoA carboxylase activity. J. Biol. Chem. 269, 22162–22168 (1994).
Kudo, N., Barr, A. J., Barr, R. L., Desai, S. & Lopaschuk, G. D. High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5′-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J. Biol. Chem. 270, 17513–17520 (1995).
Munday, M. R., Campbell, D. G., Carling, D. & Hardie, D. G. Identification by amino acid sequencing of three major regulatory phosphorylation sites on rat acetyl-CoA carboxylase. Eur. J. Biochem. 175, 331–338 (1988).
Mihaylova, M. M. & Shaw, R. J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 13, 1016–1023 (2011).
Boudeau, J. et al. MO25α/β interact with STRADα/β enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J. 22, 5102–5114 (2003).
Zeqiraj, E., Filippi, B. M., Deak, M., Alessi, D. R. & van Aalten, D. M. Structure of the LKB1-STRAD-MO25 complex reveals an allosteric mechanism of kinase activation. Science 326, 1707–1711 (2009).
Marignani, P. A. et al. Novel splice isoforms of STRADα differentially affect LKB1 activity, complex assembly and subcellular localization. Cancer Biol. Ther. 6, 1627–1631 (2007).
Shan, C. et al. Lysine acetylation activates 6-phosphogluconate dehydrogenase to promote tumor growth. Mol. Cell 55, 552–565 (2014).
Fan, J. et al. Tyrosine phosphorylation of lactate dehydrogenase A is important for NADH/NAD(+) redox homeostasis in cancer cells. Mol. Cell. Biol. 31, 4938–4950 (2011).
Fan, J. et al. Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex. Mol. Cell 53, 534–548 (2014).
Hitosugi, T. et al. Tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase 1 is important for cancer metabolism. Mol. Cell 44, 864–877 (2011).
Hitosugi, T. et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci. Signal. 2, ra73 (2009).
Hitosugi, T. et al. Tyr26 phosphorylation of PGAM1 provides a metabolic advantage to tumours by stabilizing the active conformation. Nat. Commun. 4, 1790 (2013).
Shan, C. et al. Tyr-94 phosphorylation inhibits pyruvate dehydrogenase phosphatase 1 and promotes tumor growth. J. Biol. Chem. 289, 21413–21422 (2014).
Yi, W. et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 337, 975–980 (2012).
Yanagawa, T. et al. Differential regulation of phosphoglucose isomerase/autocrine motility factor activities by protein kinase CK2 phosphorylation. J. Biol. Chem. 280, 10419–10426 (2005).
Zhao, S. et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science 324, 261–265 (2009).
Lin, R. et al. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol. Cell 51, 506–518 (2013).
Natali, F., Siculella, L., Salvati, S. & Gnoni, G. V. Oleic acid is a potent inhibitor of fatty acid and cholesterol synthesis in C6 glioma cells. J. Lipid Res. 48, 1966–1975 (2007).
Soh, Y., Song, B. J., Jeng, J. & Kallarakal, A. T. Critical role of arg433 in rat transketolase activity as probed by site-directed mutagenesis. Biochem. J. 333, 367–372 (1998).
Lee, J. Y., Cheong, D. E. & Kim, G. J. A novel assay system for the measurement of transketolase activity using xylulokinase from Saccharomyces cerevisiae. Biotechnol. Lett. 30, 899–904 (2008).
Wu, C. et al. Cordycepin activates AMP-activated protein kinase (AMPK) via interaction with the γ1 subunit. J. Cell. Mol. Med. 18, 293–304 (2014).
Acknowledgements
This work was supported in part by NIH grants CA140515, CA183594, CA174786 (J.C.), CA175316 (S.K.), GM071440 (C.H.) and the Pharmacological Sciences Training Grant T32 GM008602 (S.E.), DoD grant W81XWH-12-1-0217 (J.C.), National Natural Science Funds of China No. 20902013 (L.Zhou), Charles Harris Run For Leukemia, Inc. (H.J.K.) and the Hematology Tissue Bank of the Emory University School of Medicine and the Georgia Cancer Coalition (H.J.K.). T.H. is a Fellow Scholar of the American Society of Hematology. S.E. is a NIH pre-doctoral fellow and an ARCS Foundation Scholar. H.J.K., F.R.K., S.K. and J.C. are Georgia Cancer Coalition Distinguished Cancer Scholars. S.K. is a Robbins Scholar. S.K. and J.C. are American Cancer Society Basic Research Scholars. J.C. is a Scholar of the Leukemia and Lymphoma Society.
Author information
Authors and Affiliations
Contributions
R.L., S.E. and C.S. contributed equally to this work. J.X., T.-L.G., S.Z., K.Y., P.R.C., D.J.B., M.L.A., S.L., H.J.K., Q.L. and F.R.K. provided critical reagents. S.J.H. performed data analysis of pharmacokinetics studies. M.T. and T.-L.G. performed mass spectrometry-based assays. Q.J., L.Zhou, L.Zhang and C.H. performed biochemical analysis of lysine-acetylated 6PGD and molecular docking studies and analysed the data. J.S., L.J., M.M., R.J.D., S.W., Y.L. and H.M. performed quantitative mass spectrometry and NMR-based assays, and analysed data. B.H.L. performed the histopathological analyses. T.J.B. performed structural analyses. D.W. and G.Z.C. helped with xenograft experiments. C.S., S.E., H.-B.K., J.H.S., T.H. and J.F. performed all other experiments. R.L., S.E., C.S., S.K., J.F. and J.C. designed the study and wrote the paper. S.K., J.F. and J.C. are senior authors and jointly managed the project. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 6PGD is important for tumor growth and contributes to regulation of glycolysis.
(a) Tumor growth in xenograft nude mice injected with 6PGD KD (knockdown) K562 cells and control vector cells. (b) Left: Dissected tumors in a representative nude mouse and expression of 6PGD in tumor lysates. Scale bar represents 5 mm. Right: tumor mass in xenograft nude mice injected with 6PGD KD K562 cells and control vector cells. (c) 6PGD activity (upper) and expression (lower) in K562 cells with inducible knockdown of 6PGD in the presence and absence of Dox (doxycycline). (d) Cell proliferation rates determined by cell counting in K562 cells with induced 6PGD knockdown and control cells. (e) Tumor growth in xenograft nude mice injected with K562 cells with inducible 6PGD knockdown fed with drinking water (+/− Dox). (f) Dissected tumors in two representative nude mice (left) and tumor mass (right) of xenograft mice injected with K562 cells with inducible 6PGD knockdown fed with drinking water (+/− Dox). Scale bar represents 2 mm. (g) 6PGD activity (left) and protein expression (right) in tumor lysates from mice injected with K562 cells with inducible 6PGD knockdown fed with drinking water in the presence and absence of Dox are shown. (h–i) 6PGD KD H157 and K562 cells were tested for oxidative PPP flux (h) and NADPH/NADP+ ratio (i). (j) 6PGD KD K562 cells were tested for intracellular Ru-5-P (left) and R-5-P (right) levels. (k) 6PGD KD H157 and K562 cells were tested for RNA biosynthesis. (a,e) Mean ± S.E.M.; n = 8 tumors from 8 mice. (b,f) n = 8 tumors from 8 mice; centerlines represent means. The P values were determined by paired Student’s t-test for (b) and unpaired Student’s t-test for (a,e,f) (∗: 0.01 < p < 0.05; ∗∗: 0.001 < p < 0.01; ∗∗∗: p < 0.001). (a–d, g–k) Data are from a single experiment that is representative of 2 independent experiments for (a,b,d,g–k) and 3 independent experiments for (c). Source data for independent replications and experiments with sample size <5 are available in Supplementary Table 1. Uncropped Western blots are provided in Supplementary Figure 9.
Supplementary Figure 2 6PGD controls Ru-5-P level to inhibit LKB1.
(a) K562 cells with inducible knockdown of 6PGD were tested for oxidative PPP flux (left) and lipogenesis (right) (+/− Dox). (b) 6PGD KD H1299 cells treated with Ru-5-P were tested for intracellular R-5-P levels. (c) Western blot results show AMPK knockdown by shRNA in 6PGD KD H1299 cells. (d) Lysates of 6PGD KD H1299 cells were treated with NADPH, followed by Western blot. (e) Lysates of 6PGD KD K562 cells were treated with Ru-5-P (left) or R-5-P (right), followed by Western blot. (f) Purified active AMPK proteins from H1299 cells were incubated with Ru-5-P, followed by in vitro AMPK kinase activity assay using SAMS peptide as substrate in the presence of γ-32P-ATP. The AMPK activity was assessed by incorporated amount of γ-32P in SAMS peptide and normalized to the control sample without Ru-5-P treatment. (g) Purified active LKB1 complex was incubated with recombinant AMPK in the presence of Ru-5-P, followed by Western blot to detect AMPK phosphorylation (p-T172). (h) Purified LKB1 complex was incubated with AMPK (upper) or MBP (lower) as substrates in the presence of NADPH, followed by Western blot to detect AMPK phosphorylation (upper) and Ser/Thr phosphorylation of MBP using a pan phospho-Ser/Thr antibody (lower). (i,j) Lysates from H1299 cells treated with Ru-5-P were applied to immunoprecipitation of LKB1 and Western blot to detect co-immunoprecipitated MO25 and STRAD (i), and protein expression levels of MO25, LKB1 and STRAD (j). (k) Lysates from H1299 cells treated with NADPH were applied to LKB1 immunoprecipitation and Western blot. (l) Cell lysates of 6PGD KD K562 cells were incubated with Ru-5-P (left) or R-5-P (right), followed by MO25 immunoprecipitation and Western blot. (m) Purified active LKB1 complex were incubated with R-5-P (left) or NADPH (right) for 20 minutes, followed by immunoprecipitation of LKB1 and Western blot. Data are from a single experiment that is representative of 2 independent experiments for (a,b,d,f,h–m) and 3 independent experiments for (c,e,g). Source data for independent replications and experiments with sample size <5 are available in Supplementary Table 1. Uncropped Western blots are provided in Supplementary Figure 9.
Supplementary Figure 3 6PGD is important for regulation of lipogenesis and ROS levels in cancer cells.
(a) 6PGD KD H1299 cells were assayed for lipogenesis in the absence and presence of NAC (3 mM). The relative lipogenesis rates were normalized to the control vector cells without NAC treatment. (b) 6PGD KD 212LN cells were assayed for general ROS levels in the absence and presence of NAC by measuring ROS-mediated DCFDA oxidation to fluorescent DCF in the cells by flow cytometry. The relative general ROS levels were normalized to the control vector cells without NAC treatment. (c–e) Proliferation rates of H1299 cells treated with Compound C and/or NAC were determined using a cell number-based assay. (f) Proliferation rates of H1299 cells with AMPK knockdown by shRNA (left) were determined using a cell number-based assay. (g) Cell proliferation rates were determined by cell counting in 6PGD KD H1299 cells cultured in the absence and presence of NAC (left), Compound C (middle) or AMPK shRNA (right). Samples were prepared using same number of cells. (h) Effect of LKB1 knockdown (left) on AMPK phosphorylation in 6PGD knockdown cells was examined by Western blot. (i) H1299 cells with knockdown of 6PGD and/or LKB1 were tested for intracellular Ru-5-P levels (left) and lipogenesis (right). (j–n) 6PGD knockdown A549 cells with expression of LKB1 WT or the kinase-dead form K78M were tested for AMPK activation using Western blot (j), lipogenesis in the absence (k) or presence (l) of Ru-5-P, cell proliferation in the presence and absence of NAC treatment (m), and intracellular Ru-5-P levels (n). Data are from a single experiment that is representative of 2 independent experiments for (a,b,d,f,h–n) and 3 independent experiments for (c,e,g). Source data for independent replications and experiments with sample size <5 are available Supplementary Table 1. Uncropped Western blots are provided in Supplementary Figure 9.
Supplementary Figure 4 Physcion and S3 are 6PGD inhibitors.
(a) K562 cells treated with increasing concentrations of Physcion were assayed for 6PGD (left) and G6PD (right) enzyme activity, which was normalized to the control cells without drug treatment. (b) Cell viability was determined in 6PGD KD K562 cells and control vector cells in the presence of increasing concentrations of Physcion. (c) Purified 6PGD WT (left) and M15A mutant (right) were assayed for 6PGD enzyme activity in the presence of increasing concentrations of S3. Absolute IC50 values are shown; NR = not reached. (d) H1299 6PGD knockdown cells were transiently transfected with vectors encoding 6PGD WT and M15A mutant, followed by Western blot. (e) H1299 6PGD knockdown cells were transiently transfected with vectors encoding 6PGD WT (left) and M15A mutant (right), followed by cell proliferation assay based on cell numbers in the presence of increasing concentrations of S3. Absolute IC50 values are shown; NR = not reached. (f) Cell proliferation rates of K562, MDA-MB-231 and 212LN cells in the presence of increasing concentrations of Physcion were determined by cell counting. (g) Apoptotic cell death of H1299 cells treated with increasing concentrations of Physcion for 12 hours, which was determined by annexin V staining. (h–l) K562 cells were assayed for intracellular concentration of 6-PG (h) and Ru-5-P (i), NADPH/NADP+ ratio (j), oxidative PPP flux and lipids (k), as well as apoptosis (l) in the presence and absence of Physcion. Data are from a single experiment that is representative of 2 independent experiments for (a–f,h–l) and 3 independent experiments for (g). Source data for independent replications and experiments with sample size <5 are available Supplementary Table 1. Uncropped Western blots are provided in Supplementary Figure 9.
Supplementary Figure 5 6PGD inhibitors attenuate cancer cell proliferation.
(a,b) H1299 and K562 cells were assayed for intracellular 6-PG (a) and Ru-5-P (b) concentrations in the presence and absence of S3. (c) H1299 and K562 cells were treated with increasing concentrations of Physcion (left panels) or S3 (right panels), followed by PFK enzyme activity assay. (d,e) H1299 and K562 cells were treated with increasing concentrations of Physcion, followed by assays for lactate production (d) and intracellular ATP levels (e). (f) H1299 cells treated with or without S3 were assayed for cell proliferation rates by cell counting in the presence and absence of NAC. (g) K562 cells were treated with increasing concentrations of Physcion, followed by Western blot to detect phosphorylation levels of ACC1 (p79; upper) and AMPK (pT172; lower). 6PGD KD cells were included as a control. (h,i) K562 cells treated with or without Physcion were assayed for general ROS levels (h) and cell proliferation rates by cell counting (i) in the presence and absence of NAC. (j) K562 cells treated with or without Physcion were assayed for cell proliferation rates by cell counting in the presence and absence of Compound C (left) or lentivirus harboring AMPK shRNA (middle and right). (k–o) Effects of Physcion treatment on normal proliferating HFF cells were assayed for 6PGD activity (k), oxidative PPP flux rate (l), phosphorylation levels of AMPK (m) and lipid biosynthesis (n), as well as cell proliferation rates by cell counting (o). Data are from a single experiment that is representative of 2 independent experiments for (a,b,d,f,h–o) and 3 independent experiments for (c,e,g). Source data for independent replications and experiments with sample size <5 are available in SI Table 1. Uncropped Western blots are provided in Supplementary Information.
Supplementary Figure 6 S3 effectively inhibits tumor growth in xenograft nude mice.
(a) Cell viability of human cancer cells treated with S3 was determined by MTT assay. Normal proliferating human melanocyte PIG1 cells and human dermal fibroblasts (HDF) were included as controls. (b,c) Tumor growth (b) and tumor mass (c; left) in K562 cell-xenograft nude mice treated with S3 or DMSO. P values were determined by a two-tailed unpaired Student’s t test. c, right: Dissected tumors in representative nude mice treated with DMSO control or S3 are shown. Scale bar represents 2 mm. (d) 6PGD activity (upper) and protein expression (lower) in tumor lysates of K562 xenograft mice treated with DMSO or S3. (e) Representative images of IHC staining of Ki-67 (brown color) from K562 xenograft mice treated with DMSO or S3 are shown. Scale bars represent 50 μM. (f) 6PGD activity and cell proliferation in Tu212 cells treated with Physcion. (g) Dissected tumors in representative nude mice injected with Tu212 cells and treated with DMSO or S3. Scale bar represents 2 mm. (h,i) Effects of chronic treatment with S3 or DMSO on body weights (h) and serum chemistry (i) of nude mice are shown. ALT: alanine aminotransferase; AST: aspartate aminotransferase. (j) Peripheral blood samples from S3 or DMSO-treated H1299 xenograft nude mice (3 representative mice) were examined for hematological properties using CBC analysis. (k) Histological morphology of hematoxylin-eosin stained tissue sections of representative H1299 xenograft nude mice in S3 or DMSO-treated groups presented in Figure 8a–8d. Images were analyzed and captured using ImageScope software (Aperio Technologies Inc.) without any additional or subsequent image processing (high power images are 20×; low power images are 4×). Scale bars are indicated. (b) Mean ± S.E.M.; n = 6 tumors from 6 mice. (c,h) n = 6 tumors from 6 mice; (c) centerlines represent means. (i,j) Mean ± S.D.; n = 3 serum from 3 mice. P values were obtained by two-sided Student’s t-test for (b,c) (∗: 0.01 < p < 0.05). (a–d,f) Results of one representative experiment from two independent experiments are shown. Source data for independent replications and experiments with sample size <5 are available Supplementary Table 1. Uncropped Western blots are provided in Supplementary Figure 9.
Supplementary Figure 7 Physcion and S3 effectively inhibit cell proliferation of human primary leukemia cells from human patients with minimal toxicity.
(a) Effect of treatment with Physcion on cell proliferation was examined in human primary leukemia cells isolated from bone marrow (BM) samples from a representative AML patient. (b,c) Effect of Physcion treatment on lactate production (b) and intracellular ATP levels (c) in human primary leukemia cells isolated from PB samples from a representative CML patient. (d) Effect of S3 treatment on cell viability was examined in human primary leukemia cells isolated from PB samples from a representative B-ALL patient. (e,f) Effect of treatment with S3 on cell proliferation of human primary leukemia cells isolated from PB samples from a representative B-ALL patient (e) and cell viability human primary leukemia cells isolated from PB or BM samples from two AML and one CML patients (f) were examined. (g) S3 shows no toxicity in treatment (72h) of peripheral blood cells from a representative healthy human donor. (h) Physcion and S3 show no toxicity in treatment (72h) of CD34+ cells isolated from bone marrow samples from representative healthy human donors. (a–h) The data are obtained from peripheral blood or bone marrow of multiple human primary tissue samples. Each sample was used for a single experiment of 3 technical replicates. Source data for experiments with sample size <5 are available in Supplementary Table 1.
Supplementary Figure 8 6PGD provides an additional link between oxidative PPP and lipogenesis.
(a) Proposed working model. Left: In cancer cells, 6PGD activity is commonly upregulated by lysine acetylation, which promotes oxidative PPP and subsequent biosynthesis of nucleotide and RNA. The intracellular Ru-5-P is maintained at a physiological level that is sufficient to inhibit LKB1-AMPK signaling, leading to relief of AMPK-dependent inhibition of ACC1 and consequently high levels of lipid biosynthesis to fulfill the request of rapidly growing tumors. Right: Attenuation of 6PGD by shRNA or small molecule inhibitors results in decreased Ru-5-P levels, leading to reduced nucleotide/RNA biosynthesis and increased LKB1-AMPK signaling, which inhibits ACC1 and lipogenesis. (b,c) H1299 cells with stable knockdown of H6PD (b) or G6PD (c) were tested for AMPK phosphorylation (left), Ru-5-P levels (middle), and cell proliferation (right). Data are from a single experiment that is representative of 2 independent experiments for (b) and 3 independent experiments for (c). Source data for independent replications and experiments with sample size <5 are available in Supplementary Table 1. Uncropped Western blots are provided in Supplementary Figure 9.
Supplementary information
Supplementary Information
Supplementary Information (PDF 1393 kb)
Supplementary Table 1
Supplementary Information (XLSX 324 kb)
Supplementary Table 2
Supplementary Information (XLS 30 kb)
Supplementary Table 3
Supplementary Information (XLS 24 kb)
Rights and permissions
About this article

Cite this article
Lin, R., Elf, S., Shan, C. et al. 6-Phosphogluconate dehydrogenase links oxidative PPP, lipogenesis and tumour growth by inhibiting LKB1–AMPK signalling. Nat Cell Biol 17, 1484–1496 (2015). https://doi.org/10.1038/ncb3255
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ncb3255
This article is cited by
-
Mangiferin alleviates diabetic pulmonary fibrosis in mice via inhibiting endothelial-mesenchymal transition through AMPK/FoxO3/SIRT3 axis
Acta Pharmacologica Sinica (2024)
-
Discovery of a nitroaromatic nannocystin with potent in vivo anticancer activity against colorectal cancer by targeting AKT1
Acta Pharmacologica Sinica (2024)
-
Metabolic reprogramming in colorectal cancer: regulatory networks and therapy
Cell & Bioscience (2023)
-
New insights into activation and function of the AMPK
Nature Reviews Molecular Cell Biology (2023)
-
N6-methyladenosine modification of circ_0003215 suppresses the pentose phosphate pathway and malignancy of colorectal cancer through the miR-663b/DLG4/G6PD axis
Cell Death & Disease (2022)
