
Abstract
Cells require nucleotides to support DNA replication and repair damaged DNA. In addition to de novo synthesis, cells recycle nucleotides from the DNA of dying cells or from cellular material ingested through the diet. Salvaged nucleosides come with the complication that they can contain epigenetic modifications. Because epigenetic inheritance of DNA methylation mainly relies on copying of the modification pattern from parental strands1,2,3, random incorporation of pre-modified bases during replication could have profound implications for epigenome fidelity and yield adverse cellular phenotypes. Although the salvage mechanism of 5-methyl-2′deoxycytidine (5mdC) has been investigated before4,5,6, it remains unknown how cells deal with the recently identified oxidized forms of 5mdC: 5-hydroxymethyl-2′deoxycytidine (5hmdC), 5-formy-2′deoxycytidine (5fdC) and 5-carboxyl-2′deoxycytidine (5cadC)7,8,9,10. Here we show that enzymes of the nucleotide salvage pathway display substrate selectivity, effectively protecting newly synthesized DNA from the incorporation of epigenetically modified forms of cytosine. Thus, cell lines and animals can tolerate high doses of these modified cytidines without any deleterious effects on physiology. Notably, by screening cancer cell lines for growth defects after exposure to 5hmdC, we unexpectedly identify a subset of cell lines in which 5hmdC or 5fdC administration leads to cell lethality. Using genomic approaches, we show that the susceptible cell lines overexpress cytidine deaminase (CDA). CDA converts 5hmdC and 5fdC into variants of uridine that are incorporated into DNA, resulting in accumulation of DNA damage, and ultimately, cell death. Our observations extend current knowledge of the nucleotide salvage pathway by revealing the metabolism of oxidized epigenetic bases, and suggest a new therapeutic option for cancers, such as pancreatic cancer, that have CDA overexpression and are resistant to treatment with other cytidine analogues11.
Similar content being viewed by others


Main
Modified cytidines can enter deoxynucleotide pools, because salvage and nutrient uptake pathways can recover nucleosides, rather than simpler degradation products such as uric acid in the salvage of purines12. Previous biochemical work has suggested that 5mdC is not incorporated in the DNA, but is salvaged as thymidine4,5,6. Salvage of oxidized 5-methylcytosine variants has not been previously characterized. We rationalized that, if nucleosides are recovered in unphosphorylated forms (through import) or monophosphate forms (through intracellular hydrolysis), the barrier restricting their incorporation into the DNA may lie in the nucleotide salvage enzymes or DNA polymerases. Providing cells with a final substrate for DNA polymerases, in the form of deoxynucleoside triphosphate, would allow decoupling of DNA synthesis from salvage enzyme activity. Therefore, we transfected two human cancer cell lines—MDA-MB-231 and H1299—with 5-hydroxymethyl-2′deoxycytidine triphosphate (5hmdCTP), isolated DNA and analysed the base composition by a high-performance liquid chromatography–ultraviolet (HPLC–UV) method, using a set of nucleoside standards for calibration (Fig. 1a). After 5hmdCTP transfection, two additional nucleosides were observed in the hydrolysed DNA that correspond to 5hmdC and 5hmdU (Fig. 1b, c and Extended Data Fig. 1b). This indicates that DNA polymerases can incorporate 5hmdC into DNA, and also demonstrates strong deaminase activity acting on either the nucleotide or the incorporated base, resulting in the presence of 5-hydroxymethyluracil (5hmUra) in the DNA. The capacity for DNA polymerases to use 5hmdCTP was also evident in an in vitro replication assay13 (Fig. 1d), demonstrating that human DNA polymerases are not selective against the incorporation of 5hmdC into DNA. Therefore, if salvage pathways can convert pre-existing sources of 5hmdC into their nucleotide triphosphate forms, this could result in their incorporation into cellular DNA and potentially lead to deleterious effects on the epigenome.

a, HPLC–UV (260 nm) chromatogram of nucleosides mixed in equimolar amounts. b, HPLC–UV examination of nucleosides derived from DNA extracted from MDA-MB-231 cells nucleoporated with 5hmdCTP. c, The abundance of 5hmdC relative to dG in nucleoporated MDA-MB-231 (n = 3). d, 5hmdCTP incorporation assessed by in vitro replication assay (IVRA) performed in HeLa cells, measured by HPLC–UV (n = 5). Error bars in c and d denote s.d.; ND, not detected e, Thin-layer chromatography (TLC) separation of reaction products of DCK and CMPK1 kinases, which were supplied with different modified cytidine substrates. xDP, cytidine diphosphates; xMP, cytidine monophosphates.
The final triphosphate form of cytidine in a cell is produced by sequential phosphorylation by three classes of cytidine kinases. First, deoxycytidine kinase (DCK) produces a monophosphate, which is then converted into a diphosphate by cytidine monophosphate kinases (CMPK1 and CMPK2), and subsequently converted into a triphosphate by the family of nucleoside diphosphate kinases14. Because nucleoside diphosphate kinases phosphorylate both purine and pyrimidine nucleosides15, and CMPK2 is found in the mitochondria16, we directed our efforts towards examining the substrate selectivity of DCK and CMPK1. Recombinant DCK was able to transfer the phosphate from ATP[γ-32P] to 5mdC, 5hmdC and 5fdC, but not to 5cadC (Fig. 1e and Extended Data Fig. 1d), while CMPK1 phosphorylated only unmodified cytidine monophosphate (Fig. 1e). In agreement with previous work on 5mdC (ref. 4), we can conclude that the inability of CMPK1 to create diphosphates of modified nucleotides provides the main barrier to the formation of respective dCTPs, limiting their availability for DNA polymerases, which can instead accept modified dCTPs.
Given this inherent selectivity of the nucleotide salvage pathway kinase CMPK1 for unmodified cytidine, we proposed that the introduction of abundant biologically modified cytidine variants would have little adverse effect on the physiology of a cell, unless they significantly impaired nucleotide metabolism. First, we determined that biological cytidine variants retain 70–100% of their original form after incubation in water and cell culture media for 10 days at 37 °C, while 80% of the synthetic variant 5-aza-2′deoxycytidine (5azadC) decomposed by day 2 in agreement with previous observations17 (Extended Data Fig. 2a–c). Next, a panel of 19 human cell lines was selected, sampling various tissue origins and p53 mutation statuses18 (Extended Data Fig. 2d). When cell growth media was supplemented with 10 μM 5hmdC or dC, most of the cell lines continued to proliferate at a normal rate. However, two cell lines (HOP-92 and MDA-MB-231) unexpectedly ceased to proliferate in the presence of 5hmdC (Fig. 2a). We found that 10 μM 5hmdC was lethal and 1 μM 5hmdC caused mild growth inhibition (Fig. 2b). Interestingly, 5fdC was more potent at 1 and 10 μM doses in the MDA-MB-231 cell line, but showed the same cell line selectivity as 5hmdC (Fig. 2b).

a, Ratio of live cells after 10 days of treatment with 5hmdC versus dC (n = 3). b, Growth curves of MDA-MB-231 and H1299 cell lines treated with three different concentrations of dC, 5hmdC and 5fdC over a period of 10 days (n = 3). c, Volcano plot illustrating analysis of gene expression of MDA-MB-231 and HOP-92 versus MCF-7 and MDA-MB-435 cell lines. Shown in orange are the genes that are significantly different between the groups (P < 0.01; >2-fold change). Dots in black show nucleoside transporters (SCL29A1, SLC29A2, SLC29A3, SLC29A4, SLC28A1, SLC28A2 and SLC28A3) and kinases (DCK and CMPK1). d, CDA expression levels from NCI60 and CCLE global gene expression data sets for different cancer cell lines. e, Western blot confirming CDA overexpression in SN12C and Capan-2 cell lines. Growth curves of Capan-2 and SN12C after treatment with 10 μM 5hmdC, 5fdC and dC (n = 3). Error bars denote s.d.
Genetic alterations or gene expression differences could modify the response of a cell line to biologically modified cytidine variants. By comparing the existing gene expression profiles of the cell lines (NCI60 and CCLE projects19,20) that we established as sensitive to modified cytidine variants to two randomly chosen resistant ones, we identified 1,380 differentially expressed genes (P < 0.01, >2-fold change). Notably, by focusing on differentially expressed genes known to be involved in nucleoside metabolism, we identified cytidine deaminase (CDA) overexpression in the 5hmdC-sensitive cell types, which had the ninth lowest P value of all the genes (Fig. 2c and Supplementary Table 1). None of the other known genes involved, either in nucleoside transport or cytidine recycling, were differentially expressed (Fig. 2c). To identify other cell lines with CDA overexpression, we ranked the 21 available cell lines according to their CDA messenger RNA levels (Fig. 2d). SN12C and Capan-2 cell lines had the highest expression levels of CDA, and this was confirmed at the protein level by western blot (Fig. 2e). Examination of 5hmdC and 5fdC tolerance revealed that a 10 μM dose substantially inhibited the growth of both cell lines, suggesting that the expression level of CDA is predictive of cytotoxicity for these epigenetic cytidine variants (Fig. 2e).
To determine whether CDA overexpression is necessary for selective cytotoxicity, we manipulated CDA levels in the identified cell lines. Cell lines (MDA-MB-231 and SN12C) with stable short hairpin RNA (shRNA) knockdown of CDA were able to survive 10 μM 5hmdC (Fig. 3a and Extended Data Fig. 2e). Furthermore, stable overexpression of CDA in normally 5hmdC-resistant cell lines (H1299 and MCF-7) induced substantial growth inhibition (Fig. 3b and Extended Data Fig. 2f). These experiments clearly established that CDA overexpression is predictive, necessary and sufficient for cytotoxic activity. In vitro measurements of recombinant CDA protein activity were performed with various cytidine variants. First, we determined that CDA deaminates 5mdC, 5hmdC and 5fdC, but not 5cadC, creating thymidine and respective variants of uridine (Extended Data Fig. 2g, h). Second, reaction kinetic data fitted well with a pseudo zero-order kinetics model (R2 > 0.9) revealing that, after deoxycytidine, the second best substrate (that is, with the second highest turnover number (kcat)) for CDA is 5fdC (Fig. 3c, d and Extended Data Fig. 2h). This was unexpected, because the catalytic activity does not follow a simple relationship with the dimensions of the 5′ modification as it does in the case of AID and APOBEC enzymes21. Molecular docking of cytidine variants to the CDA structure22 suggested that 5fdC docks to the catalytic site with nearly 180° rotation when compared to unmodified cytidine, retaining the amino group position close to the active site containing Zn2+ (Extended Data Fig. 3a). By contrast, 5hmdC docks in the active site by displacing the amino group, which provides a potential explanation for the lower catalytic turnover observed (Extended Data Fig. 3a).

a, Western blot showing knockdown of CDA by shRNA (using sh-CDA) in the MDA-MB-231 cell line. Right panel illustrates growth curves of derived stable cell lines after treatment with 10 μM 5hmdC (n = 3). (0) and (8) indicate two different shRNA constructs used for the experiments, and sh-luc denotes a non-targeting control shRNA that targets luciferase. WT, wild type. b, Western blot showing overexpression of CDA after lentiviral transduction of H1299 cells with a construct overexpressing CDA (CDA_dsRed). Right panel shows the growth curve after treatment with 10 μM 5hmdC (n = 3). c, CDA activity fitted to the Michaelis–Menten model. Right panel shows a zoomed-in curve, when 5hmdC was used as a substrate. d, kcat values of CDA supplied with cytidine variants. e, TLC separation of reaction products of thymidine kinase 1 (TK1) and thymidylate kinase (DTYMK), which were exposed to different modified uridine substrates. xDP indicates diphosphates; xMP, monophosphates. f, HPLC–UV chromatogram of nucleosides from DNA of MDA-MB-231 cells treated with 10 μM 5hmdC or dC for 3 days. Right panel shows the abundance of 5hmdU relative to T (n = 3, t-test, **P = 0.0057). g, γH2AX immunofluorescence in MDA-MB-231 and H1299 cell lines at day 3 after treatment with 10 μM 5hmdC or dC. Scale bars, 50 μm. Below are quantifications of cells showing positive signals (n = 3 , t-test, P = 0.0017). DAPI, 4′,6-diamidino-2-phenylindole. All error bars denote s.d.
The deamination of dC and 5mdC results in dU and T, which are the normal precursors for thymidine triphosphate synthesis. Conversely, deamination of 5hmdC and 5fdC produces 5hmdU and 5fdU, respectively, which are not canonical nucleosides. When phosphorylated and incorporated into DNA, 5hmdU and 5fdU are toxic to the cells (Extended Data Fig. 3b) as they are recognized as damaged bases and trigger extensive uracil glycosylase activity resulting in DNA breaks23. Therefore, we asked whether the uptake of 5hmdC in CDA-overexpressing cells leads to its conversion into 5hmdU and to its incorporation into DNA, potentially explaining cell-type-specific lethality. First, we determined activities of thymidine kinase and thymidylate kinase on 5hmdU and 5fdU. In contrast to the inability of CMPK1 to act on equivalent cytidine variants, thymidine kinase and thymidylate kinase phosphorylated both uridine variants (Fig. 3e). Notably, the corresponding triphosphates are not substrates nor potent inhibitors of dUTPase, a robust enzyme that removes dUTP from cells (Extended Data Fig. 3c). Finally, analysis of the genomic DNA composition of 5hmdC- and 5fdC-treated MDA-MB-231 cells identified 5hmUra and 5fUra, but no detectable change in 5-hydroxymethylcytosine (5hmCyt) or 5-formylcytosine (5fdCyt) levels in the DNA (Fig. 3f, Extended Data Figs 3d, e and 4a–c). Overall, in all the cell lines examined, a linear correlation was observed between CDA expression and the amount of 5hmUra in the DNA after treatment with 5hmdC (Extended Data Fig. 4d). Signs of extensive DNA damage were detected by phosphorylated H2AX (γH2AX) staining in 5hmdC-treated CDA-overexpressing cells (MDA-MB-231). By contrast, a cell line expressing low CDA levels (H1299) had no obvious γH2AX staining (Fig. 3g and Extended Data Fig. 5c, d). Also, increased numbers of cells in S and G2 phases of the cell cycle were observed in CDA-overexpressing cell lines, consistent with cell cycle arrest triggered by a DNA damage response (Extended Data Fig. 5a). We did not observe deviations in the dNTP pools of treated cells, indicating that the cell death is likely to be caused by extensive base excision by SMUG1 DNA glycosylase, which recognizes 5hmUra and 5fUra triggering repair and DNA double-stranded breaks (Extended Data Fig. 6). Together, these observations demonstrate that CDA deaminates 5hmdC and 5fdC, creating 5hmdU and 5fdU, respectively, which are incorporated into the DNA, leading to cell cycle arrest and eventually death.
CDA overexpression has been linked to resistance to cytidine analogues—such as gemcitabine, cytosine arabinoside or 5-azacytidine—that are currently used in cancer treatment, presenting a major obstacle to their use11,24,25,26. Our observations about biological nucleoside variants demonstrate an opposite effect: CDA overexpression sensitizes cells to otherwise non-toxic 5hmdC and 5fdC. Because cancers originating in the pancreas27, stomach, testis and vagina have upregulated CDA expression28 (Extended Data Fig. 7a, b), we postulated that the administration of 5hmdC and 5fdC could have a selective activity against these tumour cells. We first tested whether cytotoxic activity is cell autonomous for CDA-overexpressing H1299 cells in the presence of wild-type (CDA-low) H1299 cells. Both 5hmdC and 5fdC were able to eliminate CDA-overexpressing cells selectively, suggesting that secreted CDA or 5hmdU is insufficient for cytotoxicity (Fig. 4a). Tolerance to and the stability of 5hmdC and 5fdC in vivo was determined in immunocompromized BALB/cOlaHsd-Foxn1nu/nu mice after they received a range of doses (12.5 to 100 mg kg−1) of 5hmdC and 5fdC by intraperitoneal injection. Half an hour after injection, we were able to detect 5hmdC and 5fdC in the bloodstream, and to quantify 5hmdC using RapidFire mass spectrometry (Extended Data Fig. 7c, d). We observed no adverse effects on behaviour, injection site, weight or histology in the panel of tissues studied, even though some tissues (kidney and intestine) express CDA (Extended Data Fig. 7e–h, data not shown). To determine whether cytidine variants have an effect on tumour growth in proliferating cells, we subcutaneously injected H1299 wild-type and CDA-overexpressing cells into each side of an animal, which was later treated with 5hmdC or 5fdC (Fig. 4b). Xenografts with CDA overexpression grew slightly slower (reaching 64% of wild-type tumour volume), and the volume of the tumour was further reduced twofold in animals treated with 5hmdC or 5fdC (Fig. 4c). CDA-overexpressing tumours showed a twofold decrease in the number of proliferating cells and a threefold increase in the number of cells with DNA damage in 5fdC-injected animals, but smaller differences in animals that were injected with 5hmdC (Fig. 4d). Similar CDA-dependent effects on tumour volume and proliferation were observed when SN12C wild-type and SN12C CDA knockdown cells were used in the xenograft assay (Extended Data Fig. 8).

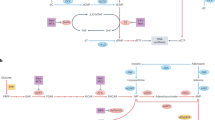
a, Wild-type and CDA-overexpressing H1299 cells were mixed at equal ratios and exposed to the indicated variants of cytidine. Representative histogram (left) and quantification of the results (right) are shown (n = 3, 10,000 events recorded). Lower concentrations of 5fdC were used to demonstrate higher cytotoxic potency. b, Schematic illustration of xenograft establishment and treatment with nucleoside variants. D, days. c, Volume of tumours, calculated by assuming that tumours were spheres with their diameters measured using Vernier calipers (n = 8 in 5fdC and n = 7 in 5hmdC experiments, two-way analysis of variance (ANOVA) with repeated measures Holm–Sidak correction, P < 0.0001). Dissected tumours are illustrated below. d, Evaluation of proliferation (immunofluorescence, H3PS10) and DNA damage (immunofluorescence, γH2AX) in dissected tumour samples. Scale bar, 50 μm (n = 4, one-way ANOVA, H3PS10: **P = 0.0057, γH2AX: *P = 0.0491 (5hmdC versus PBS), ***P = 0.0001 (5fdC versus PBS)). Error bars denote s.d. e, Model of metabolism of epigenetic nucleoside variants.
Here, we have characterized the metabolism of newly discovered biologically modified nucleosides, leading to a model in which the selectivity of CMPK1 prevents random incorporation of modified cytosines (Fig. 4e). Notably, we have discovered that 5hmdC and 5fdC, but not 5cadC, are deaminated by CDA at different rates, resulting in the formation of cytotoxic 5hmdU and 5fdU. Our data on oxidized epigenetic bases are similar to the proposed mechanism of 5mdC salvage, in which CMPK1 is rate-limiting in the production of the diphosphate, whereas 5mdC deamination produces a normal T (refs 4, 5, 6). We did not observe any adverse effects during the administration of 5hmdC and 5fdC in mice, presumably because the cytotoxic threshold is only reached in highly proliferating and CDA-overexpressing cells, in which there is substantial incorporation of nucleoside variants in the DNA, reflected by the CDA-dependent regression of xenografts. Together with recent publications demonstrating the importance of and therapeutic opportunities targeting MTH1, which surveys damaged nucleosides29,30, our data extend the current understanding of the metabolism of biological cytidine variants and provide a novel avenue for cancer therapy.
Methods
Purification of DCK, CMPK1, CDA, TMPK and DUT
Human DCK with a carboxy-terminal 6×His tag was cloned in pET28a(+) and expressed in Escherichia coli BL21 RIPL (Life Technologies) for 4 h at 37 °C following induction with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) in LB. The bacterial pellet was resuspended in 50 mM sodium phosphate, pH 8, 300 mM NaCl and protease inhibitors (Complete EDTA-free, Roche). The protein was bound to a Hi-TRAP HP 5 ml column (GE Healthcare) and eluted with a linear gradient of 0–500 mM imidazole in the lysis buffer, supplemented with 10% glycerol. The fractions were assessed by electrophoresis and ones containing the protein were pooled, concentrated with Amicon 3-kDa centrifugal filter units (Millipore) and separated on a HiPrep 16/60 Sephacryl S-200 gel filtration column (GE Healthcare). The protein was again concentrated using Amicon columns, supplemented with final 10 mM dithiothreitol (DTT) and 40% of glycerol, snap-frozen and stored in aliquots at −80 °C. Human CMPK1 was tagged at the C terminus with 6×His and purified using a similar workflow to DCK with the following exceptions: the lysis buffer was 50 mM Tris, pH 7.5, 10 mM NaCl and protease inhibitors (Complete Mini, Roche); after the gel-filtration step the protein was bound to an anion exchange column HiTrap Q HP 5 ml (GE Healthcare) and eluted with a 20-column-volumes linear gradient of 0–1 M NaCl. The salt was removed by dialysis in 50 mM Tris, pH 8, the protein concentrated and 10 mM DTT added to the final preparation, before storage in 40% glycerol at −80 °C. C-terminal 6×His-tagged CDA was purified in a similar workflow to CMPK1 with the following modifications: the protein was expressed for 19 h at 37 °C; the lysis buffer was 50 mM Tris, pH 7.5, 1 mM DTT, 1 mM EDTA and protease inhibitors (Complete Mini, Roche); following HiTRAP purification, 6×His tag was cleaved by thrombin (Sigma); cleaved tag and uncleaved protein was removed by separation using a HiTRAP HP 5 ml column and collection of the flow-through. Subsequently, the protein was purified using gel filtration as indicated above and stored in aliquots at −80 °C. Protein purity was assessed by electrophoresis and CDA was additionally identified by mass spectrometry. Human TMPK1 with a C-terminal 6×His tag was cloned in pET28a(+) and expressed in E. coli BL21 RIPL (Life Technologies) for 4 h at 37 °C following induction with 1 mM IPTG in LB. The bacterial pellet was resuspended in 50 mM sodium phosphate, pH 7, 300 mM NaCl and protease inhibitors (Complete EDTA-free, Roche) and lysed with a French press (EmulsiFlex C5, Avestin) at ∼100 MPa equipped with a recirculating cooler (F250, Julabo) set at 4 °C. The protein was bound to a Hi-TRAP HP 5 ml column (GE Healthcare) and eluted with a linear gradient of 0–500 mM imidazole in the lysis buffer, supplemented with 10% glycerol. The fractions were assessed by electrophoresis and ones containing the protein were pooled, concentrated with Amicon 3 kDa centrifugal filter units (Millipore), supplemented with 40% of glycerol, snap frozen in aliquots and stored at −80 °C. Human DUT with a C-terminal 6×His tag was cloned in pET28a(+) and expressed in E. coli BL21 RIPL (Life Technologies) for 4 h at 37 °C following induction with 0.2 mM IPTG in LB. The bacterial pellet was resuspended in 20 mM sodium phosphate pH 7.3, 150 mM NaCl, 1% Triton X-100 and protease inhibitors (Complete EDTA-free, Roche) and lysed with a French press (EmulsiFlex C5, Avestin) at ∼100 MPa equipped with a recirculating cooler (F250, Julabo) set at 4 °C. The protein was bound to a Hi-TRAP HP 5 ml column (GE Healthcare) and eluted with a linear gradient of 0–500 mM imidazole in the lysis buffer, supplemented with 10% glycerol. The fractions were assessed by electrophoresis and ones containing the protein were pooled, concentrated with Amicon 3 kDa centrifugal filter units (Millipore), supplemented with 40% of glycerol, snap frozen and stored in aliquots at −80 °C. Thymidine kinase was purchased and the purity assessed by SDS–PAGE (8180-TK-050, R&D Systems).
Nucleoside stability
Nucleosides were obtained from the following sources: 5hmdC (PY-7588, Berry & Associates), 5fdC (PY-7589, Berry & Associates), 5cadC (PY-7593, Berry & Associates), 5azadC (A3656, Sigma Aldrich), ATP solution (Thermo Fisher), [γ-32P]ATP (Perkin Elmer), dC (Sigma Aldrich, D3897), dCMP (Sigma Aldrich, D7625), 5hmdCTP (Bioline, BIO-39046). 100 μM solutions of 5hmdC, 5fdC and 5azadC were prepared in HPLC-grade water (Thermo Fisher) or in DMEM (Lonza). The solutions were incubated at 37 °C for 10 days. A sample was taken every 24 h and subjected to HPLC–UV analysis.
Enzyme assays
The substrate selectivity of DCK and CMPK1 kinases were measured by 32P transfer and detection using 1D or 2D TLC. 1 μg of DCK was incubated in 100 mM Tris, pH 7.5, 100 mM KCl, 10 mM MgCl2, 1 mM [γ-32P]ATP and 200 μM of the respective nucleoside in a 50 μl reaction volume at 37 °C for 2 h. 1 μl of products was separated via 2D TLC on glass-backed AVICEL cellulose plates (Analtech) as described31. CMPK1 was assayed through a coupled assay with DCK following the conditions described previously32 with 1 μg DCK, 1 μg CMPK1 and 1 mM substrate. Thymidine kinase (8180-TK-050, R&D Systems) and TMPK1 were assayed through a coupled assay with 1 μg thymidine kinase, 1 μg TMPK1 and 1 mM substrate in 50 mM Tris, pH 7.4, 50 mM KCl, 5 mM MgCl2, 1 mM ATP and 2.5 μCi [γ-32P]ATP at 37 °C. 1D TLC was performed using glass-backed TLC sheets (PEI cellulose F, Millipore) as described previously32. The plates were exposed to a storage phosphor screen (GE Heathcare), which was scanned using Phosphoimager (Biorad) and images analysed with ImageLab software (Biorad). CDA kinetic activity data was collected as described33 by monitoring the absorbance at 260 nm with a spectrophotometer (SpectraMax M2, Molecular devices) using 45 ng of enzyme (500 ng for 5hmdC) and the data fitted according to pseudo zero order Michaelis–Menten enzyme kinetic model by Prism software (GraphPad). 1 μg DUT was assayed in 50 mM Tris, pH 7.5, 4 mM MgCl2, 1 mM DTT, 0.1 mg ml−1 BSA with 5 μM of substrate in 40 μl reaction volume for 10 min at 37 °C. The generated pyrophosphate was detected with a bioluminescent coupled assay (PPiLight inorganic pyrophosphate assay LT07-500, Lonza). The plate was then read in a GloMax instrument (Promega).
Molecular docking
A tetramer was generated with CDA structure 1MQ0 (ref. 22) and subject to DockPrep in Chimera 1.8 (http://www.cgl.ucsf.edu/chimera). Substrates were dC (ZINC18286013)34, 5hmdC (ZINC77300654)34 and 5fdC (CSID:10291642) (http://www.chemspider.com) downloaded as .mol files and subjected to .mol2 files conversion in Chimera. Docking was subsequently performed with SwissDock (http://www.swissdock.ch/docking)35. The model with the lowest ΔG of ligand was then visualized and analysed with Chimera.
In vitro replication assay
The assay was carried out following protocols for nuclear extract and cytoplasmic fraction preparation and for the replication assay13,36. The reaction contained 0.3 mM of each canonical nucleotide, except dCTP which was substituted by 5hmdCTP. The reaction was stopped with the addition of 0.1 M EDTA final. DNA was extracted with phenol and chloroform, treated with RNase A/T1 (Thermo Fisher) and free nucleotides removed with a Mini Quick Spin DNA column (Roche) before HPLC assay.
DNA glycosylase assay
The single stranded DNA oligonucleotide substrates (5′-FAM CATAAAGTGXAAAGCCTGGA, in which X denotes uracil, 5hmUra or 5fUra) were purchased from AtdBio and their complementary strand from IDT (all HPLC purified). Recombinant human SMUG1 (NEB) was incubated with annealed oligonucleotides as described before37. The reaction products were resolved on a 15% denaturing polyacrylamide TBE–urea gel (Invitrogen) and quantified using ChemiDoc (BioRad) with blot detection protocol for Alexa 488.
Quantification of nucleosides by HPLC
Genomic DNA was extracted with Gene Jet Genomic DNA extraction Kit (Thermo Fisher) or TRI Reagent (Sigma Aldrich), incubated with RNase A/T1 (Thermo Fisher) in buffer 2 (NEB), phenol/chloroform extracted and precipitated with ethanol. 1–10 μg of DNA was hydrolysed as described before7. Nucleosides were resolved with an Agilent UHPLC 1290 instrument fitted with Eclipse Plus C18 RRHD 1.8 μm, 2.1 × 150 mm column and detected with Agilent 1290 DAD fitted with a Max-Light 60 mm cell. Buffer A was 100 mM ammonium acetate, pH 6.5; buffer B was 40% acetonitrile, and the flow rate 0.4 ml min−1. The gradient was between 1.8–100% of 40% acetonitrile with the following steps: 1–2 min, 100% A; 2–16 min 98.2% A, 1.8% B; 16–18 min 70% A, 30% B; 18–20 min 50% A, 50% B; 20–21.5 min 25% A, 75% B; 21.5–24.5 min 100% B.
Quantification of nucleotides by HPLC
MDA-MB-231 and H1299 cells were treated with 10 μM dC, 10 μM 5hmdC and 1 μM 5fdC. Metabolites were extracted at day 3 as described before38. In brief, cells were washed in PBS and scraped on ice. The pellet was washed again in cold PBS and extraction was done with 50 μl of ice-cold 50% ACN per mg of pellet. The samples were vortexed and incubated on ice for 10 min. Insoluble material was pelleted at 20,000g for 10 min and supernatants were dried using a SpeedVac (Thermo Scientific). Metabolites were dissolved in 30 μl of buffer A and 20 μl was used for chromatography. HPLC was performed as described38 with some minor modifications as listed below. Nucleotides were resolved with an Agilent UHPLC 1290 instrument fitted with Eclipse Plus C18 RRHD 1.8 μm, 2.1 × 150 mm column and detected with Agilent 1290 DAD fitted with a Max-Light 60 mm cell at 254, 260 and 280 nm. Buffer A consisted of 100 mM KH2PO4 (60221, Sigma) with 8 mM tetrabutylammoniumbisulfate (98479, Sigma) set at pH 5.5. Buffer B consisted of buffer A with 25% methanol. After 8 min at 0% buffer B, the gradient started with a linear increase of buffer B to 35% in 19 min, followed by a linear increase from 35% to 38% buffer B in 5 min and from 38% to 100% buffer B in 22 min. After an 8-min hold at 100% buffer B, the gradient was reversed from 100% to 0% buffer B in 2 min, followed by a hold at 0% buffer B for 2 min. The column temperature was set at 30 °C and the flow rate was 0.4 ml min−1. The compounds were identified by comparing their retention times and their UV spectra with those of known standards, which were purchased from Sigma-Aldrich. The integrated area was used to quantify the relative abundance of nucleotides by normalizing each peak area to the ADP area as an indication of loaded amount.
Cell culture and transfections
Cell lines were routinely tested for mycoplasma contamination using Lonza Mycoalert kit. Cell proliferation assays were done by seeding cells in p60 plates or in a T25 flask with appropriate concentrations of 5hmdC, 5fdC or dC in the growth media. The cells were passaged, counted and the media was replaced every 2 days. Before counting, 1 volume of Trypan blue solution (Lonza) was added to an aliquot of single cell suspension. The live cells were counted by TC-20 Cell Counter (Bio-Rad). NTPs were introduced by nucleofection. One million MDA-MB-231 cells were nucleofected with 50 mM 5hmdC in a 100-µl volume using an Amaxa nucleofector kit (Lonza), following the manufacturer’s instructions. After transfection, cells were seeded in a 6-well plate, 24 h later washed twice with PBS, and 48 h later DNA extracted for HPLC analysis.
Production of stable cell lines
Stable cell lines were generated via lentiviral infection using a standard protocol39 with second generation packaging plasmids (pCMV-VSVG, pCMV-dR8.9, a gift from B. Amati). CDA knockdown was achieved by infecting MDA-MB-231 and SN12C cell lines with pLKO.1 vectors containing five different shRNA constructs (SHCLND-NM_001785, Sigma-Aldrich) and a control pLKO.1 containing shRNA silencing luciferase (a gift from X. Lu). Infected cells were selected by incubation with 1.5 μg ml−1 puromycin (Sigma) for 60 h. Two cell lines with the lowest CDA mRNA levels (shRNA TRCN0000051290 and TRCN0000051288, designated (0) and (8), respectively) were further assessed by immunoblotting and used for experiments. Lentivirus for CDA overexpression was generated with pLenti-puro (39481, Addgene, I.-M. Shih laboratory) expressing dsRed-IRES-CDA. H1299 and MCF-7 were infected as above. Infected cells were selected with puromycin at 2 μg ml−1 for 60 h.
Immunoblotting, FACS and immunofluorescence
For western blot analysis, 106 cells were lysed with RIPA buffer (20 mM HEPES at pH 7.5, 300 mM NaCl, 5 mM EDTA, 10% glycerol, 1% Triton X-100, supplemented with protease inhibitors (Complete EDTA-free, Roche)) and sonicated. Cleared lysates were electrophoresed and immunoblotted with the following primary antibodies: anti-CDA (Sigma, SAB1300717 1:250), anti-actin (Abcam, ab185058 1:75000). Chemiluminescent detection, after incubation of the membranes with appropriate secondary antibodies, was done through a CCD camera using the ChemiDoc System (Bio-Rad) with Image Lab software (Bio-Rad, version 4.0). For FACS analysis, 5 × 105 cells were trypsinized, washed in PBS and fixed in 70% ethanol for 1 h on ice. The pelleted cells were resuspended in 250 μl of staining solution (50 μg ml−1 propidium iodide (P4864, Sigma), 0.1 mg ml−1 RNaseA and 0.05% Triton X-100) and incubated at 37 °C for 40 min. Controls were used for G1 (serum starvation overnight) and G2 (0.1 μg μl−1 nocodazole overnight). Fluorescence of 10,000 cells was recorded with a FACS Canto flow cytometer (BD Biosciences) and analysed using FlowJo software (Version 7.6.5, TreeStar). For immunofluorescence, cells were grown on coverslips and fixed with 4% paraformaldehyde for 20 min at room temperature. Cells were washed twice in PBS and permeabilized for 10 min in 0.2% Triton X-100. After two washes in PBS, cells were blocked for 1 h in 3% BSA (Sigma Aldrich), dissolved in PBS and incubated with γH2A.X antibody (Millipore, 05-636, 1:500) overnight at 4 °C in a humidified chamber. Cells were then washed three times in PBS and incubated with anti-mouse secondary antibody conjugated with Alexa546 (1:400, Life Technologies) and DAPI (Sigma Aldrich). Coverslips were then washed three times in PBS and mounted with mounting media (Vectashield). Tiled pictures were automatically taken with a Zeiss 710 microscope with a 20× lens. The amount of nuclear fluorescence was quantified using ImageJ.
Gene expression analysis and public data sets
Data sets used in the study: Gene Expression Omnibus (GEO) accessions GSE36139 (GPL15308)20 and GSE32474 (GPL570)40. Gene expression analysis comparing was done on the data from the NCI-60 panel19 as follows. Affy HG-U133 Plus 2.0 microarray data was downloaded from CellMiner database (http://discover.nci.nih.gov/cellminer/loadDownload.do) and cel files were extracted for triplicate experiments done on BR:MCF7, ME:MDA_MB_435, BR:MDA_MB_231 and LC:HOP_92 cell lines. Data was then imported into ArrayStar v11 (DNAStar) and signal normalization and intensity correction was done using RMA Quantile method. Experiment was designed by grouping BR:MCF7, ME:MDA_MB_435 cell lines into ‘resistant’ group and BR:MDA_MB_231 and LC:HOP_92 cell lines into ‘sensitive’ group. Differential expression between the groups was determined using the Student’s t-test with Benjamini–Hochberg multiple testing correction. Genes were called as differentially expressed when P < 0.01 and fold change >2. The full data set is included in Supplementary Table 1. To derive CDA expression values in tumours, GPL15308 (ref. 20) and GPL570 (ref. 40) were analysed directly on the NCBI portal with GEO2R. P values were adjusted with Benjamini–Hochberg correction.
Toxicology and dose determination in animal experiments
Animal work was done after approval by the UK Home Office and University of Oxford Local Ethical review. Three 5–7-week-old BALB/cOlaHsd-Foxn1nu/nu (Harlan) mice per dose were injected (intraperitoneally) with 25, 50 and 100 mg kg−1 of 5hmdC and 12.5, 25, 50 and 100 mg kg−1 of 5fdC. Animals were monitored for any deviations from normal behaviour. At 30 min post-injection, a few drops of blood were collected through tail vein bleeding using Microvette CB300 (Sarstedt) to assess the amounts of the compounds in the bloodstream.
RapidFire mass spectrometry analysis of serum samples
Serum was isolated by centrifugation of Microvettes according to the recommendations of the manufacturer (Sarstedt). The samples were brought up to 200 μl with water and three volumes of methanol, and 150 μl of chloroform was added. After intense vortexing, 450 μl of water was added, samples were vortexed again and centrifuged at 14,000g for 1 min. The aqueous phase containing the soluble molecules was collected and dried in a Speedvac (Thermo Scientific). The dried pellets were then resuspended in 10 μl water, then 3 μl diluted further into 50 μl of water to load on a RapidFire 360 high throughput sample delivery system coupled to a 6530 quadrupole time-of-flight (QTOF) mass spectrometer (Agilent). The samples were aspirated by vacuum at −40 bar for 400 ms into a 10-μl sample loop and loaded onto a graphitized carbon solid phase extraction cartridge running buffer 5 mM ammonium formate at a flow of 1.5 ml min−1. The matrix components not retained on the cartridge were diverted to waste for 4,500 ms, and the retained components eluted with 95% acetonitrile 5 mM ammonium formate for 4,500 ms at a flow of 1 ml min−1. The SPE was then re-equilibrated for 4,500 ms with 5 mM ammonium formate. Data were collected in positive electrospray ionisation (ESI) mode using a 2 Gb data configuration, gas temperature 300 °C, drying gas 8 l min−1, nebuliser gas ∼240 MPa, Vcap 3,500 V and fragmentor voltage 175 V. The amount of nucleoside was measured against a standard curve produced by dissolving known amounts of 5hmdC and 5fdC in serum and processed as indicated above. Data were analysed using an Agilent Mass Hunter Qualitative (vB.06) and Quantitative (vB.05) analysis software. Standard curve analysis was determined by using a quadratic curve fit algorithm for each nucleoside with an R2 > 0.98 in all instances.
Nucleoside analysis by mass spectrometry (HPLC–QTOF)
Samples were dried in a speed-vac and re-suspended in 10 µl of water. For the analysis by HPLC–QTOF mass spectrometry, a 1290 Infinity UHPLC was fitted with a BEH C18 XP Column, (130Å, 1.7 µm, 2.1 mm × 150 mm; Waters) and coupled to a 6560 Ion mobility QTOF LC/MS mass spectrometer (Agilent Technologies) equipped with a Jetstream ESI-AJS source. The data were acquired in QToF mode using positive electrospray ionisation (ESI+). Two reference ions, m/z 121.0508 and 922.0097 were used as internal standards. The Dual AJS ESI settings were as follows: gas temperature: 150 °C, the drying gas: 5 l min−1, nebulizer 240 MPa, sheath gas temperature 360 °C, sheath gas flow 12 l min−1, Vcap 4,000 V and nozzle voltage 300 V. The fragmentor of the mass spectrometer TOF was set to 275 V.
The gradient used to elute the nucleosides started by a 1-min isocratic gradient composed with 99.5% buffer A (10 mM ammonium acetate, pH 6) and 0.5% buffer B (composed of 40% CH3CN) with a flow rate of 0.350 ml min−1 and was followed by the subsequent steps: 1–2 min, 98.2% A; 2–16 min 80% A; 16–18 min 50% A; 18–20 min 25% A; 20.20–21.5 min 0% A; 21.5–22.5 min 100% B; 22.5–24.5 min 99.5% B. The gradient was followed by a 5 min post time to re-equilibrate the column.
The raw mass spectrometry data was analysed using the MassHunter Qual Software package (Agilent Technologies, version B7.0), and the masses/retention times used for the characterization of nucleosides and their adducts are summarized in Supplementary Table 2. For the identification of compounds, raw mass spectrometry data were processed using the molecular feature extraction function in the MassHunter software, followed by metabolite searching through mass/isotope matching using the PCDL software (version B.07.00 build 7024.0) and the METLIN database (https://metlin.scripps.edu/index.php). For each nucleoside, precursor ions corresponding to the M+H, M+Na, M+K, 2M and base only species were extracted, and the most intense ion species observed for each nucleoside was used for quantification. Identities of peaks eluting at 4.5 and 5.1 min (Figs 1b and 3f) are shown in Extended Data Figs 9 and 10.
Subcutaneus xenografts
Animal work was done after approval by the UK Home Office and University of Oxford Local Ethical review. Power calculations suggested 9–6 animals per group if we were to observe a significant 50% difference in tumour size with power of 90% and s.d. between 40 and 30%. One million cells in a 50% suspension of MatriGel (200 μl) were injected into 5–7-week-old BALB/cOlaHsd-Foxn1nu/nu (Harlan) mice, 8 animals per group in each flank following the scheme: SN12C/H1299 left, SN12C shCDA8/H1299 dsRedCDA right. When the tumours reached palpable size, 8 mice were assigned randomly to each treatment group: PBS, 100 mg kg−1 of 5hmdC and 100 mg kg−1 5fdC. The compounds were administered every 72 h (four doses in total). Tumour size was measured every 3 days by Vernier caliper and the animal cohort euthanized when the cumulative tumour diameter in the first animal reached 12 mm. The experimenter was unaware of the cell line genotypes during the measurements. Tumour volume was calculated assuming that the tumours were spheres with the following formula: 4/3π (D/2)3, in which D represents the diameter of the tumour.
Histology
Organs and tumours were collected and immediately fixed in 10% formalin for 48 h. They were then embedded in wax and 4-μm thick sections cut. All sections were stained with H&E. Tumours were additionally stained with a Masson’s Trichrome Stain Kit (Sigma Aldrich) according to the manufacturer instructions.
Immunofluorescence of tissues and tumours
The 4-μm thick sections were subjected to antigen retrieval with a pressure cooker in Tris buffer, pH 9 (10 mM Tris base, 0.05% Tween 20). They were then blocked in 3% BSA in PBS for 30 min and incubated overnight in a humidified chamber at 4 °C with the following antibodies: γH2A.X (Millipore, 05-636, 1:200) and PH3 (Millipore, 06-570, 1:200) or β-catenin (BD Transduction Laboratories, 610153, 1:250) and CDA (Sigma Aldrich, SAB1300717, 1:100). The slides were then washed vigorously three times in PBS and incubated for 1 h at room temperature with an appropriate secondary antibody, Alexa546 and Alexa488 conjugated (1:400, Life Technologies) and DAPI (Sigma Aldrich). Coverslips were then washed three times in PBS and mounted with mounting media (Vectashield). Images were acquired with a Zeiss 710 confocal microscope with a ×20 objective. For quantification of DNA damage and proliferation in tumours, tiled images with Z stacks were acquired to cover the entire central section of the tumour. Image J was used to quantify the immunofluorescence signal.
References
Riggs, A. D. X inactivation, differentiation, and DNA methylation. Cytogenet. Cell Genet. 14, 9–25 (1975)
Wigler, M., Levy, D. & Perucho, M. The somatic replication of DNA methylation. Cell 24, 33–40 (1981)
Gruenbaum, Y., Cedar, H. & Razin, A. Substrate and sequence specificity of a eukaryotic DNA methylase. Nature 295, 620–622 (1982)
Vilpo, J. A. & Vilpo, L. M. Nucleoside monophosphate kinase may be the key enzyme preventing salvage of DNA 5-methylcytosine. Mutat. Res. 286, 217–220 (1993)
Jekunen, A., Puukka, M. & Vilpo, J. Exclusion of exogenous 5-methyl-2′-deoxycytidine from DNA in human leukemic cells. A study with [2(-14)C]- and [methyl-14C]5-methyl-2′-deoxycytidine. Biochem. Pharmacol. 32, 1165–1168 (1983)
Jekunen, A. & Vilpo, J. A. 5-Methyl-2′-deoxycytidine. Metabolism and effects on cell lethality studied with human leukemic cells in vitro. Mol. Pharmacol. 25, 431–435 (1984)
Kriaucionis, S. & Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324, 929–930 (2009)
Tahiliani, M. et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324, 930–935 (2009)
Ito, S. et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333, 1300–1303 (2011)
Pfaffeneder, T. et al. The discovery of 5-formylcytosine in embryonic stem cell DNA. Angew. Chem. 50, 7008–7012 (2011)
Frese, K. K. et al. nab-Paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Disov. 2, 260–269 (2012)
Loffler, M., Fairbanks, L. D., Zameitat, E., Marinaki, A. M. & Simmonds, H. A. Pyrimidine pathways in health and disease. Trends Mol. Med. 11, 430–437 (2005)
Hershey, H. V., Stieber, J. F. & Mueller, G. C. DNA synthesis in isolated HeLa nuclei. A system for continuation of replication in vivo. Eur. J. Biochem. 34, 383–394 (1973)
Arner, E. S. & Eriksson, S. Mammalian deoxyribonucleoside kinases. Pharmacol. Ther. 67, 155–186 (1995)
Boissan, M. et al. The mammalian Nm23/NDPK family: from metastasis control to cilia movement. Mol. Cell. Biochem. 329, 51–62 (2009)
Xu, Y., Johansson, M. & Karlsson, A. Human UMP-CMP kinase 2, a novel nucleoside monophosphate kinase localized in mitochondria. J. Biol. Chem. 283, 1563–1571 (2008)
Lin, K. T., Momparler, R. L. & Rivard, G. E. High-performance liquid chromatographic analysis of chemical stability of 5-aza-2′-deoxycytidine. J. Pharm. Sci. 70, 1228–1232 (1981)
Petitjean, A. et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum. Mutat. 28, 622–629 (2007)
Ross, D. T. et al. Systematic variation in gene expression patterns in human cancer cell lines. Nature Genet. 24, 227–235 (2000)
Barretina, J. et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012)
Nabel, C. S. et al. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nature Chem. Biol. 8, 751–758 (2012)
Chung, S. J., Fromme, J. C. & Verdine, G. L. Structure of human cytidine deaminase bound to a potent inhibitor. J. Med. Chem. 48, 658–660 (2005)
Boorstein, R. J., Chiu, L. N. & Teebor, G. W. A mammalian cell line deficient in activity of the DNA repair enzyme 5-hydroxymethyluracil-DNA glycosylase is resistant to the toxic effects of the thymidine analog 5-hydroxymethyl-2'-deoxyuridine. Mol. Cell. Biol. 12, 5536–5540 (1992)
Weizman, N. et al. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 33, 3812–3819 (2013)
Eliopoulos, N., Cournoyer, D. & Momparler, R. L. Drug resistance to 5-aza-2'-deoxycytidine, 2',2'-difluorodeoxycytidine, and cytosine arabinoside conferred by retroviral-mediated transfer of human cytidine deaminase cDNA into murine cells. Cancer Chemother. Pharmacol. 42, 373–378 (1998)
Qin, T. et al. Mechanisms of resistance to decitabine in the myelodysplastic syndrome. PLoS ONE 6, e23372 (2011)
Pei, H. et al. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 16, 259–266 (2009)
Shin, G. et al. GENT: gene expression database of normal and tumor tissues. Cancer Inform. 10, 149–157 (2011)
Gad, H. et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 508, 215–221 (2014)
Huber, K. V. et al. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature 508, 222–227 (2014)
Ramsahoye, B. H. Nearest-neighbor analysis. Methods Mol. Biol. 200, 9–15 (2002)
Van Rompay, A. R., Johansson, M. & Karlsson, A. Phosphorylation of deoxycytidine analog monophosphates by UMP-CMP kinase: molecular characterization of the human enzyme. Mol. Pharmacol. 56, 562–569 (1999)
Vincenzetti, S., Cambi, A., Neuhard, J., Garattini, E. & Vita, A. Recombinant human cytidine deaminase: expression, purification, and characterization. Protein Expr. Purif. 8, 247–253 (1996)
Irwin, J. J., Sterling, T., Mysinger, M. M., Bolstad, E. S. & Coleman, R. G. ZINC: a free tool to discover chemistry for biology. J. Chem. Inf. Model. 52, 1757–1768 (2012)
Grosdidier, A., Zoete, V. & Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 39, W270–W277 (2011)
Planck, S. R. & Mueller, G. C. DNA chain growth in isolated HeLa nuclei. Biochemistry 16, 2778–2782 (1977)
Kemmerich, K., Dingler, F. A., Rada, C. & Neuberger, M. S. Germline ablation of SMUG1 DNA glycosylase causes loss of 5-hydroxymethyluracil- and UNG-backup uracil-excision activities and increases cancer predisposition of Ung−/−Msh2−/− mice. Nucleic Acids Res. 40, 6016–6025 (2012)
Dietmair, S., Timmins, N. E., Gray, P. P., Nielsen, L. K. & Kromer, J. O. Towards quantitative metabolomics of mammalian cells: development of a metabolite extraction protocol. Anal. Biochem. 404, 155–164 (2010)
MacKenzie, C. J. & Shioda, T. COS-1 cells as packaging host for production of lentiviruses. Curr. Protocols Cell Biol. Chapter 26, Unit–26.27 (2011)
Pfister, T. D. et al. Topoisomerase I levels in the NCI-60 cancer cell line panel determined by validated ELISA and microarray analysis and correlation with indenoisoquinoline sensitivity. Mol. Cancer Ther. 8, 1878–1884 (2009)
Acknowledgements
We acknowledge Ludwig Cancer Research and BBSRC for funding; R. Klose, T. Milne, G. Bond, M. Muers and members of the Kriaucionis laboratory for the discussions and critical reading of the manuscript. M. Shipman for his technical assistance in image acquisition and analysis; I. Ratnayaka and R. Lisle for assistance with histology; S. Laird and J. Tanner for help with animal experiments; X. Lu, S. Lunardi and G. Bond for providing cell lines; P. Filippakopoulos for advice on molecular docking. B.M.K. was supported by the John Fell Fund 133/075 (BMK) and the Wellcome Trust 097813/Z/11/Z.
Author information
Authors and Affiliations
Contributions
M.Z. performed all experiments, with the following exceptions: mass spectrometry was done by G.B., M.-L.T., K.M.P. and B.M.K.; gene expression analysis was done by S.K.; analysis of tissue pathology was done by R.G. S.K. conceived the study, S.K. and M.Z. designed experiments and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Extended data figures and tables
Extended Data Figure 1 DNA polymerase and nucleoside kinase activities on modified nucleosides.
a, Mass spectrometry confirmation of 5hmdC, 5fdC and 5cadC in the purchased nucleosides. b, HPLC–UV chromatogram of nucleosides from DNA extracted from H1299 cells transfected with 5hmdCTP. The abundance of 5hmdC relative to dG is illustrated in the right panel (n = 3). n.d., not detected. Error bars denote s.d. c, Coomassie-stained SDS–PAGE gel of recombinant purified DCK and CMPK1 enzymes used in the study. d, Two-dimensional TLC images of DCK reaction products. Dotted lines indicate reference points, which aid in tracking the migration localization of the nucleosides. The monophosphate in each reaction is circled in red (representative picture, n = 3). e, Schematic map of nucleoside migration on two-dimensional TLC plate (asterisk indicates a background spot coming from ATP and used as a reference point)
Extended Data Figure 2 Stability of the nucleosides and CDA activity.
a, b, Quantification of nucleosides by HPLC–UV during 10 days of incubation in water (a) and DMEM (b) at 37 °C (n = 3). c, Representative HPLC–UV chromatograms at days 0, 2 and 10 with retention times indicated above each peak. d, Cell lines used in the study and their characteristics. e, Western blot showing knockdown of CDA by shRNA in the SN12C cell line. Right panel illustrates the growth of the cell line during treatment with 10 μM 5hmdC (n = 3). f, Western blot showing expression of CDA in wild-type and lentivirally transduced MCF7 cell line. Growth curve after treatment with 10 μM 5hmdC is shown on the right (n = 3). g, Coomassie-stained SDS–PAGE gel of recombinant purified CDA enzyme used in this study. h, HPLC–UV chromatograms showing the retention times and identity of substrates and CDA-catalysed products. i, List of Km, kcat and vmax values of catalytic activity of CDA catalysing the deamination of cytidine variants. All error bars denote s.d.
Extended Data Figure 3 Mechanism of CDA catalysed deamination of epigenetic nucleosides, their cytotoxicity and dUTPase activity.
a, Molecular docking of dC, 5hmdC and 5fdC on the CDA active site (Protein Data Bank (PDB) accession 1MQ0). The detailed view of the catalytic pocket is shown with the modified nucleoside in the centre. Chains A, B and C indicate units of the tetramer, which CDA forms to deaminate four nucleosides. Thin yellow lines show compatible distances for the formation of hydrogen bonds. b, Growth curves of H1299 and MCF7 cell lines treated with 10 and 1 μM of dC, 5hmdU and 5fdU over a period of 10 days (n = 3). c, Coomassie-stained gel demonstrating recombinant purified DUT (molecular mass, 18 kDa) and in vitro measurements of dUTPase activity using non-canonical uridine triphosphates (n = 3). d, Extracted ion chromatogram of nucleoside standards analysed by HPLC–QTOF mass spectrometry. Each nucleoside intensity was measured using the merged m/z values of the [M+H]+, [M+Na]+, [M+H]+, [2M+H]+ and [base+H]+ and a symmetric single m/z expansion of ±0.02. e, The most prominent ion of 5hmdU was identified in 5hmdC-treated MDA-MB-231 cells. All error bars denote s.d.
Extended Data Figure 4 Mass spectrometry identification of 5fUra and ultraviolet quantification of 5hmdU in the DNA.
a, Extracted ion chromatogram of nucleoside standards with 5fdU analysed by HPLC–QTOF mass spectrometry (as in Extended Data Fig. 3d). b, Weak, but consistent signal of 5fUra is identified in DNA of 5fdC-treated MDA-MB-231 cells, but not dC-treated cells or buffer alone. Two representative examples are shown. c, Relative quantification of 5fUra signal from three biological mass spectrometry replicates. d, Relationship between measured 5hmdU/T in the DNA of cell lines treated with 10 μM 5hmdC for 3 days and CDA expression levels. The cell lines used in this study are in coloured font (n = 3). All error bars denote s.d.
Extended Data Figure 5 Effect of 5hmdC administration on the cell cycle and DNA damage.
a, b, Propidium iodide FACS assay of the cell cycle. Shown are two representative plots of MDA-MB-231 cells at day 3 of treatment with dC and 5hmdC (10 μM) (a) and quantification for all the cell lines analysed (n = 3) (b). Two-way ANOVA: P = 0.0027 (S: 5hmdC versus dC MDA-MB-231), P = 0.0149 (G2-M: 5hmdC versus dC MDA-MB-231). HOP-92 P < 0.0001, P = 0.0005 (S: 5hmdC versus dC Capan-2), P < 0.0001 (G2-M: 5hmdC versus dC Capan-2) (n = 3; 10,000 events acquired). c, γH2AX immunofluorescence in MDA-MB-231 and H1299 cell lines at day 3 after treatment with 10 μM 5hmdC or dC. Scale bar, 50 μm. d, Fraction of cells showing a γH2AX signal above background (n = 3). ANOVA with Sidak correction for multiple comparisons: P = 0.0208 (5hmdC versus dC MDA-MB-231), P = 0.0135 (5hmdC versus dC HOP-92). Error bars denote s.d.
Extended Data Figure 6 Quantification of intracellular nucleotides by ion-pair HPLC and SMUG1 glycosylase activity.
a, Illustrative chromatogram of all standards indicated in b mixed together. b, Retention times of nucleotides were determined by analysing each standard separately and are indicated in the table. c, An average relative abundance of NTP and dNTP levels in cells treated with dC, 5hmdC and 5fdC (n = 3). d, Representative chromatograms of indicated experiments (blue) overlaid with standards separated on the same run (red). e, Typical image of denaturing PAGE electrophoresis of DNA incubated with SMUG1 and cleaved with APE1. f, Quantification of the DNA oligonucleotides with excised bases. g, Expression of SMUG1 and uracil DNA glycosylase (UNG) in MDA_MB_231, SN12C and Capan-2 cell lines (Genevestigator). Error bars denote s.d.
Extended Data Figure 7 CDA expression in human cancer and normal tissues, and toxicity evaluation of 5hmdC and 5fdC in mice.
a, CDA overexpression in pancreatic cancer (t-test, P < 0.0001). b, CDA expression across a panel of cancer (red) versus normal (green) tissues (GENT database). Arrows indicate cancer types with an evident difference between normal (N) and cancerous tissues (C). c, 5hmdC and 5fdC detection in the blood (mass spectrometry) of intraperitoneally injected mice at 30 min after injection. d, Label-free mass spectrometry quantification of 5hmdC in the blood of animals injected with doses of 25, 50 and 100 mg kg−1 (n = 3 (100 mg ml−1) and n = 4 (25 and 50 mg ml−1)). Error bars denote s.e.m. e, Immunohistochemistry showing CDA expression in the intestine. f, Haematoxylin and eosin staining of the intestine of mice injected with PBS and 100 mg kg−1 of 5hmdC and 5fdC. Tissue was removed 5 days after the injection. g, Immunofluorescence evaluation of proliferation (H3PS10) and DNA damage (γH2AX) in the intestine of mice treated with PBS and 100 mg kg−1 of 5hmdC and 5fdC 5 days after treatment. In parallel, the protocol was done on testis of irradiated mice, where positive signals for γH2AX were observed (data not shown). Scale bars, 50 μm (e–g). h, Weight of the mice plotted over the treatment period (n = 16 per group).
Extended Data Figure 8 Evaluation of wild-type SN12C cell line and CDA knockdown in a mouse xenograft model.
a, Schematic illustration of xenograft establishment and treatment with nucleoside variants. b, Tumour diameter was measured by Vernier caliper and volume calculated by assuming that tumours were spheres (n = 8, two-way ANOVA with repeated measures and Holm–Sidak correction, P < 0.0001). c, Photos of the dissected tumours (asterisks indicate dissected lymph nodes found after histological analysis). d, Western blot showing CDA expression in tumours extracted from mice. e, Quantification of proliferation (H3PS10) and DNA damage (γH2AX) using confocal microscopy and ImageJ of the central section of the tumour. Scale bar, 50 μm (n = 4, one-way ANOVA, SN12C H3PS10: P = 0.0033 (PBS versus 5hmdC), P = 0.0046 (PBS versus 5fdC); γH2AX: P = 0.0003 (PBS versus 5hmdC), P = 0.0436 (PBS versus 5fdC); SN12CshCDA_8: P = 0.0130 (PBS versus 5hmdC)). f, 5hmdU quantified from a HPLC–UV chromatogram of nucleosides from DNA extracted from tumours of mice treated with 5hmdC and PBS (n = 4, one-way ANOVA P = 0.0041). Error bars denote s.d.
Extended Data Figure 9 Identification and quantification of compounds' resulting peaks in HPLC–UV.
a, The abundance of molecule eluting at 5.1 min (5.7 min on the HPLC–QTOF) is not significantly different between dC- and 5hmdC-treated samples. It is a common component of DNA hydrolysis buffer. b, 5-methylcytosine in the DNA does not change after treatment with 5hmdC. Identity of 5mdC in the samples was confirmed by HPLC–QTOF mass spectrometry.
Extended Data Figure 10 Identification and quantification of compounds' resulting peaks in HPLC–UV.
Compound eluting at 4.5 min (5.0 min on the HPLC–QTOF) is an abundant component of DNA hydrolysis buffer, generating a m/z of 202.18.
Supplementary information
Supplementary Table 1
This table contains the gene expression analysis of 5hmdC sensitive and resistant cell lines. (XLSX 9160 kb)
Supplementary Table 2
This table contains the Mass spectrometry data of nucleosides. (XLSX 9 kb)
Rights and permissions
About this article

Cite this article
Zauri, M., Berridge, G., Thézénas, ML. et al. CDA directs metabolism of epigenetic nucleosides revealing a therapeutic window in cancer. Nature 524, 114–118 (2015). https://doi.org/10.1038/nature14948
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature14948
This article is cited by
-
Small molecule inhibitors for cancer metabolism: promising prospects to be explored
Journal of Cancer Research and Clinical Oncology (2023)
-
Upregulation of cytidine deaminase in NAT1 knockout breast cancer cells
Journal of Cancer Research and Clinical Oncology (2023)
-
FANCD2 maintains replication fork stability during misincorporation of the DNA demethylation products 5-hydroxymethyl-2’-deoxycytidine and 5-hydroxymethyl-2’-deoxyuridine
Cell Death & Disease (2022)
-
DNA methylome and single-cell transcriptome analyses reveal CDA as a potential druggable target for ALK inhibitor–resistant lung cancer therapy
Experimental & Molecular Medicine (2022)
-
Metabolic syndrome related gene signature predicts the prognosis of patients with pancreatic ductal carcinoma. A novel link between metabolic dysregulation and pancreatic ductal carcinoma
Cancer Cell International (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
