
Abstract
Chromatin and metabolic states both influence lifespan, but how they interact in lifespan regulation is largely unknown. The COMPASS chromatin complex, which trimethylates lysine 4 on histone H3 (H3K4me3), regulates lifespan in Caenorhabditis elegans. However, the mechanism by which H3K4me3 modifiers affect longevity, and whether this mechanism involves metabolic changes, remain unclear. Here we show that a deficiency in H3K4me3 methyltransferase, which extends lifespan, promotes fat accumulation in worms with a specific enrichment of mono-unsaturated fatty acids (MUFAs). This fat metabolism switch in H3K4me3 methyltransferase-deficient worms is mediated at least in part by the downregulation of germline targets, including S6 kinase, and by the activation of an intestinal transcriptional network that upregulates delta-9 fatty acid desaturases. Notably, the accumulation of MUFAs is necessary for the lifespan extension of H3K4me3 methyltransferase-deficient worms, and dietary MUFAs are sufficient to extend lifespan. Given the conservation of lipid metabolism, dietary or endogenous MUFAs could extend lifespan and healthspan in other species, including mammals.
Similar content being viewed by others


Main
Chromatin state, which encompasses post-translational modification of histone proteins, integrates environmental signals to influence gene expression and downstream cellular processes. Enzymes that deposit or remove histone modifications regulate lifespan in several species1,2. For example, methyltransferase or demethylase complexes that modify H3K4me3, H3K27me3 and H3K36me3 modulate lifespan in yeast, worms and flies1,2. Several chromatin-modifying enzymes require intracellular metabolites as co-factors (for example, NAD+, α-ketoglutarate or S-adenosyl methionine), rendering their activity highly sensitive to metabolic states1,2. A subset of these chromatin-modifying enzymes, such as deacetylases, in turn influence metabolism1,2. However, the impact of methyltransferases and demethylases on metabolism remains unclear.
Fat metabolism plays an important role in many physiological and pathological processes. It influences long-term energy storage, intercellular and intracellular signalling and membrane homeostasis. In humans, excessive fat storage in the form of triglycerides (TAGs) is associated with diseases such as atherosclerosis and type 2 diabetes3. On the other hand, studies in both invertebrates and mammals have suggested that specific alterations in fat profiles, and even elevated fat storage, can be associated with longevity4. Whether fat metabolism links chromatin modifiers to lifespan extension is unknown.
H3K4me3 modifiers alter fat metabolism
The COMPASS chromatin complex, which catalyses trimethylation of lysine 4 on histone H3 (H3K4me3)5, regulates lifespan in C. elegans6. The H3K4me3 global landscape is remodelled during ageing and in cellular senescence in mammals1,2. However, the mechanism by which H3K4me3 modifiers influence longevity remains unclear. To determine whether the COMPASS H3K4me3 methyltransferase complex affects fat metabolism, we used Oil-Red-O (ORO) staining. In fixed tissues, ORO stains neutral lipids such as TAGs7. Worms with a deficiency in the COMPASS H3K4me3 methyltransferase SET-2 (homologue of mammalian SET1), which are long-lived6 (Supplementary Table 1), exhibited increased ORO staining (Fig. 1a). Conversely, worms deficient for the H3K4me3 demethylase RBR-2 (homologue of mammalian JARID1), which are slightly short-lived6 (Supplementary Table 1), displayed decreased ORO staining (Fig. 1a). RNA interference (RNAi)-mediated knockdown of ash-2 (homologue of mammalian ASH2L) and wdr-5.1 (homologue of mammalian WDR5), which are shared components between the COMPASS and Trithorax-related H3K4me3 complexes5, and of set-2, a specific component of the COMPASS H3K4me3 complex5, also extended lifespan6 (Supplementary Table 1) and increased ORO staining (Fig. 1b and Extended Data Fig. 1a). By contrast, knockdown of set-16 (homologue of mammalian MLL) and utx-1 (homologue of mammalian UTX, also known as KDM6A), specific components of the Trithorax-related H3K4me3 complex5, did not significantly affect ORO levels (Fig. 1b). Thus, the COMPASS H3K4me3 methyltransferase complex specifically influences fat levels.

a, ORO staining images and quantification. Mean ± s.d., n ≥ 55 worms per condition. b, As in a, n ≥ 16 worms per condition. c, As in a, n ≥ 29 worms (upper panels) or n ≥ 25 intestinal cells (lower panels) per condition. d, GC–MS quantification. Mean ± s.e.m. of two independent experiments, each with three biological replicates. e, As in a, n ≥ 26 worms per condition. f, ash-2 RNAi extends lifespan in wild-type (P < 0.0001, log-rank) but not in rde-1(ne219); Is[Pnhx-2::rde-1] worms. g, As in a, n ≥ 18 worms per condition. h, ash-2 RNAi extends lifespan in both wild-type and rde-1(ne219); Si[Pmex-5::rde-1] worms (P < 0.0001, log-rank). b, c, e–h, Representative of two experiments. P values: a, b, e, g, Kruskal-Wallis with Dunn’s correction; c, d, two-tailed Mann-Whitney.
Fat accumulated most prominently in the intestine in ash-2 knockdown worms (Fig. 1c), and this was observed at the initiation of reproduction, during reproduction and after reproduction (Fig. 1c and Extended Data Fig. 1b, c). The intestinal fat accumulation in ash-2 knockdown worms was also observed using Nile red, which stains neutral lipids in fixed tissues8 (Extended Data Fig. 1d–f). Furthermore, gas chromatography coupled to mass spectrometry (GC–MS) showed that ash-2 knockdown worms had significantly higher levels of TAGs than control worms, but similar levels of phospholipids (Fig. 1d). Fat accumulation in ash-2 knockdown worms is unlikely to be caused by a shift in fat resources resulting from reduced fertility, because ash-2 knockdown worms had a similar number of progeny to wild-type worms6 (Extended Data Fig. 1g). Thus, H3K4me3 methyltransferase-deficient worms, which are long-lived and fertile, accumulate intestinal fat in the form of TAGs.
H3K4me3 modifiers act mostly in the germline to extend lifespan6, but fat accumulation occurs in the intestine (Fig. 1c). To determine whether H3K4me3 modifiers act in the germline or intestine to regulate fat accumulation, we performed tissue-selective RNAi knockdown and transgenic rescue experiments. Knockdown of ash-2 in the intestine (rde-1(ne219);Is[Pnhx-2::rde-1]9) did not promote fat accumulation or lifespan extension (Fig. 1e, f and Extended Data Fig. 1h, i). Likewise, knockdown of set-2 in the intestine did not increase fat accumulation (Extended Data Fig. 1j), and overexpression of set-2 in the intestine of set-2(ok952) mutants did not abrogate the elevated fat levels found in set-2(ok952) mutants (Extended Data Fig. 1k–m). These results suggest that H3K4me3 modifiers act mostly outside of the intestine to influence fat accumulation. As strains that allow germline-specific RNAi knockdown do not currently exist in C. elegans, we used the rde-1(ne219);Si[Pmex-5::rde-1] strain, which restricts RNAi to the germline and intestine10. Interestingly, ash-2 knockdown in the germline and intestine promoted fat accumulation and extended lifespan (Fig. 1g, h and Extended Data Fig. 1i). Similarly, set-2 knockdown in the germline and intestine also increased fat levels (Extended Data Fig. 1j). ash-2 knockdown in rrf-1(pk1417) mutants, in which RNAi is restricted to the germline, intestine and some hypodermal seam cells11, also increased fat levels and extended lifespan (Extended Data Fig. 1n and Supplementary Table 1). Thus, the COMPASS H3K4me3 methyltransferase complex acts mostly in the germline to regulate intestinal fat accumulation and lifespan, implying that there is communication between the germline and intestine.
A switch to MUFA metabolism
Like mammals, worms have different categories of fatty acids: saturated fatty acids (SFAs), mono-unsaturated fatty acids (MUFAs) and poly-unsaturated fatty acids (PUFAs) (Fig. 2a). To identify specific lipid species associated with longevity in H3K4me3 methyltransferase-deficient worms, we profiled long-chain fatty acids by GC–MS. At all ages tested, ash-2 knockdown increased MUFAs, while SFAs and PUFAs remained mostly unchanged (Fig. 2b). The MUFAs palmitoleic acid and cis-vaccenic acid were significantly elevated at all ages, whereas the MUFA oleic acid was raised after reproduction in ash-2 knockdown worms and in worms deficient for set-2 (Fig. 2b, c). By contrast, MUFAs remained largely unaffected in worms deficient for set-16 and utx-1 (Extended Data Fig. 2a). Thus, the COMPASS H3K4me3 methyltransferase complex specifically influences MUFA metabolism.

a, Fat metabolism pathways with genes used in RT–qPCR. b, c, GC–MS quantification of fatty acids. Mean ± s.e.m. of two or three independent experiments, each with three biological replicates. d, RT–qPCR. Mean ± s.e.m. of two independent experiments, each with three biological replicates. e, RT–qPCR. Mean ± s.e.m. of two independent experiments, each with two or three biological replicates. f, Images and quantification of GFP fluorescence. Mean ± s.d., n ≥ 18 worms per condition. Representative of two (FAT-5) or three (FAT-7) experiments. Scale bars, 100 μm. g, h, GC–MS quantification of MUFAs. Mean ± s.e.m. of three (g) or two (h) independent experiments, each with two or three biological replicates. P values: two-tailed Mann–Whitney (with Benjamini–Hochberg correction for >2 conditions). *P < 0.05, **P < 0.01, ***P < 0.001.
To determine whether this switch to MUFA accumulation arises from changes in fat synthesis, storage or breakdown, we examined the expression of the specific enzymes that control these steps (Fig. 2a). Knockdown of ash-2 strongly increased the expression of the delta-9 fatty acid desaturase genes fat-5 and fat-7 (Fig. 2d). FAT-5, a palmitoyl-coenzyme A (CoA) desaturase, preferentially converts palmitic acid into the MUFA palmitoleic acid, whereas FAT-7 and FAT-6, two stearoyl-CoA desaturases, convert stearic acid into the MUFA oleic acid12 (Fig. 2a). The expression of fat-5 and fat-7 was also upregulated in set-2 mutant or knockdown worms (Extended Data Fig. 2b). By contrast, the expression of fat-2, which catalyses the first step in converting MUFAs to PUFAs13, and the expression of other fat metabolism enzymes (for example, lipases), was not consistently affected by ash-2 knockdown (Fig. 2d). The expression of fat-5 and fat-7 was elevated in the intestine but not in the germline of ash-2 knockdown worms (Fig. 2e), and ash-2 acts mostly in the germline to promote this intestinal fat-5 and fat-7 elevation (Fig. 2e). Transgenic worms expressing green fluorescent protein (GFP) translational reporters for FAT-5 or FAT-7 also showed increased GFP fluorescence in the intestine upon ash-2 knockdown (Fig. 2f). Thus, a deficiency in germline H3K4me3 methyltransferase leads to the upregulation of delta-9 desaturases in the intestine, suggesting that MUFA accumulation occurs at the level of synthesis.
We next examined the role of delta-9 desaturases in the accumulation of MUFA in H3K4me3 methyltransferase-deficient worms. Knockdown of fat-7 in ash-2-deficient worms specifically decreased oleic acid levels at the initiation of reproduction and reduced all three MUFAs (oleic, palmitoleic and cis-vaccenic acid) after reproduction (Fig. 2g, h and Extended Data Fig. 2c). By contrast, fat-5 knockdown in ash-2-deficient worms increased palmitoleic and cis-vaccenic acid (Extended Data Fig. 2d), probably owing to compensatory upregulation of fat-7 (Extended Data Fig. 2e). These results suggest that the MUFA switch in H3K4me3 methyltransferase-deficient worms is driven by FAT-7 upregulation in the intestine.
Germline targets of H3K4me3 modifiers
The H3K4me3 methyltransferase complex acts mostly in the germline to trigger intestinal upregulation of delta-9 desaturases. To understand the germline-to-intestine communication better, we identified germline targets of the H3K4me3 methyltransferase complex. We generated an RNA sequencing (RNA-seq) dataset using micro-dissected germlines and intestines from ash-2 knockdown worms and analysed this dataset (Extended Data Fig. 3a, b and Supplementary Table 2–5), as well as other large-scale datasets6,14,15. Because H3K4me3 methyltransferase complexes generally activate gene expression5, we selected targets that were downregulated in H3K4me3 methyltransferase-deficient worms (Extended Data Table 1). Using quantitative PCR with reverse transcription (RT–qPCR), we confirmed that ash-2 knockdown led to the significant downregulation of four candidate genes in the germline (rsks-1, tbh-1, nhr-10 and unc-132), one candidate in both the germline and intestine (F21A3.11) and two candidates in the intestine (asm-2 and dod-23) (Fig. 3a and Extended Data Fig. 3c).

a, RT–qPCR. Mean ± s.e.m. of two independent experiments, each with three biological replicates. b, ORO quantification. Mean ± s.d., n ≥ 23 worms per condition. Representative of two experiments. c, GC–MS profiles of fatty acids. Mean ± s.e.m. of two independent experiments, each with two or three biological replicates. d, RT–qPCR. Mean ± s.e.m. of two independent experiments, each with three biological replicates. P values: b, Kruskal–Wallis with Dunn’s correction; a, c, d, two-tailed Mann–Whitney with Benjamini–Hochberg correction. *P < 0.05, **P < 0.01.
We next assessed the functional involvement of these candidates in fat metabolism. Knockdown of rsks-1, tbh-1, F21A3.11 or unc-132 led to increased ORO staining (Fig. 3b and Extended Data Fig. 3d–g). Among these four targets, rsks-1 was the only one that mostly functioned in the germline to regulate fat accumulation (Extended Data Fig. 3d–g). RSKS-1 (homologue of mammalian S6 kinase) is a key conserved substrate of mechanistic target of rapamycin (mTOR) complex 116. A deficiency in RSKS-1 extends lifespan in C. elegans and mice17 and promotes fat accumulation in C. elegans18. We investigated whether rsks-1 could mediate the fat metabolism switch of H3K4me3 methyltransferase-deficient worms. Knockdown or mutation of rsks-1 led to increased fat accumulation, MUFA levels and fat-7 expression (Fig. 3b–d and Extended Data 3h–j). Notably, ash-2 knockdown could not further increase fat accumulation and fat-7 expression in rsks-1-deficient worms (Fig. 3d and Extended Data 3i, j). Likewise, knockdown of the RSKS-1 upstream regulator let-363 (homologue of mammalian mTOR) also led to increased fat accumulation, MUFA levels and fat-7 expression (Extended Data Fig. 3k–n), and ash-2 knockdown did not further increase fat-7 expression in these worms (Extended Data Fig. 3k). Deficiencies in RSKS-1 or its upstream regulators (let-363 or daf-15, homologue of mammalian Raptor) resulted in lifespan extension, and the ability of ash-2 or set-2 deficiency to extend lifespan was diminished in the context of rsks-1 deficiency (Extended Data Fig. 3o, p). Collectively, these data suggest that RSKS-1 is a germline target of the H3K4me3 methyltransferase complex that mediates at least part of the intestinal upregulation of delta-9 desaturases leading to MUFA accumulation.
MUFAs mediate longevity by H3K4me3 modifiers
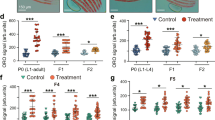
To examine the role of MUFAs in the modulation of longevity by H3K4me3 modifiers, we first investigated the molecular mechanisms that underlie MUFA accumulation in worms with H3K4me3 methyltransferase deficiency. Delta-9 desaturases are known targets of a highly conserved intestinal transcriptional network that involves the transcription factor SBP-1 (homologue of mammalian SREBP)19 and the Mediator complex subunit MDT-15 (homologue of mammalian MED15)20, as well as other transcription factors, such as NHR-49 (functional homologue of mammalian PPARα)21 and NHR-80 (homologue of mammalian HNF4)22 (Fig. 4a). Knockdown of ash-2 and set-2, as well as mutation of set-2, increased nuclear accumulation of SBP-1 in intestinal cells (Fig. 4b and Extended Data Fig. 4a), and this accumulation was reduced in rsks-1-deficient worms (Extended Data Fig. 4b–d). We next tested whether SBP-1 and MDT-15 are necessary for upregulation of delta-9 desaturases and accumulation of MUFAs in H3K4me3 methyltransferase-deficient worms. Knockdown of sbp-1 or mdt-15 decreased the basal levels of fat-5 and fat-7 mRNA and reduced their upregulation by ash-2 knockdown (Fig. 4c and Extended Data Fig. 4e). Knockdown of sbp-1 or mdt-15 also blocked the elevation of MUFAs (palmitoleic, cis-vaccenic and oleic acid) in ash-2-deficient worms after reproduction (Fig. 4d and Extended Data Fig. 4f). Thus, SBP-1 and MDT-15 are critical for delta-9 desaturase upregulation and MUFA accumulation in H3K4me3 methyltransferase-deficient worms.

a, Conserved transcriptional network regulating delta-9 desaturase genes. b, Images and quantification of SBP-1 nuclear accumulation. Mean ± s.d. of two independent experiments, each with n = 4–6 nuclei per worm and ≥ 8 worms per condition. c, RT–qPCR. Mean ± s.e.m. of three independent experiments, each with three biological replicates. d, GC–MS. Mean ± s.e.m. of two independent experiments, each with two or three biological replicates. e, ORO quantification. Mean ± s.d., n ≥ 32 worms per condition. f, Lifespan extension by ash-2 RNAi is reduced in sbp-1 RNAi (9.78%) worms compared to control worms (21.90%) (P = 0.0003, two-way ANOVA). g, As in e, n ≥ 13 worms per condition. h, Dietary oleic acid extends lifespan in the context of fat-7 and ash-2 double RNAi (P < 0.0001, log-rank). g, h, Representative of two experiments. P values: b, two-tailed Mann-Whitney; e, g, Kruskal-Wallis with Dunn’s correction; c, d, two-tailed Mann-Whitney with Benjamini–Hochberg correction. *P < 0.05, **P < 0.01.
We next used this molecular knowledge to assess whether the MUFA metabolic switch causes the lifespan extension of H3K4me3 methyltransferase-deficient worms. Interestingly, knockdown or mutation of sbp-1 or mdt-15 abolished fat accumulation and lifespan extension in ash-2 knockdown worms (Fig. 4e, f, Extended Data Fig. 4g, h and Supplementary Table 1). Likewise, sbp-1 knockdown abrogated the longevity of set-2 mutant worms (Supplementary Table 1). By contrast, nhr-49 mutation only partially diminished the ability of ash-2 deficiency to promote fat accumulation and lifespan extension (Extended Data Fig. 4i, j) and nhr-80 mutation had no effect (Extended Data Fig. 4k, l). We also tested whether delta-9 desaturases are required for the longevity of H3K4me3 methyltransferase-deficient worms. Knockdown of fat-7 or double mutations in fat-6 and fat-7, which produce a strong deficiency in oleic acid synthesis23, blocked the fat accumulation and longevity of ash-2-deficient worms (Fig. 4g, h and Extended Data Fig. 5a, b). By contrast, single hypomorphic mutations in fat-7 or fat-6 (or double mutations in fat-5 and fat-7 or fat-5 and fat-6), which produce only mild deficiencies in MUFA synthesis22,23, did not abolish the longevity of ash-2-knockdown worms (Extended Data Fig. 5c–f). Thus, a conserved pathway involving SBP-1, MDT-15 and the downstream delta-9 desaturase FAT-7 is critical for the longevity of H3K4me3 methyltransferase-deficient worms.
To assess the specific role of MUFAs in H3K4me3 methyltransferase-deficient worms, we supplemented oleic acid in the diet (Extended Data Fig. 5g, h). Dietary oleic acid largely restored the high-fat phenotype and lifespan extension in ash-2 and fat-7 double knockdown worms (Fig. 4g, h and Extended Data Fig. 5i, j). Similarly, dietary oleic acid restored high fat levels in ash-2 and sbp-1 double knockdown worms (Extended Data Fig. 5k). Furthermore, knockdown of fat-2, which normally converts oleic acid into downstream PUFA13, increased fat accumulation and lifespan extension, and ash-2 deficiency did not further increase fat levels or extend lifespan in fat-2 knockdown worms (Extended Data Fig. 5l–o). Thus, the MUFA oleic acid plays a key role in the longevity of H3K4me3 methyltransferase-deficient worms.
Dietary MUFAs extend lifespan
The role of oleic acid in lifespan regulation in the context of H3K4me3 methyltransferase deficiency led us to examine the importance of MUFAs and their downstream PUFAs in the regulation of lifespan under physiological conditions (Fig. 5a). Dietary supplementation of individual MUFAs (oleic, palmitoleic or cis-vaccenic acid; Fig. 5b, d and Extended Data Fig. 6a) was sufficient to extend lifespan (Fig. 5c, e, f and Extended Data Fig. 6b). By contrast, dietary supplementation of the PUFAs linoleic and alpha-linolenic acid (Extended Data Fig. 6c, e) did not significantly affect lifespan (Extended Data Fig. 6d, f and Fig. 5f). All MUFAs and PUFAs tested increased total fat accumulation, except for the PUFA alpha-linolenic acid (Fig. 5c, e and Extended Data Fig. 6b, d, f). Furthermore, overexpression of the oleic acid-synthesizing enzyme FAT-7 in the intestine increased fat accumulation and extended lifespan (Fig. 5g and Extended Data Fig. 6g–i). Oleic acid supplementation did not further extend the long lifespan of FAT-7-overexpressing worms (Fig. 5g and Extended Data Fig. 6i). Collectively, these results suggest that dietary supplementation and endogenous accumulation of MUFAs, but not downstream PUFAs, are beneficial for lifespan.

a, Chemical structure of fatty acids used in supplementation experiments. b, GC–MS. Mean ± s.e.m. of three independent experiments, each with two or three biological replicates. c, Oleic acid supplementation extends lifespan in wild-type worms (P < 0.0001, log-rank). Inset: ORO quantification. Mean ± s.d., n ≥ 27 worms per condition. d, GC–MS. Mean ± s.e.m. of two independent experiments, each with two or three biological replicates. e, Palmitoleic acid supplementation extends lifespan in wild-type worms (P < 0.0001, log-rank). Inset: as in c, n ≥ 45 worms per condition. f, Mean lifespan changes in independent supplementation experiments (P values: log-rank). g, FAT-7 overexpression extends lifespan (P < 0.0001, log-rank), and this lifespan is not extended further by dietary oleic acid. P values: two-tailed Mann-Whitney (Benjamini–Hochberg correction for more than one comparison). *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Our study shows that histone methylation modifiers influence MUFA metabolism. We propose a model in which H3K4me3 methyltransferase deficiency causes the downregulation of specific targets (for example, RSKS-1) in the germline, which in turn triggers a germline-to-intestine signal leading to activation of SBP-1, expression of delta-9 desaturases and accumulation of MUFAs in the intestine (Extended Data Fig. 6j). The nature of this germline-to-intestine signal is unknown, but it could be a diffusible lipid signal (‘lipokine’), a signal from the maturing eggs or an alteration in levels of lipid-binding proteins. Deficiency in the H3K4me3 methyltransferase complex might also affect fat metabolism by altering the methyl pool available for other enzymatic reactions or by methylating non-histone substrates that have not yet been identified.
Our work also shows that dietary MUFAs, but not their downstream PUFAs, positively affect lifespan. Previous studies have tested supplementation of individual fatty acids on lifespan24,25,26,27,28 and implicated other PUFAs in lifespan regulation24,26. Dietary oleic acid did not extend the lifespan of wild-type worms in previous studies25,27,28, although these studies used different oleic acid supplementation protocols. MUFAs could contribute to longevity by promoting membrane fluidity, minimizing oxidative stress, enhancing energy storage or activating signalling pathways. The beneficial effects of MUFAs are exciting because of their dietary availability (for example, oleic acid in olives) and microbiotic sources (for example, palmitoleic acid in E. coli)29. In humans, diets rich in MUFAs have been associated with a decreased risk of cardiovascular disease and diabetes30. The general conservation of lipid metabolism between worms and mammals suggests that the benefits of MUFAs on healthspan and lifespan could be conserved in other species.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment except where indicated.
Worm strains
All strains were maintained on standard nematode growth medium (NGM) plates with Escherichia coli (OP50-1, streptomycin-resistant). Unless otherwise noted, worms were grown on the RNAi strain HT115 (DE3) for all experiments. The wild-type N2 strain (ABR) was provided by M.-W. Tan. The NL2098 rrf-1(pk1417) strain, which restricts RNAi to the germline, intestine and partially the hypodermal seam cells11,31, was provided by A. Fire. The germline and intestine RNAi strain AMJ345 rde-1(ne219);jamSi2[Pmex-5::rde-1(+)] and corresponding wild-type N2 (AMJ) were provided by A. Jose10. The intestine-specific RNAi strain VP303 rde-1(ne219);kbIs7[nhx-2p::rde-1 + rol-6(su1006)] was obtained from the Caenorhabditis Genetics Center (CGC). Other strains obtained from the CGC were as listed: RB1025 set-2(ok952), ZR1 rbr-2(tm1231), DA1116 eat-2(ad1116), XA7702 mdt-15(tm2182), STE68 nhr-49(nr2041), BX165 nhr-80(tm1011), BX153 fat-7(wa36), BX106 fat-6(tm331), BX160 fat-7(wa36); fat-5(tm420), BX110 fat-6(tm331);fat-5(tm420), BX156 fat-6(tm331);fat-7(wa36), RB1206 rsks-1(ok1255), and fluorescence translational fusion reporters BX150 lin-15B&lin-15A(n765);waEx18[fat-5::GFP + lin15(+)], BX113 lin-15B&lin-15A(n765);waEx15[fat-7::GFP + lin-15(+)] and CE548 sbp-1(ep79);epEx141[sbp-1::GFP::SBP-1 + rol-6(su1006)]. Mutants set-2(ok952), rbr-2(tm1213), eat-2(ad1116), and transgenic strain CE548 were outcrossed to our laboratory N2 (ABR) at least six times. During backcrossing, the sbp-1(ep79) mutation in CE548 was removed to generate a line expressing a translational fusion between GFP and SBP-1 in a wild-type background. See Supplementary Table 6 for a list of strains. All experiments were performed using hermaphrodites.
RNA interference
RNAi was performed as described6. HT115 (DE3) bacteria were transformed with vectors expressing double-stranded (ds)RNA targeting the following genes of interest: ash-2, set-2, wdr-5.1, set-16, utx-1, jmjd-3.1, fat-2, fat-5, fat-7, mdt-15, sbp-1, tbh-1, nhr-10, F21A3.11, dod-23 (Ahringer Library, A. Fire), let-363 (X. Long), daf-15 (A. Hsu), rsks-1, asm-2 and unc-132 (Vidal Library, Dharmacon). All vectors were confirmed by sequencing. Empty vector bacteria, containing the RNAi plasmid without an RNAi insert, served as a control for all RNAi experiments. RNAi culture was concentrated 30-fold, and stored at 4 °C for no more than 2 weeks. For double RNAi treatments, two cultures were mixed in a 1:1 ratio by volume. To obtain a synchronized worm population, egglay was performed on appropriate RNAi plates for 1–4 h. All RNAi treatments began at hatching, except for ORO and lifespan experiments using sbp-1, mdt-15, fat-2, let-363 or daf-15 RNAi, which began at the initiation of reproduction (day 2.5 of life) to minimize the effects on development.
Lifespan assays
Worm lifespan assays were performed at 20 °C as described32. Kaplan–Meir lifespan curves were generated using GraphPad Prism, and statistical results were obtained by log-rank analysis using JMP software. Two-way ANOVA was used to assess lifespan interactions between conditions. Lifespan assays that were blinded or repeated by an independent investigator are indicated in Supplementary Table 1. Representative Kaplan–Meir lifespan curves are shown in the figures and all statistics are included in Supplementary Table 1.
Oil Red O and Nile Red staining and quantification
ORO staining of fixed worms was conducted as described7, except that 2% paraformaldehyde in PBS was used to fix worm tissues. ORO staining of dissected intestines was performed for 1 h. Nile Red staining of fixed worms was performed as described33, except that staining was conducted in 1.5-ml Eppendorf tubes. ORO- or Nile Red-stained worms were mounted onto 2% agar pads, and imaged at 10× magnification using a Zeiss AxioSkop 2 Plus. Images were acquired using an AxioCam MRc camera with AxioVision 4.7 software and saved as TIF files. The same exposure settings were used across all conditions within each experiment. Using FIJI image processing software, raw images were subtracted from background, converted to greyscale, inverted and thresholded to outline the worm body. The same threshold values were used across all conditions within each experiment. ‘Analyze Particles’ was used to measure mean ORO intensity per worm. To quantify anterior intestinal cells of whole worms or extruded intestines of dissected worms, specific regions in the raw images were selected using the oval brush tools (intestinal cells) or the segmented line tool (extruded intestines), saved to the Region of Interest (ROI) manager, processed as described above, and quantified for mean ORO intensity per selected region. Mean intensity values in arbitrary units (a.u.) were graphed using Graphpad Prism, and statistical significance was determined using a two-tailed Mann–Whitney test (two conditions) or the Kruskal–Wallis test with Dunn’s correction for multiple comparisons (more than two conditions). For figures containing straightened whole worms or intestines, unmodified images were selected and straightened, and representative worms or intestines from each condition were concatenated into montages.
GFP quantification
For quantification of FAT-5 and FAT-7 expression, transgenic lines expressing a translational fusion between FAT-5 and GFP or FAT-7 and GFP were used. Worms at active reproduction (day 3 of life) were anaesthetized in M9 (22 mM KH2PO4, 42 mM Na2HPO4, 86 mM NaCl and 1mM MgSO4, dissolved in water) containing 100 μM levamisole and mounted on 2% agar pads. All images were acquired on Zeiss AxioSkop 2 Plus using an AxioCam MRc camera with AxioVision 4.7 software with identical exposure settings for the same experiment. Mean fluorescence intensity in arbitrary units (a.u.) of the whole worm was quantified in FIJI as described above. These experiments were conducted in a blinded manner. For visualization of intestinal nuclear SBP-1 expression, a transgenic line expressing a translational fusion between GFP and SBP-1 was used. Worms at active reproduction (day 3 of life) were mounted on 2% agarose pads and anaesthetized with 20 mM tetramisole in M9. Images were acquired using a Zeiss Axio Imager.M2 microscope equipped with an ApoTome.2 system and an AxioCam MRc camera using identical exposure settings for the same experiment. Images showing the maximum number of defined intestine nuclei were selected in each animal and saved as TIF files. The mean fluorescence intensity in arbitrary units (a.u.) of each nucleus was quantified using the oval brush tool in FIJI as described above.
Worm tissue micro-dissection
Synchronized worms grown on appropriate RNAi bacteria were collected at specified stages of life. At least 20 worms from each condition were washed three times with M9 buffer and deposited onto a glass slide in drops of M9 (one worm per drop) with a glass Pasteur pipette. Under the dissection light microscope, individual worms were decapitated using 22.5° knives (Surgistar) to extrude their anterior intestines and full germlines (including developing oocytes). For ORO or Nile Red staining, reproductive worms (day 5 of life) with extruded intestines were used for staining. For RT-qPCR or RNA-seq, fully detached intestines and germlines from worms during active reproduction (day 3 of life) or at initiation of reproduction (day 2.5 of life) were harvested. To isolate the full germline containing maturing oocytes, an incision was made before the spermatheca to minimize sperm contamination. Dissected germlines were immediately collected in 500 μl Trizol LS reagent (Invitrogen) using a glass Pasteur pipette whose tip was pulled under the flame to thin out the opening. To isolate the anterior intestine from the same worms, an incision was made near the 10 pairs of anterior intestinal cells. The anterior intestinal samples were collected in Trizol in separate tubes. All samples were stored at −80 °C until RNA extraction.
Quantitative RT–PCR
For experiments using whole worms, at least 100 age-synchronized worms in biological triplicate for each condition were harvested, washed three times with M9 and resuspended in 500 μl Trizol. For experiments using dissected tissues, 20–40 germlines or intestines in biological triplicate for each condition were harvested. To extract total RNA, worm or tissue pellets in Trizol underwent six freeze-thaw cycles in a dry ice-ethanol bath. RNA was extracted according to the Trizol procedure, and resuspended in RNase- and DNase-free water. RNA from whole worms was quantified using the Qubit 2.0 fluorometer (Invitrogen), and at least 500 ng total RNA per condition was used for cDNA synthesis. RNA from dissected tissues was assessed for quality and quantified using the Agilent Bioanalyzer (RNA 6000 Pico), and at least 2 ng total RNA per condition was used for cDNA synthesis.
Total RNA was treated with DNase I Amp Grade (Invitrogen), then reverse-transcribed using Oligo-dT primers (Invitrogen) and Superscript III reverse transcriptase (Invitrogen), based on the manufacturer’s instructions. RT–qPCR was performed using diluted cDNA on an ABI7900HT or a Bio-Rad C1000 thermal cycler using iTaq master mix (Bio-Rad) containing a passive reference dye (ROX). Primers used in RT–qPCR were designed to span exon–exon junctions towards the 3′ end of the gene and were used at a final concentration of 250 nM each. Primers are listed in Supplementary Table 7. Melt curves were examined to ensure primer specificity. Results were analysed using the standard ΔΔCT method. For each biological replicate, the mean CT value of 3–4 technical replicates was analysed. act-1 served as the reference gene in all analyses, and changes in mRNA levels relative to act-1 were confirmed using cdc-42, ama-1 or pmp-3 as alternate reference genes in independent experiments (data not shown).
RNA-seq on micro-dissected tissues
For each of the five replicates, full germlines and anterior intestines were collected from 20 worms treated with ash-2 RNAi or empty vector control. At the initiation of reproduction (day 2.5 of life), worms from each pair of ash-2 RNAi and empty vector plates were dissected within the same hour. Tissue dissection and RNA extraction procedures were the same as described above and 20 full germlines or approximately 200 intestinal cells were used as the initial input for individual RNA-seq libraries. For cDNA synthesis, 150 pg (intestine) or 750 pg (germline) of extracted RNA was treated with RQ1 RNase-free DNase (Promega), which was later removed, and reverse transcribed using an oligo(dT)-primed, Clontech SMART-Seq v4 Ultra Low Input RNA Kit (634889) according to the manufacturer’s instructions. Poly-A selected RNA-seq libraries were generated using Illumina Nextera XT DNA Library Prep Kit (FC-131-1024). All 20 libraries were processed together to minimize batch effects. The libraries were uniquely barcoded and multiplexed onto one lane, and 150 base pair paired-end reads were sequenced on NextSeq 500 (Illumina).
RNA-seq analysis
RNA-seq reads were aligned to the WBcel235 genome and gene read counts were calculated using STAR v2.5.1b34. Low-coverage genes were filtered out: only genes with at least one read count per million mapped reads, in at least five samples, were included in the analysis, resulting in a dataset of 10,442 genes and 20 samples. More genes were detected in the germline samples (185 genes with a median count number of 0) than in the intestinal samples (682 genes with a median count number of 0), with 76% of the genes having a higher median read count in germline samples than in intestinal samples. Because of these tissue-specific differences, we did not perform normalization across samples (which assumes that most genes are not differentially expressed) and instead used FPKM values to compare germline and intestinal samples. The read counts were transformed to log2(FPKM+1) values, which were used for principal component analysis (PCA) on the samples from both tissues together. A gene was determined to be germline-enriched if the lowest expression value (log2(FPKM+1)) in the germline empty-vector samples was at least twofold higher than the highest expression value in the intestine empty-vector samples (5,494 germline-enriched genes). A gene was determined to be intestine-enriched if the lowest expression value (log2(FPKM+1)) in the intestine empty-vector samples was at least twofold higher than the highest expression value in the germline empty-vector samples (1,418 intestine-enriched genes).
We then compared empty-vector and ash-2 RNAi samples for each tissue separately. Low-coverage genes were filtered out: only genes with at least one read count per million mapped reads in at least five samples were included in the analysis. This resulted in a germline dataset of 9,073 genes and 10 samples, and an intestine dataset of 7,288 genes and 10 samples. Using DESeq2 v1.10.135, the read counts were normalized using the variance stabilizing transformation (VST) within each tissue and PCA was performed on the germline and intestine datasets. For each tissue, differentially expressed genes between empty-vector and ash-2 RNAi were examined, accounting for a plate effect (GE ~ condition plus plate). Gene-wise P values were corrected for multiple hypothesis testing using Benjamini–Hochberg and FDR = 0.05 was used as a significance threshold.
Selection of ASH-2 candidate targets
ASH-2 candidate targets were selected using the following datasets: 1) our RNA-seq dataset, 2) microarrays on ash-2 knockdown larval worms6 and ash-2 mutant adult germlines14, and 3) ASH-2 chromatin immunoprecipitation followed by DNA microarray (ChIP–chip) analysis on worm embryos15. Predicted candidate expression in the germline or intestine for datasets 2 and 3 was based on published gene expression datasets36,37,38 (A. Rechtsteiner and S. Strome, personal communication) and pre-publication RNA-seq datasets (R. Waterston, personal communication). The following criteria were used to select potential targets: i) downregulation upon ash-2 deficiency, as the H3K4me3 methyltransferase complex is generally associated with gene activation; ii) expression in the germline or intestine, as these are the two key tissues involved in this fat metabolic switch; and/or iii) previously identified role in lifespan or fat metabolism.
Generation of transgenic plasmids
The Pges-1::fat-7 plasmid (pSH1) contains 2 kb of ges-1 promoter region (Pges-1) upstream of a fat-7 full genomic DNA sequence followed by a fat-7 3′ UTR in the P4-P1R vector. The plasmid containing Pges-1 in the P4-P1R vector was a gift from F. Mann. The fragment containing fat-7 full genomic DNA (containing the coding region and 3′UTR) was generated by PCR amplification from wild-type (N2) genomic DNA using the following primers (pSH1_fat-7_F: 5′-CTATTACATATCTTATCTTTGAATTCAGATGACGGTAAAAACTCGCGC GAGCATTGCCAA-3′ and pSH1_fat-7_R: 5′-GCCGCCCTGCAGCTCTAGAGCTCGAATTCTTAGAATTAACCAAATTTATTCAGGAATAA-3′). The fragment containing Pges-1 and the P4-P1R vector was PCR-amplified using the following primers (pSH1_Pges-1_F: 5′-TTGGCAATGCTCGCGCGAGTTTTTACCGTCATCTGAATTCAAAGATAAGATATGTAATAG-3′and pSH1_Pges-1_R: 5′-TTTATTCCTGAATAAATTTGGTTAATTCTAAGAATTCGAGCTCTAGAGCTGCAGGGCGGC-3′). The fragments were assembled via Gibson assembly39.
The Pges-1::set-2a cDNA::SL2::GFP plasmid (pSH9) contains 2 kb of ges-1 promoter region (Pges-1) upstream of the full-length cDNA of set-2.a (the long isoform of set-2) followed by the SL2 trans-splicing sequence, GFP cDNA and unc-54 3′ UTR in the pIM26 vector. pIM26 containing SL2 upstream of GFP and the unc-54 3′ UTR was a gift from Y. Zhang. The 2-kb Pges-1 fragment upstream of ges-1 ATG was generated by PCR amplification from wild-type (N2) genomic DNA using the following primers (pSH9_ Pges-1_F: 5′-ATGCCTGCAGGTCGACTCTAGAGGATCCCCAAACTCCGAACTATGATGACGAAAAAATGT-3′ and pSH9_Pges-1_R: 5′-AGGATGATGGTTCATATCATGTGTGGACATCTGAATTCAAAGATAAGATATGTAATAGAT-3′). The set-2.a cDNA fragment was generated by PCR amplification from pAS2.1 (a gift from F. Palladino) using the following primers (pSH9_set-2.a_F: 5′-ATCTATTACATATCTTATCTTTGAATTCAGATGTCCACACATGATATGAACCATCATCCT- 3′ and pSH9_set-2.a_R: 5′-AACTAGGTGAAAGTAGGATGAGACAGCCCCTCAATTAAGATATCCACGACACGTCTTCGC-3′), and was corrected for a short missing sequence. The pIM26 fragment was amplified by PCR using the following primers (pSH9_pIM26_F: 5′-GCGAAGACGTGTCGTGGATATCTTAATTGAGGGGCTGTCTCATCCTACTTTCACCTAGTT-3′ and pSH9_pIM26_R: 5′-ACATTTTTTCGTCATCATAGTTCGGAGTTTGGGGATCCTCTAGAGTCGACCTGCAGGCAT-3′). Three fragments were assembled via Gibson assembly39.
All fragments generated via PCR amplification were sequence-verified in the final plasmid to ensure no mutations were introduced.
Generation of transgenic worm strains
The strain carrying extra-chromosomal arrays of [Pges-1::fat-7;myo-3::mCherry] was generated by injecting wild-type N2 (ABR) worms with pSH1 and a co-injection plasmid pCFJ104, with a muscle-specific myo-3 promoter driving the mCherry gene (Pmyo-3::mCherry, made by E. Jorgensen, Addgene plasmid number 19328). The strain carrying extra-chromosomal arrays of [Pges-1::set-2a cDNA;myo-3::mCherry] was generated by injecting set-2(ok952) (ABR10) worms with pSH9 and Pmyo-3::mCherry. The injection mixture of both plasmids had a final concentration of 50 ng μl-1. Control strains were generated by injecting wild-type N2 (ABR) worms with the co-injection marker (Pmyo-3::mCherry) at a final concentration of 50 ng μl-1.
Worms containing extra-chromosomal arrays were selected on the basis of mCherry fluorescence in the body wall muscles. In transgenic worms also carrying a GFP reporter, GFP fluorescence was used as a primary marker for strain maintenance. Overexpression of fat-7 in wild-type transgenic worms was confirmed by RT–qPCR with primers designed to detect fat-7 mRNA. Overexpression of set-2.a in set-2(ok952) mutant transgenic worms was confirmed by RT–qPCR with primers designed to detect transgenic (wild-type) but not mutant set-2.a mRNA. Functional validation of set-2 intestine rescue transgenic lines was conducted by RT–qPCR analysis of two intestinal targets of COMPASS H3K4me3 complex, dod-23 and asm-2, which are normally downregulated in set-2(ok952) mutants. Primer sequences are listed in Supplementary Table 7.
GC–MS quantification of fatty acids
Quantification of total triglycerides and total phospholipids levels, and profiling of long-chain fatty acids, were performed as described33 with the following modifications. For each condition, 500–1,000 age-synchronized animals were used. For GC–MS, an Agilent 7890A gas chromatograph equipped with an HP-5MS column (30 m × 0.25 mm I.D. × 0.25 μm, Agilent) using helium as a carrier gas was used, and 1 μl of each sample was injected at a 10:1 split ratio. The initial oven temperature was set to 150 °C, increased to 220 °C at 20 °C per min, increased to 230 °C at 2 °C per min, increased to 320 °C at 50 °C per min, and held for 2 min at this temperature. Fatty acid abundance was expressed as a ratio (μg per mg) of fatty acid concentration (μg per ml) to protein concentration (mg per ml) for each sample. In all GC–MS figure panels involving comparisons to the empty vector control (unless otherwise mentioned), raw fatty acid concentration (μg per mg) of each condition was normalized to the raw fatty acid concentration (μg per mg) of the empty vector control. The final ratio is expressed as relative fatty acid levels. Raw GC–MS data for all figure panels are provided in Supplementary Table 8.
Fertility assessment
The fertility of N2 worms treated with ash-2 RNAi or the empty vector control was assayed at 20 °C. After a 1-h egglay, eggs were maintained at 20 °C until worms reached the initiation of reproduction (2.5 days of life). Each of the 30 reproductive worms from each condition was transferred onto an individual 3.5-cm plate seeded with corresponding bacteria. Individual worms were transferred onto a new plate every day, and the number of fertilized eggs (eggs with visible shell) and unfertilized oocytes (eggs without visible shell) were assessed on the old plate. The old plate with eggs was incubated at 20 °C for an additional day, and the number of hatched progeny (live brood size) was counted. Fertility was assessed daily until no more hatched worms, fertilized eggs or unfertilized oocytes were seen on each plate. This experiment was conducted in a blinded manner.
Fatty acid supplementation
The fatty acid supplementation protocol was adapted from a previous study40 with several modifications. To facilitate fatty acid dissolution, the detergent Tergitol (type NP-40, Sigma-Aldrich) was added to a final concentration of 0.001% in liquid NGM agar medium for both non-supplemented and supplemented plates before autoclaving. NGM plates for RNAi experiments were prepared as described6, and HT115 bacteria were used for all supplementation experiments. A final concentration ranging from 0.1 mM to 4 mM of oleic acid was used during the initial experimental optimization, and a final concentration of 0.8 mM was used for all fatty acids in subsequent supplementation experiments. HT115 bacteria expressing the empty vector or the appropriate RNAi were seeded onto plates containing the respective fatty acids at room temperature for 24–48 h before the addition of worms to ensure incorporation of fatty acids into feeding bacteria. Successful incorporation of supplemented fatty acids into the HT115 bacteria and into the worms was confirmed by GC–MS.
Data availability
RNA-seq data are available at Sequence Read Archive (SRA) under accession number PRJNA343151. The code for RNA-seq analysis is available on the Brunet Laboratory Github: https://github.com/brunetlab.
References
Benayoun, B. A., Pollina, E. A. & Brunet, A. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 16, 593–610 (2015)
Sen, P., Shah, P. P., Nativio, R. & Berger, S. L. Epigenetic mechanisms of longevity and aging. Cell 166, 822–839 (2016)
Miller, M. et al. Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation 123, 2292–2333 (2011)
Hansen, M., Flatt, T. & Aguilaniu, H. Reproduction, fat metabolism, and life span: what is the connection? Cell Metab. 17, 10–19 (2013)
Shilatifard, A. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 81, 65–95 (2012)
Greer, E. L. et al. Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature 466, 383–387 (2010)
O’Rourke, E. J., Soukas, A. A., Carr, C. E. & Ruvkun, G. C. elegans major fats are stored in vesicles distinct from lysosome-related organelles. Cell Metab. 10, 430–435 (2009)
Brooks, K. K., Liang, B. & Watts, J. L. The influence of bacterial diet on fat storage in C. elegans. PLoS One 4, e7545 (2009)
Espelt, M. V., Estevez, A. Y., Yin, X. & Strange, K. Oscillatory Ca2+ signaling in the isolated Caenorhabditis elegans intestine: role of the inositol-1,4,5-trisphosphate receptor and phospholipases C beta and gamma. J. Gen. Physiol. 126, 379–392 (2005)
Marré, J ., Traver, E. C. & Jose, A. M. Extracellular RNA is transported from one generation to the next in Caenorhabditis elegans. Proc. Natl Acad. Sci. USA 113, 12496–12501 (2016)
Kumsta, C. & Hansen, M. C. elegans rrf-1 mutations maintain RNAi efficiency in the soma in addition to the germline. PLoS One 7, e35428 (2012)
Watts, J. L. & Browse, J. A palmitoyl-CoA-specific delta9 fatty acid desaturase from Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 272, 263–269 (2000)
Peyou-Ndi, M. M., Watts, J. L. & Browse, J. Identification and characterization of an animal delta(12) fatty acid desaturase gene by heterologous expression in Saccharomyces cerevisiae. Arch. Biochem. Biophys. 376, 399–408 (2000)
Robert, V. J. et al. The SET-2/SET1 histone H3K4 methyltransferase maintains pluripotency in the Caenorhabditis elegans germline. Cell Reports 9, 443–450 (2014)
Pferdehirt, R. R., Kruesi, W. S. & Meyer, B. J. An MLL/COMPASS subunit functions in the C. elegans dosage compensation complex to target X chromosomes for transcriptional regulation of gene expression. Genes Dev. 25, 499–515 (2011)
Laplante, M. & Sabatini, D. M. mTOR signaling in growth control and disease. Cell 149, 274–293 (2012)
Kapahi, P. et al. With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab. 11, 453–465 (2010)
Shi, X. et al. Regulation of lipid droplet size and phospholipid composition by stearoyl-CoA desaturase. J. Lipid Res. 54, 2504–2514 (2013)
Yang, F. et al. An ARC/Mediator subunit required for SREBP control of cholesterol and lipid homeostasis. Nature 442, 700–704 (2006)
Taubert, S., Van Gilst, M. R., Hansen, M. & Yamamoto, K. R. A Mediator subunit, MDT-15, integrates regulation of fatty acid metabolism by NHR-49-dependent and -independent pathways in C. elegans. Genes Dev. 20, 1137–1149 (2006)
Van Gilst, M. R., Hadjivassiliou, H., Jolly, A. & Yamamoto, K. R. Nuclear hormone receptor NHR-49 controls fat consumption and fatty acid composition in C. elegans. PLoS Biol. 3, e53 (2005)
Brock, T. J., Browse, J. & Watts, J. L. Genetic regulation of unsaturated fatty acid composition in C. elegans. PLoS Genet. 2, e108 (2006)
Brock, T. J., Browse, J. & Watts, J. L. Fatty acid desaturation and the regulation of adiposity in Caenorhabditis elegans. Genetics 176, 865–875 (2007)
Shmookler Reis, R. J. et al. Modulation of lipid biosynthesis contributes to stress resistance and longevity of C. elegans mutants. Aging (Albany, N.Y.) 3, 125–147 (2011)
Goudeau, J. et al. Fatty acid desaturation links germ cell loss to longevity through NHR-80/HNF4 in C. elegans. PLoS Biol. 9, e1000599 (2011)
O’Rourke, E. J., Kuballa, P., Xavier, R. & Ruvkun, G. ω-6 Polyunsaturated fatty acids extend life span through the activation of autophagy. Genes Dev. 27, 429–440 (2013)
Ratnappan, R. et al. Germline signals deploy NHR-49 to modulate fatty-acid β-oxidation and desaturation in somatic tissues of C. elegans. PLoS Genet. 10, e1004829 (2014)
Lee, D. et al. SREBP and MDT-15 protect C. elegans from glucose-induced accelerated aging by preventing accumulation of saturated fat. Genes Dev. 29, 2490–2503 (2015)
Magnuson, K., Jackowski, S., Rock, C. O. & Cronan, J. E., Jr. Regulation of fatty acid biosynthesis in Escherichia coli. Microbiol. Rev. 57, 522–542 (1993)
Gillingham, L. G., Harris-Janz, S. & Jones, P. J. Dietary monounsaturated fatty acids are protective against metabolic syndrome and cardiovascular disease risk factors. Lipids 46, 209–228 (2011)
Sijen, T. et al. On the role of RNA amplification in dsRNA-triggered gene silencing. Cell 107, 465–476 (2001)
Greer, E. L. et al. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr. Biol. 17, 1646–1656 (2007)
Pino, E. C., Webster, C. M., Carr, C. E. & Soukas, A. A. Biochemical and high throughput microscopic assessment of fat mass in Caenorhabditis elegans. J. Vis. Exp. 73, 50180 (2013)
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013)
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014)
Reinke, V., Gil, I. S., Ward, S. & Kazmer, K. Genome-wide germline-enriched and sex-biased expression profiles in Caenorhabditis elegans. Development 131, 311–323 (2004)
Wang, X. et al. Identification of genes expressed in the hermaphrodite germ line of C. elegans using SAGE. BMC Genomics 10, 213 (2009)
Gerstein, M. B. et al. Comparative analysis of the transcriptome across distant species. Nature 512, 445–448 (2014)
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009)
Deline, M. L., Vrablik, T. L. & Watts, J. L. Dietary supplementation of polyunsaturated fatty acids in Caenorhabditis elegans. J. Vis. Exp. 81, 50879 (2013)
Acknowledgements
We thank A. Jose, A. Rechtsteiner, S. Strome and R. Waterston for sharing expression data and strains pre-publication; A. Fire, M. Hansen, S. Kim, F. Palladino, D. Pattabiraman, Y. Zhang and the Caenorhabditis Genetics Center for plasmids and strains; M. Hansen, E. O’Rourke, L. Booth, C-K. Hu, D. Leeman, J. Lim and S. Mahmoudi for reading the manuscript; A. Fire, O. Gozani, S. Kim and Brunet laboratory members for discussions; A. Chien for GC–MS consulting; and J. Coller at the Stanford Functional Genomics Facility. Supported by NIH DP1AG044848 (A.B.), NIH R01AG054201 (A.B. and W.B.M.), NIH R01AG044346 (W.B.M.), a Stanford Mass Spectrometry grant (S.H. and A.B.), NSF Graduate Research Fellowship, Stanford Graduate Fellowship and NIH T32AG047126 (S.H.), and NIH T32AG047126 and NIH F32AG051337 (E.A.S.).
Author information
Authors and Affiliations
Contributions
S.H. conceived the study under the guidance of A.B. S.H. performed all the experiments except those specified below. E.A.S. planned and performed Nile Red and FAT-5/FAT-7 reporter imaging, time-course RT–qPCR and one oleic acid supplementation lifespan experiment. C.G.S.-G. planned and performed SBP-1 localization experiments and generated set-2 transgenic lines under the guidance of W.B.M. K.H. analysed the RNA-seq data. S.H. wrote the paper with the help of A.B. and E.A.S. C.G.S.-G., K.H. and W.B.M. provided feedback.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information Nature thanks H. Aguilaniu, M. Kaeberlein and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Figure 1 Deficiency in H3K4me3 modifiers leads to fat accumulation in the intestine without altering fertility.
a, ORO quantification. Mean ± s.d., n ≥ 17 worms per condition. b, ORO images. Scale bars, 100 μm. c, ORO quantification. Mean ± s.d., n ≥ 9 dissected intestines per condition. d, e, Nile Red staining and quantification. Mean ± s.d., n ≥ 7 dissected intestines (d) or n ≥ 11 worms (e) per condition. Scale bars, 100 μm. f, Autofluorescence and quantification. Mean ± s.d., n ≥ 17 worms per condition. Scale bars, 100 μm. g, Fertility quantification of live brood size (i), fertilized eggs (ii), and unfertilized oocytes (iii) laid per worm. Mean ± s.d., n ≥ 25 worms per condition. h, RT–qPCR. Mean ± s.e.m. of two independent experiments, each with two or three biological replicates. i, RT–qPCR. Mean ± s.e.m. from two independent experiments, each with two or three biological replicates. j, ORO quantification. Mean ± s.d., n ≥ 26 worms per condition. k, ORO quantification. Mean ± s.d., n ≥ 29 worms per condition. l, RT–qPCR. Mean ± s.e.m. of three biological replicates. m, Differential interference contrast (DIC) (Nomarski) and GFP fluorescence images. n, ORO quantification. Mean ± s.d., n ≥ 26 worms per condition. a–f, k (rescue line number 1), n, Representative of two experiments. P values: a, c–g, two-tailed Mann–Whitney; h, i, two-tailed Mann–Whitney with Benjamini–Hochberg correction; j, k, n, Kruskal–Wallis with Dunn’s correction; l, two-tailed t-test with Benjamini–Hochberg correction. *P < 0.05, **P < 0.01.
Extended Data Figure 2 The delta-9 desaturases FAT-5 and FAT-7 support MUFA accumulation in H3K4me3 methyltransferase-deficient worms.
a, GC–MS quantification of MUFAs. Mean ± s.e.m. of two independent experiments, each with three biological replicates. b, c, RT–qPCR. Mean ± s.e.m. of two independent experiments, each with three biological replicates. d, GC–MS quantification of MUFAs. Mean ± s.e.m. of two independent experiments, each with two or three biological replicates. e, RT–qPCR. Mean ± s.e.m. of two independent experiments, each with three biological replicates. P values: a–e, two-tailed Mann–Whitney with Benjamini–Hochberg correction. *P < 0.05, **P < 0.01.
Extended Data Figure 3 RNA-seq on micro-dissected germlines and intestines and functional validation of ASH-2 targets.
a, RNA-seq tissue sample collection pipeline. b, Principal component analysis (PCA) with both intestinal and germline samples (left), only intestinal samples (middle) or only germline samples (right). c, RT–qPCR. Mean ± s.e.m. of two independent experiments, each with two or three biological replicates. d – g, ORO quantification. Mean ± s.d., n ≥ 15 worms per condition. h, RT–qPCR. Mean ± s.e.m. of two independent experiments, each with three biological replicates. i, j, ORO quantification. Mean ± s.d., n ≥ 21 (i) and n ≥ 27 (j) worms per condition. k, RT–qPCR. Mean ± s.e.m. of two independent experiments, each with three biological replicates. l, GC–MS. Mean ± s.e.m. of two independent experiments, each with three biological replicates. m, ORO quantification. Mean ± s.d., n ≥ 19 worms per condition. n, RT–qPCR. Mean ± s.e.m. of two independent experiments, each with three biological replicates. o, Lifespan extension by ash-2 RNAi is reduced in rsks-1 mutants (13.12%) compared to wild-type worms (29.20%) (P < 0.0001, two-way ANOVA). p, let-363 RNAi and daf-15 RNAi extend lifespan in wild-type worms (P < 0.0001, log-rank), but not in set-2 mutants. d (except rrf-1 data), i, j, Representative of two experiments. P values: c, k, l, n, two-tailed Mann–Whitney with Benjamini–Hochberg correction; h, two-tailed Mann-Whitney; d–g, i, j, m, Kruskal–Wallis with Dunn’s correction. *P < 0.05, **P < 0.01.
Extended Data Figure 4 Role of SBP-1, MDT-15, NHR-49 and NHR-80 in the fat accumulation and longevity of H3K4me3 methyltransferase-deficient worms.
a–d, Images and quantification of SBP-1 nuclear accumulation. Mean ± s.d. of two independent experiments, each with 4–6 nuclei per worm of ≥ 8 worms per condition. e, RT–qPCR. Mean ± s.e.m. of two independent experiments, each with three biological replicates. f, GC–MS quantification of MUFAs. Mean ± s.e.m. of two independent experiments, each with two or three biological replicates. g, ORO quantification. Mean ± s.d., n ≥ 21 worms per condition. h, ash-2 RNAi extends lifespan in wild-type worms (P < 0.0001, log-rank), but not in mdt-15 RNAi worms. i, ORO quantification, Mean ± s.d., n ≥ 24 worms per condition. j, ash-2 RNAi extends lifespan in both wild-type worms and nhr-49 mutants (P < 0.0001, log-rank). k, ORO quantification. Mean ± s.d., n ≥ 27 worms per condition. l, ash-2 RNAi extends lifespan in both wild-type worms and nhr-80 mutants (P < 0.0001, log-rank). i–k, Representative of two experiments. P values: a, two-tailed Mann–Whitney; b–d, g, i, k, Kruskal–Wallis with Dunn’s correction; e, f, two-tailed Mann–Whitney with Benjamini–Hochberg correction. *P < 0.05.
Extended Data Figure 5 Delta-9 desaturases FAT-6 and FAT-7 and MUFA oleic acid mediate the longevity of H3K4me3 methyltransferase-deficient worms.
a, ORO quantification. Mean ± s.d., n ≥ 29 worms per condition. b, ash-2 RNAi leads to lifespan extension in control (P < 0.0001, log-rank), but not in fat-6 and fat-7 double mutants. c, ash-2 RNAi extends lifespan in both wild-type worms and fat-7 single mutants (P < 0.0001, log-rank). d, ash-2 RNAi extends lifespan in fat-6 single mutants (P = 0.0162, log-rank), but lifespan extension by ash-2 RNAi is reduced in fat-6 mutants (9.26%) compared to wild-type worms (20.46%) (P = 0.0072, two-way ANOVA). e, ash-2 RNAi extends lifespan in fat-5 and fat-7 double mutants (P < 0.0001, log-rank). f, ash-2 RNAi extends lifespan in fat-5 and fat-6 double mutants (P = 0.0002, log-rank), but lifespan extension by ash-2 RNAi is reduced in fat-5 and fat-6 double mutants (14.03%) compared to wild-type worms (20.46%) (P = 0.0358, two-way ANOVA). g, h, GC–MS quantification of oleic acid. Mean ± s.e.m. of three biological replicates. i, ORO quantification. Mean ± s.d., n ≥ 13 worms per condition. Boxed conditions are identical to Fig. 4g. j, Oleic acid supplementation extends lifespan (P < 0.0001, log-rank), which is not further extended by ash-2 RNAi. Oleic acid supplementation extends lifespan in ash-2 and fat-7 double RNAi worms (P < 0.0001, log-rank). Boxed conditions are identical to Fig. 4h. k, ORO quantification. Mean ± s.d., n ≥ 21 worms per condition. l, GC–MS quantification of MUFAs. Mean ± s.e.m. of two independent experiments, each with two or three biological replicates. m, RT–qPCR. Mean ± s.e.m. of two independent experiments, each with three biological replicates. n, ORO quantification. Mean ± s.d., n ≥ 25 worms per condition. o, ash-2 RNAi extends lifespan in control (P < 0.0001, log-rank) but not fat-2 RNAi worms. c, i, j, n, o, Representative of two experiments. P values: a, g–i, k, n, Kruskal–Wallis with Dunn’s correction; l, m, two-tailed Mann–Whitney with Benjamini–Hochberg correction. *P < 0.05, **P < 0.01.
Extended Data Figure 6 Dietary supplementation with MUFAs, but not PUFAs, extends lifespan in wild-type worms.
a, GC–MS. Mean ± s.e.m. of two independent experiments, each with two or three biological replicates. b, Cis-vaccenic acid supplementation extends lifespan in wild-type worms (P < 0.0001, log-rank). Inset: ORO quantification. Mean ± s.d., n ≥ 20 worms per condition. c, GC–MS. Mean ± s.e.m. of two independent experiments, each with three biological replicates. d, Linoleic acid supplementation does not extend lifespan in wild-type worms. Inset: ORO quantification. Mean ± s.d., n ≥ 23 worms per condition. e, GC–MS. Mean ± s.e.m. of two independent experiments, each with three biological replicates. f, Alpha-linolenic acid supplementation does not increase the lifespan of wild-type worms. Inset: ORO quantification. Mean ± s.d., n ≥ 23 worms per condition. g, RT–qPCR. Mean ± s.e.m. of three biological replicates. h, ORO quantification. Mean ± s.d., n ≥ 54 worms per condition. i, Overexpression of FAT-7 extends lifespan (P < 0.0001, log-rank), but this lifespan is not extended further by dietary oleic acid. Boxed regions are identical to Fig. 5g. j, Proposed model by which ash-2 deficiency in the germline could lead to the fat metabolic switch in the intestine. b, d, f, Representative of two experiments. P values: a, c, e, two-tailed Mann–Whitney with Benjamini–Hochberg correction; b inset, d inset, f inset, two-tailed Mann–Whitney; g, Kruskal–Wallis test with Dunn’s correction (non-significant, probably owing to small sample size). One-way ANOVA with Bonferroni’s correction. h, Kruskal–Wallis with Dunn’s correction; *P < 0.05, **P < 0.01, ***P < 0.001.
Supplementary information
Supplementary Tables
This file contains Supplementary Tables 1-8 and a Supplementary Table guide. (ZIP 3003 kb)
Rights and permissions
About this article

Cite this article
Han, S., Schroeder, E., Silva-García, C. et al. Mono-unsaturated fatty acids link H3K4me3 modifiers to C. elegans lifespan. Nature 544, 185–190 (2017). https://doi.org/10.1038/nature21686
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature21686
This article is cited by
-
Evolutionarily related host and microbial pathways regulate fat desaturation in C. elegans
Nature Communications (2024)
-
The chromatin factors SET-26 and HCF-1 oppose the histone deacetylase HDA-1 in longevity and gene regulation in C. elegans
Nature Communications (2024)
-
Reducing the metabolic burden of rRNA synthesis promotes healthy longevity in Caenorhabditis elegans
Nature Communications (2024)
-
Fatty acid- and retinol-binding protein 6 does not control worm fatty acid content in Caenorhabditis elegans but might play a role in Haemonchus contortus parasitism
Parasites & Vectors (2023)
-
Early life changes in histone landscape protect against age-associated amyloid toxicities through HSF-1-dependent regulation of lipid metabolism
Nature Aging (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
