
Abstract
Photoluminescence blinking—random switching between states of high (ON) and low (OFF) emissivities—is a universal property of molecular emitters found in dyes1, polymers2, biological molecules3 and artificial nanostructures such as nanocrystal quantum dots, carbon nanotubes and nanowires4,5,6. For the past 15 years, colloidal nanocrystals have been used as a model system to study this phenomenon5,6. The occurrence of OFF periods in nanocrystal emission has been commonly attributed to the presence of an additional charge7, which leads to photoluminescence quenching by non-radiative recombination (the Auger mechanism)8. However, this ‘charging’ model was recently challenged in several reports9,10. Here we report time-resolved photoluminescence studies of individual nanocrystal quantum dots performed while electrochemically controlling the degree of their charging, with the goal of clarifying the role of charging in blinking. We find that two distinct types of blinking are possible: conventional (A-type) blinking due to charging and discharging of the nanocrystal core, in which lower photoluminescence intensities correlate with shorter photoluminescence lifetimes; and a second sort (B-type), in which large changes in the emission intensity are not accompanied by significant changes in emission dynamics. We attribute B-type blinking to charge fluctuations in the electron-accepting surface sites. When unoccupied, these sites intercept ‘hot’ electrons before they relax into emitting core states. Both blinking mechanisms can be electrochemically controlled and completely suppressed by application of an appropriate potential.
Similar content being viewed by others
Main
In the conventional blinking model (Fig. 1a), ON and OFF periods correspond to a neutral nanocrystal and a charged nanocrystal, respectively, and photo-assisted charging/discharging causes random switching between these two states. The dynamics of the bright state is dominated by radiative recombination of the neutral exciton, X0 (Fig. 1b), which is characterized by a long, mono-exponential decay (15–30 ns in CdSe nanocrystals11,12,13). For a charged exciton (trion), X−, three-particle Auger recombination opens a fast, non-radiative channel, resulting in a shorter lifetime (a few nanoseconds or less) and, consequently, a reduced photoluminescence quantum yield. As illustrated in Figs 1c, d, this model predicts correlated fluctuations of the photoluminescence intensity and lifetime (referred to here as A-type blinking) that have indeed been observed experimentally11,12,14. Photocharging can lead to ‘binary’ switching between the ON and the OFF states (Fig. 1c) when the timescale of charge fluctuations is longer than the experimental binning time (typically at least tens of milliseconds). As we discuss below, the same discrete charging process can also produce quasi-continuous photoluminescence fluctuations, referred to as ‘flickering’ (Fig. 1d). In this case, the data within each bin represents an average over the neutral and charged nanocrystal states, which results in photoluminescence intensities and lifetimes that vary continuously according to the relative times spent by the nanocrystal in each charge state.

a, In the conventional photoluminescence (PL) blinking model, ON and OFF periods correspond to a neutral nanocrystal (X0) and a charged nanocrystal (X−), respectively. b, Schematic photoluminescence decay of the ON and the OFF states on a logarithmic scale. The dynamics of the ON state is dominated by the radiative rate γr. In the charged state, the increase in the number of recombination pathways leads to a higher radiative rate, 2γr, responsible for the higher emission intensity at short delays. Simultaneously, the onset of three-particle Auger recombination with the rate γA  γr opens a new, non-radiative, channel, leading to faster photoluminescence decay and reduced photoluminescence quantum yield. c, When the timescale of charging and discharging is longer than the experimental binning time, binary blinking is observed. d, For fluctuations much faster than the bin size, a continuous distribution of intensities and lifetimes is obtained, often referred to as flickering. The insets in c and d show corresponding schematic fluorescence lifetime–intensity distributions (FLIDs).
γr opens a new, non-radiative, channel, leading to faster photoluminescence decay and reduced photoluminescence quantum yield. c, When the timescale of charging and discharging is longer than the experimental binning time, binary blinking is observed. d, For fluctuations much faster than the bin size, a continuous distribution of intensities and lifetimes is obtained, often referred to as flickering. The insets in c and d show corresponding schematic fluorescence lifetime–intensity distributions (FLIDs).
A convenient tool for the analysis of correlations between photoluminescence intensities and lifetimes is a fluorescence lifetime–intensity distribution (FLID) representation. In this representation, the probability of occupying a given state in the two-dimensional lifetime–intensity space is shown by false colour, which changes from blue to red as the probability increases. As illustrated in the insets of Figs 1c, d, the use of FLIDs allows ready identification of different types of charge state as well as different types of blinking behaviour (for example binary blinking versus flickering).
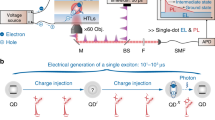
In our work, to verify the validity of the charging model of photoluminescence intermittency, we combine single-nanocrystal spectroscopy with an electrochemical approach for controlling the extent of nanocrystal charging15,16,17. Specifically, we conduct single-nanocrystal, time-tagged, time-resolved, single-photon counting studies of samples incorporated into a three-electrode electrochemical cell (Fig. 2a). We investigate core–shell CdSe/CdS nanocrystals synthesized according to ref. 18. In the case of exceptionally thick, 16–19-monolayer shells, they show nearly complete suppression of blinking18. Here we use nanocrystals with intermediate shell thicknesses (7–9 monolayers) that show typical blinking behaviour but have quantum yields during OFF periods that are considerably higher than those of standard nanocrystals. This allows us to investigate in detail the properties of the OFF state and the effect of controlled charging on its emissivity and dynamics. All changes in photoluminescence intensity and dynamics induced by the applied potential are reversible, indicating no permanent chemical or photochemical modifications of the nanocrystals and suggesting that such changes are due to controlled charging/discharging. To verify that the observed photoluminescence originates from a single nanocrystal, we measure the second-order intensity correlation function19, g2, and ensure that g2(0) < 0.5 (Methods Summary; Fig. 2b, inset). All experiments are performed under ambient conditions at room temperature. All potentials are reported with respect to a silver wire quasi-reference (see Methods for further experimental details).

a, Set-up of a single-nanocrystal spectroelectrochemical experiment. APD, avalanche photodiode; BS, 50/50 beam splitter; ITO, indium tin oxide; TCSPC, time-correlated single-photon counting. b, Series of photoluminescence decays for a single nanocrystal for increasingly negative applied potentials. The thin grey lines show the best global triple-exponential fits with the shared time constants, yielding the lifetimes τd = 2 ns, τs = 5 ns and τn = 24 ns. Top inset: the second-order photoluminescence intensity correlation function measured for this nanocrystal indicates that g2(0) = 0.08. Bottom inset: residuals of the global fit indicate very high fidelity of the fitting procedure, with deviations within the noise level and below 1% of the maximum photoluminescence signal.
Electrochemical control of emission intensity from individual nanocrystals has been demonstrated previously17. In this study, we analyse the effect of charging on both photoluminescence intensity and photoluminescence dynamics. Figure 2b shows photoluminescence time transients recorded for a single nanocrystal under increasing negative potential, V, which corresponds to electron injection. The photoluminescence decay becomes progressively faster as V becomes more negative. All decays can be fitted globally to a triple-exponential function (Fig. 2b, grey lines) The high fidelity of the fit (see residuals in the lower inset) suggests that only three distinct emitting states are involved, each having a well-defined photoluminescence lifetime: τd = 2 ns, τs = 5 ns and τn = 24 ns. As V is decreased from 0 to −0.7 V, the weight of the τs component gradually increases relative to that of the τn component. At more-negative values of V, the component with the fastest decay (corresponding to τd) emerges and becomes dominant at −1.1 V. We assign the lifetimes τn, τs and τd to three distinct states, respectively neutral excitons (X0) and singly (X−) and doubly (X2−) charged excitons. Owing to increased rates of radiative decay, singly charged excitons (negative trions) show increased signal at short times relative to neutral excitons (Fig. 2b); however, the time-integrated photoluminescence signal is reduced because of Auger recombination. The observed quickening of photoluminescence decay on charging is due to enhancement in both the radiative decay rate and the non-radiative decay rate.
To confirm the above assignments and investigate the relationship between charging and blinking, we analyse the correlations in the temporal variations of photoluminescence decay time and intensity. In Fig. 3, we plot photoluminescence intensity and average lifetime trajectories (calculated for a 50-ms bin size; Supplementary Information) along with corresponding FLIDs for the nanocrystal shown in Fig. 2. To illustrate the variability in blinking behaviours, we present the data collected for this nanocrystal on two different days. These data, representing examples of binary ON–OFF switching (Fig. 3a) and flickering (Fig. 3b), indicate a strong correlation between the photoluminescence intensity and the photoluminescence lifetime during the fluctuations, in agreement with the conventional charging model. We call this A-type blinking.

a, Photoluminescence intensities (black lines) and average lifetimes (red lines), and corresponding FLIDs, for the nanocrystal shown in Fig. 2 at three different potentials. Binary blinking seen at V = 0 V is largely suppressed at V = +0.6 V, whereas electron injection is achieved at V = −0.6 V. In the FLID colour scale, red corresponds to the most frequently occurring lifetime–intensity pair, and probabilities less than 1% of this maximum are indicated by dark blue. A linear scaling from blue to red is used between these extremes. b, Data from the same nanocrystal, acquired on a different day, display continuous photoluminescence intensity and lifetime fluctuations, typical of flickering. At V = −1.1 V, we observe emission from a doubly charged exciton, X2−. All data were analysed with a bin size of 50 ms. Full time trajectories for a and b are shown in Supplementary Fig. 1.
At 0 V (Fig. 3a, middle), the nanocrystal shows binary blinking between the neutral state (X0) and the singly charged state (X−). The average photoluminescence lifetime of X0 (the ON state) is τn ≈ 24 ns, which corresponds to a radiative lifetime of ∼60 ns (Supplementary Information) and is in agreement with previous ensemble studies of this type of nanocrystal13. Application of a positive potential, V = +0.6 V (Fig. 3a, left), drastically suppresses charge fluctuations and results in almost non-blinking emission from the neutral exciton (see corresponding FLID and Supplementary Figs 1 and 2; another example is shown in Supplementary Fig. 12). At a negative potential, V = −0.6 V (Fig. 3a, right), the peak of the photoluminescence distribution shifts to the lower-emissivity state, X−, characterized by a lifetime of ∼5 ns. Assuming a ‘statistical’ scaling of recombination rates with the number of charges (Supplementary Information and ref. 20), we deduce the Auger lifetime for X− to be ∼3.5 ns. This is much shorter than the radiative lifetime of X− (∼30 ns; Supplementary Information), which explains the relatively low photoluminescence quantum yield of the negative trion. The existence of fluctuations between X0 and X− is indicated by a well-resolved trace in the FLID connecting the two states. We simulate the FLID data assuming that the photoluminescence intensity during a given time bin is determined by the relative times spent by the nanocrystal in the states X− and X0 (Supplementary Information). A very good agreement, without any adjustable parameters, between the simulated trace (Fig. 3a, white lines) and the measured FLID provides strong support for both the assignment of emitting states and the model used in the analysis.
We note that the same nanocrystal measured on a different day (Fig. 3b) shows a more continuous distribution of photoluminescence intensities and lifetimes, typically referred to as flickering. This change in the blinking behaviour probably occurs as a result of the shortening of time spent by the nanocrystal in a given charge state, which leads to fast switching between X0 and X− within the bin time used in the measurements. Photoluminescence from X− becomes dominant at V = −0.7 V (Fig. 3b, middle FLID). By applying a more negative potential, V = −1.1 V, we detect a new state with lifetime τd ≈ 2 ns, associated with the formation of a doubly charged exciton, X2−, with an Auger lifetime of ∼1.2 ns. Judging from the FLID at this potential (Fig. 3b, right), fluctuations occur also between the states X2− and X−.
Figure 4a shows data from a different nanocrystal, which has distinct blinking behaviour that we refer to as B-type blinking. Specifically, at V = 0 V (Supplementary Fig. 3) and V = +0.8 V (Fig. 4a, left), we observe periods of low photoluminescence intensity that are not accompanied by significant shortening of photoluminescence lifetimes. In fact, the photoluminescence time constant measured for the B-type OFF state is identical to that of the ON state (X0). These B-type blinking events were observed in 20 of the 23 dots we studied (Supplementary Table 1) and usually coexisted with A-type fluctuations (Supplementary Fig. 3). Notably, at V = −1 V there is complete suppression of blinking but the long photoluminescence lifetime (∼26 ns) typical of a neutral exciton is preserved. This suppression could be achieved in the majority of the nanocrystals with B-type blinking; however, the potential required to obtain the suppression varied widely from dot to dot (from −0.6 to −1.4 V; Supplementary Table 1). For some nanocrystals, the elimination of B-type blinking occurred simultaneously with the onset of A-type fluctuations between X0 and X− (see below). At a more negative potential (V = −1.2 V; Fig. 4a, right), we observe clear signatures of electron injection into the nanocrystal. The photoluminescence decay becomes bi-exponential, with an increasing contribution from the negative trion, which in this quantum dot has a lifetime of ∼6 ns (Supplementary Fig. 4). In this case, switching between X0 and X− occurs on a much shorter timescale than the bin time, which gives rise to a narrow photoluminescence lifetime–intensity distribution. As with the data in Fig. 3, we can closely reproduce this pattern using the charging model (simulated white lines in FLID).

a, Photoluminescence intensities (black lines) and average lifetimes (red lines), and corresponding FLIDs, for a nanocrystal showing the B-type OFF state; analysis done with a 10-ms bin. Full time trajectories are shown in Supplementary Fig. 6. b, The model of B-type blinking. The B-type OFF state is due to the activation of recombination centres (R) that capture hot electrons at a rate, γD, that is higher than the intraband relaxation rate, γB (the ground and the excited electron states are shown as 1Se and 1Pe, respectively; 1Sh is the band-edge hole state). The position of the Fermi level, EF, relative to the trap energy, ER, is determined by the electrochemical potential and controls the occupancy of the surface trap R. This, in turn, allows for electrochemical control of B-type blinking.
To explain B-type blinking, we invoke the activation and deactivation of non-radiative recombination centres (denoted R) that efficiently capture ‘hot’ electrons before they relax into the lowest-energy emitting state (Fig. 4b). Such processes of hot-electron trapping have been recently observed for both nanocrystals in solutions21 and surface-dispersed particles22,23. In this picture, photoluminescence dynamics during the OFF periods should be similar to that of a neutral exciton whereas the emission intensity will be reduced according to the ratio between the rates of intraband relaxation, γB, and hot-electron capture by the recombination centre, γD. Because the frequency of B-type blinking events is controlled by the electrochemical potential, the activation and deactivation of the bypass channel are probably associated with emptying and, respectively, filling of the corresponding surface trap state. For a positive potential (V = +0.8 V; Fig. 4a, left), the Fermi level decreases in energy, which increases the relative time spent by the trap in the unoccupied (that is, active) state and leads to increased occurrence of B-type OFF events (Fig. 4b, left). The trapped electron can recombine non-radiatively with a valence-band hole before the next photoexcitation event, leaving behind a neutral dot. Occasionally, photon absorption occurs before reneutralization of the dot, resulting in a positive trion, X+; Auger decay of X+ could explain observations of shorter photoluminescence lifetimes within the B-type OFF periods illustrated in Supplementary Fig. 5.
For an increasingly negative potential, the Fermi level increases in energy and eventually a regime is reached where the trap states become populated and γD → 0 owing to Coulomb blockade (V = −1 V; Fig. 4a, b, middle). In this case, B-type blinking is completely suppressed. Application of an even more negative potential leads to charging of the nanocrystal core with an extra electron and emission from negative trions (V = −1.2 V; Fig. 4a, b, right).
Blinking suppression due to filling of electron-accepting trap sites is consistent with previous observations that electron-donating thiolates enhance ensemble photoluminescence emission24 and reduce blinking25. Similar phenomena were observed for other electron-donating molecules26 as well as n-doped substrates27. These observations of a significant effect of surface species on photoluminescence intensity and intermittency imply that the trap sites responsible for B-type blinking are probably of surface origin. Recent ultrafast studies of carrier surface trapping in ensembles of CdSe nanocrystals also suggest that this process is directly relevant to the problem of nanocrystal blinking28. Finally, our model of B-type hot-electron surface traps provides an explanation of previously reported properties of the nanocrystal OFF state, such as low emission quantum yields9 and the lack of a systematic size dependence of photoluminescence lifetimes10, that could not be explained by the traditional charging model (Supplementary Information, section IV).
The distinct nature of the processes responsible for A- and B-type blinking is evident from the effect of increasing shell thickness on photoluminescence intermittency. Specifically, we observe that as the outer shell gets thicker, the B-type type blinking events become less frequent until they are completely eliminated for shells with 15 or more CdS monolayers. By contrast, the A-type blinking can still be observed even in the case of the extremely thick 19-monolayer shells. The analysis of photoluminescence intermittency in more than twenty nanocrystals with 15-monolayer shells (Supplementary Table 2 and Supplementary Figs 7 and 8) indicates that ∼70% of these dots are non-blinking and that the rest have A-type blinking behaviour; none of the nanocrystals showed any detectable B-type blinking. By contrast, B-type blinking is clearly the dominant behaviour in nanocrystals with 7–9-monolayer shells (Supplementary Table 1). The fact that B-type blinking is quickly suppressed as shell thickness increases is consistent with the proposed mechanism of hot-electron tunnelling outside the nanocrystal, because this process is expected to be extremely (in fact exponentially) sensitive to the thickness of the tunnelling barrier.
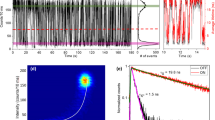
The studies of statistics of ON and OFF times also indicate a clear distinction between the A-type and B-type blinking mechanisms. In Fig. 5a, we show a nanocrystal with B-type blinking at −0.8 V, which switches to A-type blinking at −1 V. Remarkably, whereas the B-type ON and OFF times both follow a power-law distribution over almost three decades, the distributions of ON and OFF times in the A-type blinking regime are quasi-exponential with a cut-off time of ∼70 ms (Fig. 5b). This electrochemically controlled switching between different blinking regimes in the same nanocrystal is another strong indication that the difference between A-type and B-type blinking is linked to the distinct nature of the underlying physical mechanisms but not to dot-to-dot variations. Furthermore, the fact that the cut-off time measured in the case of A-type blinking is close to a typical bin size used in the measurements suggests that relatively small changes in the timescale of charge fluctuations can result in switching between binary blinking and flickering as seen, for example, in Fig. 3.

a, FLIDs indicating a nanocrystal switching from B-type blinking at −0.8 V (left) to A-type blinking at −1 V (right). Details of the analysis are given in Supplementary Fig. 9. b, Statistics for ON (red circles) and OFF (black squares) times for the FLIDs in a, in the log–log representation. At −0.8 V (B-type blinking), the data can be fitted to a power-law distribution, ∝t−α, with α = 1.17 for the ON times (red line) and α = 1.00 for the OFF times (black line). At −1 V (A-type blinking), this description is no longer valid; however, the data can be closely fitted by introducing an exponential cut-off such that the distribution is ∝t−αexp(−t/tc), where α = 0.54 and tc = 73.4 ms for the ON times (red line) and α = 0.37 and tc = 70.8 ms for the OFF times (black line).
Methods Summary
We used a home-built electrochemical cell with a three-electrode configuration. The nanocrystals were directly deposited onto an ITO-coated transparent working electrode from a very dilute hexane or water solution. As a counterelectrode, we used platinum gauze attached to a platinum wire. All potentials reported in the main text are measured relative to a silver wire quasi-reference. The electrochemical experiments were performed using several combinations of solvents (acetonitrile and propylene carbonate) and supporting electrolytes (all concentrations, 0.1 M): tetrabutylammonium hexafluorophosphate (TBAPF6), tetrabutylammonium perchlorate (TBAClO4) and lithium perchlorate (LiClO4). We note that the results presented here are not dependent on the identities of the solvent, supporting electrolyte or surface ligands used.
The nanocrystals were excited by a pulsed diode laser at a wavelength of 405 nm using low fluences (the average number of excitons per nanocrystal per pulse, 〈N〉 ≤ 0.2) to avoid multiexcitonic effects and to limit photocharging. The photoluminescence was collected confocally and sent to a Hanbury Brown/Twiss set-up (time resolution, 300 ps) to measure the second-order intensity correlation function, g2. The area of the central peak normalized to the area of a side peak is a measure of multiphoton emission probability during a single excitation cycle. Any g2(0) value less than 0.5 implies that the measured signal originates from a single quantum emitter (a single nanocrystal). For lifetime and blinking analyses, we used a time-tagged, time-resolved mode, in which we recorded the delay time of each photoluminescence photon with regard to the laser pulse. These data were analysed with the SYMPHOTIME software. All subsequent analysis and plotting were performed in ORIGIN 8.0.
Online Methods
Materials
Cadmium oxide, oleic acid (90%), 1-octadecene (ODE, 90%), 1-octadecane (OD, 90%), oleylamine, sulphur, selenium pellet and trioctylphosphine (TOP) were purchased from Aldrich and used without further purification. Trioctylphosphine oxide (TOPO, 90%) was purchased from Strem and used without further purification.
Nanocrystal synthesis
A 100-ml round-bottomed flask equipped with a reflux condenser and a thermocouple probe was charged with 1 g TOPO, 8 ml ODE and 0.38 mmol Cd-oleate under standard air-free conditions. The reaction system was evacuated for 30 min at room temperature (22°C) and 30 min at 80 °C, and then the temperature was raised to 300 °C under argon. Following this, a mixture of 4 mmol of TOP-Se, 3 ml oleylamine and 1 ml of ODE was quickly injected into the reaction system. The temperature was then lowered to 270 °C for CdSe nanocrystal growth. After several minutes, the solution was cooled to room temperature and CdSe nanocrystals (diameter, d = 3 nm) were collected by precipitation with ethanol and centrifugation. The CdSe nanocrystals were redispersed in hexane.
The synthesis of core–shell CdSe/nCdS nanocrystals followed the successive ionic layer adsorption and reaction (SILAR) approach18,29,30 with modifications. A 250-ml round-bottomed flask was charged with ∼2 × 10−7 mol pre-washed CdSe cores, 5 ml oleylamine and 5 ml OD. Here OD was chosen as the solvent because it alleviated the problem of precipitation observed during later stages of thick-shell growth. Elemental sulphur (0.2 M) dissolved in OD and 0.2 M Cd-oleate in ODE were used as precursors for shell growth. The quantity of precursor used for each addition of shell monolayer was calculated to account for the successive increases in particle volume as a function of increasing shell thickness. The reaction temperature was 240 °C and growth times were 1 h for sulphur and 2.5 h for Cd2+ precursors. Reactions were continued until a desired shell thickness was achieved. The core–shell nanocrystals were washed in a similar fashion as the CdSe cores, by precipitating two to three times with ethanol and redispersing in hexane. Relative photoluminescence quantum yields were determined by comparison with a standard dye (rhodamine 6G, 99%; Acros) and were observed to vary as a function of shell thickness. For the CdSe/9CdS nanocrystals used in the present study, the photoluminescence quantum yield was ∼30%. The purified core–shell nanocrystals were studied using transmission electron microscopy to determine their shapes and sizes and to confirm the growth of a thick, high-quality CdS shell over the CdSe core.
Ligand exchange
Core–shell nanocrystals were precipitated with ethanol then centrifuged for approximately 5 min (5,000 r.p.m., or 2,450g). The resulting pellet was redispersed in toluene. This procedure was repeated twice. The nanocrystal concentrations were calculated according to ref. 31. An amount of ligand (mercaptoundecanoic acid) equivalent to twice the number of moles of Cd-chalcogenide in the sample was added to the toluene solution. After 2 h, a solution of tetramethylammonium hydroxide in water (four times the number of moles of Cd-chalcogenide) was added dropwise. The nanocrystals were transferred from the toluene phase to the water phase. The water phase was separated from the toluene phase and precipitated with isopropanol, followed by centrifugation (∼5 min at 5,000 r.p.m., or 2,450g). Finally, the pellet was redispersed in distilled water.
Electrochemical cell
A home-built electrochemical cell with a three-electrode configuration was used. As working electrode, we used an ITO-coated glass slide with sheet resistance of ∼50 Ω (SPI Supplies). Before use, the electrode was sonicated in acetone and isopropanol baths, rinsed with deionized water, dried and plasma-etched for 10 min. The nanocrystals were directly deposited onto the electrode from a very dilute hexane or water solution. We note that plasma etching significantly improves the attachment of water-soluble nanocrystals by providing a hydrophilic surface. As a counterelectrode, we used platinum gauze attached to a platinum wire. The high-surface-area gauze was used to achieve uniform current density across the working electrode. A silver wire was used as a quasi-reference electrode. This electrode was calibrated using the Ru3+/2+ redox-couple of [Ru(bpy)3](PF6)2 (refs 32, 33). By comparison of the half-wave potentials obtained with the silver wire, standard calomel electrode and Ag/Ag(NO3) reference electrodes, we found that the silver quasi-reference is offset from the normal hydrogen electrode by 0.31 ± 0.01 V. All potentials reported in the main text are measured relative to the silver quasi-reference. The electrochemical experiments were performed using several combinations of solvents (acetonitrile and propylene carbonate) and supporting electrolytes (all concentrations, 0.1 M): tetrabutylammonium hexafluorophosphate (TBAPF6), tetrabutylammonium perchlorate (TBAClO4) and lithium perchlorate (LiClO4). The results presented here are not dependent on the identities of the solvent, supporting electrolyte or surface ligands used.
Optical set-up
The excitation source was a PicoQuant pulsed diode laser producing ∼30-ps pulses at 405 nm with a repetition rate of 2.5–40 MHz. Most of the experiments were performed at 2.5 MHz, which corresponds to pulse-to-pulse separation of 400 ns, an order of magnitude greater than the longest photoluminescence lifetimes. This allows us to minimize ‘pile-up’ effects and parasitic charge accumulation due to possible photocharging. The average nanocrystal excitonic occupancies generated per pulse, 〈N〉, were estimated from absorption cross-sections calculated using nanocrystal sizes derived from transmission electron microscopy data and were independently verified by photoluminescence saturation and intensity-dependent g2 measurements34. Photoluminescence was excited and collected through an oil-immersion Olympus objective with a numerical aperture of 1.3. After reflection from a dichroic mirror (Semrock), photoluminescence then went through a long-pass or band-pass filter (Semrock). A flip mirror was used to send emission to a 500-mm spectrometer equipped with a liquid-nitrogen-cooled silicon charge-coupled device. Emission from the nanocrystals typically peaked around 620 nm with a full-width of ∼25 nm at half maximum. A Hanbury Brown/Twiss set-up was realized using a 50/50 beam splitter and two avalanche photodiodes (APDs; SPCM-AQRH-14, Perkin Elmer) with a quantum efficiency of ∼50% at the photoluminescence wavelength, a time jitter of ∼300 ps and a dark count rate of <100 Hz. The single-photon counting device was a PicoHarp 300 stand-alone module (PicoQuant). Two APDs were used to produce start and stop signals in the measurements of the second-order intensity correlation function, whereas the synchronization pulse of the laser provided the start signal in the time-tagged, time-resolved mode. Photon arrival times were recorded from one of the APDs (stop signal).
Analysis
For the analysis of raw time-tagged, time-resolved data, we used the SYMPHOTIME software. All subsequent analysis and plotting were performed in ORIGIN 8.0. For the dynamical correlated lifetime–intensity analysis, we chose a bin time, corresponding to more than 100 photons per bin on average, to ensure a reliable bi-exponential fitting for each decay curve. We fixed the lifetimes on the basis of the values produced by the global fit procedure and constrained the amplitudes to be positive numbers. To enhance the precision, we used a Poissonian maximum-likelihood estimator. To confirm the validity of the multi-exponential approach, we also constructed FLIDs for which the lifetime for each bin was calculated as a weighted average of photoluminescence photon arrival times, that is, without any fitting procedure. The resulting FLIDs were similar to those produced by a multi-exponential fit, as illustrated in Supplementary Fig. 13.
References
Hoogenboom, J. P., Hernando, J., van Dijk, E. M. H. P., van Hulst, N. F. & García-Parajó, M. F. Power-law blinking in the fluorescence of single organic molecules. ChemPhysChem 8, 823–833 (2007)
Bout, D. A. V. et al. Discrete intensity jumps and intramolecular electronic energy transfer in the spectroscopy of single conjugated polymer molecules. Science 277, 1074–1077 (1997)
Riley, E. A., Bingham, C., Bott, E. D., Kahr, B. & Reid, P. J. Two mechanisms for fluorescence intermittency of single violamine R molecules. Phys. Chem. Chem. Phys. 13, 1879–1887 (2011)
Frantsuzov, P., Kuno, M., Janko, B. & Marcus, R. A. Universal emission intermittency in quantum dots, nanorods and nanowires. Nature Phys. 4, 519–522 (2008)
Nirmal, M. et al. Fluorescence intermittency in single cadmium selenide nanocrystals. Nature 383, 802–804 (1996)
Fernando, D. Stefani, J. P. H. & Barkai, E. Beyond quantum jumps: blinking nanoscale light emitters. Phys. Today 6, 34–39 (2009)
Efros, A. L. & Rosen, M. Random telegraph signal in the photoluminescence intensity of a single quantum dot. Phys. Rev. Lett. 78, 1110–1113 (1997)
Klimov, V. I., Mikhailovsky, A. A., McBranch, D. W., Leatherdale, C. A. & Bawendi, M. G. Quantization of multiparticle Auger rates in semiconductor quantum dots. Science 287, 1011–1013 (2000)
Zhao, J., Nair, G., Fisher, B. R. & Bawendi, M. G. Challenge to the charging model of semiconductor-nanocrystal fluorescence intermittency from off-state quantum yields and multiexciton blinking. Phys. Rev. Lett. 104, 157403 (2010)
Rosen, S., Schwartz, O. & Oron, D. Transient fluorescence of the off state in blinking CdSe/CdS/ZnS semiconductor nanocrystals is not governed by Auger recombination. Phys. Rev. Lett. 104, 157404 (2010)
Fisher, B. R., Eisler, H.-J., Stott, N. E. & Bawendi, M. G. Emission intensity dependence and single-exponential behavior in single colloidal quantum dot fluorescence lifetimes. J. Phys. Chem. B 108, 143–148 (2004)
Zhang, K., Chang, H., Fu, A., Alivisatos, A. P. & Yang, H. Continuous distribution of emission states from single CdSe/ZnS quantum dots. Nano Lett. 6, 843–847 (2006)
García-Santamaría, F. et al. Breakdown of volume scaling in Auger recombination in CdSe/CdS heteronanocrystals: the role of the core−shell interface. Nano Lett. 11, 687–693 (2011)
Gómez, D. E., van Embden, J., Mulvaney, P., Fernee, M. J. & Rubinsztein-Dunlop, H. Exciton−trion transitions in single CdSe–CdS core–shell nanocrystals. ACS Nano 3, 2281–2287 (2009)
Jha, P. P. & Guyot-Sionnest, P. Trion decay in colloidal quantum dots. ACS Nano 3, 1011–1015 (2009)
Houtepen, A. J. & Vanmaekelbergh, D. Orbital occupation in electron-charged CdSe quantum-dot solids. J. Phys. Chem. B 109, 19634–19642 (2005)
Jha, P. P. & Guyot-Sionnest, P. Electrochemical switching of the photoluminescence of single quantum dots. J. Phys. Chem. C 114, 21138–21141 (2010)
Chen, Y. et al. “Giant” multishell CdSe nanocrystal quantum dots with suppressed blinking. J. Am. Chem. Soc. 130, 5026–5027 (2008)
Wang, X. et al. Non-blinking semiconductor nanocrystals. Nature 459, 686–689 (2009)
Klimov, V. I., McGuire, J. A., Schaller, R. D. & Rupasov, V. I. Scaling of multiexciton lifetimes in semiconductor nanocrystals. Phys. Rev. B 77, 195324 (2008)
McGuire, J. A. et al. Spectroscopic signatures of photocharging due to hot-carrier transfer in solutions of semiconductor nanocrystals under low-intensity ultraviolet excitation. ACS Nano 4, 6087–6097 (2010)
Tisdale, W. A. et al. Hot-electron transfer from semiconductor nanocrystals. Science 328, 1543–1547 (2010)
Li, S., Steigerwald, M. L. & Brus, L. E. Surface states in the photoionization of high-quality CdSe core/shell nanocrystals. ACS Nano 3, 1267–1273 (2009)
Jeong, S. et al. Effect of the thiol−thiolate equilibrium on the photophysical properties of aqueous CdSe/ZnS nanocrystal quantum dots. J. Am. Chem. Soc. 127, 10126–10127 (2005)
Hohng, S. & Ha, T. Near-complete suppression of quantum dot blinking in ambient conditions. J. Am. Chem. Soc. 126, 1324–1325 (2004)
Fomenko, V. & Nesbitt, D. J. Solution control of radiative and nonradiative lifetimes: a novel contribution to quantum dot blinking suppression. Nano Lett. 8, 287–293 (2008)
Jin, S., Song, N. & Lian, T. Suppressed blinking dynamics of single QDs on ITO. ACS Nano 4, 1545–1552 (2010)
Tyagi, P. & Kambhampati, P. False multiple exciton recombination and multiple exciton generation signals in semiconductor quantum dots arise from surface charge trapping. J. Chem. Phys. 134, 094706 (2011)
Li, J. J. et al. Large-scale synthesis of nearly monodisperse CdSe/CdS core/shell nanocrystals using air-stable reagents via successive ion layer adsorption and reaction. J. Am. Chem. Soc. 125, 12567–12575 (2003)
Vela, J. et al. Effect of shell thickness and composition on blinking suppression and blinking mechanism in ‘giant’ CdSe/CdS nanocrystal quantum dots. J. Biophoton. 3, 706–717 (2010)
Yu, W. W., Qu, L., Guo, W. & Peng, X. Experimental determination of the extinction coefficient of CdTe, CdSe, and CdS nanocrystals. Chem. Mater. 15, 2854–2860 (2003)
Juris, A. et al. Ru(II) polypyridine complexes: photophysics, photochemistry, eletrochemistry, and chemiluminescence. Coord. Chem. Rev. 84, 85–277 (1988)
DeLaive, P. J., Foreman, T. K., Giannotti, C. & Whitten, D. G. Photoinduced electron transfer reactions of transition-metal complexes with amines. Mechanistic studies of alternate pathways to back electron transfer. J. Am. Chem. Soc. 102, 5627–5631 (1980)
Park, Y. S. et al. Near-unity quantum yields of biexciton emission from CdSe/CdS nanocrystals measured using single-particle spectroscopy. Phys. Rev. Lett. 106, 187401 (2011)
Acknowledgements
C.G. and V.I.K. acknowledge support of the Center for Advanced Solar Photophysics, an Energy Frontier Research Center funded by the US Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences (BES). Y.G. and A.S. are supported by Los Alamos National Laboratory Directed Research and Development Fund. M.S., J.A.H. and H.H. are supported by NIH-NIGMS grant 1R01GM084702–01. This work was conducted, in part, at the Center for Integrated Nanotechnologies, a DOE/BES user facility.
Author information
Authors and Affiliations
Contributions
C.G., M.S., J.A.H., V.I.K. and H.H. conceived the study. C.G., M.S. and H.H. designed the experiments. C.G. constructed the experimental set-up and performed the measurements under the guidance of M.S., V.I.K. and H.H. Y.G. synthesized and A.S. modified quantum dot materials under the guidance of J.A.H. C.G., V.I.K. and H.H. analysed and interpreted the data, and wrote the manuscript with the assistance of all other co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
This file contains Supplementary Text and Data 1-4, additional references, Supplementary Figures 1-13 with legends and Supplementary Tables 1-2. (PDF 1111 kb)
Rights and permissions
About this article
Cite this article
Galland, C., Ghosh, Y., Steinbrück, A. et al. Two types of luminescence blinking revealed by spectroelectrochemistry of single quantum dots. Nature 479, 203–207 (2011). https://doi.org/10.1038/nature10569
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature10569
This article is cited by
-
Single quantum dot spectroscopy for exciton dynamics
Nano Research (2024)
-
Electric-field-induced colour switching in colloidal quantum dot molecules at room temperature
Nature Materials (2023)
-
Post-processing of real-time quantum event measurements for an optimal bandwidth
Scientific Reports (2023)
-
Unveiling non-radiative center control in CsPbBr3 nanocrystals: A comprehensive comparative analysis of hot injection and ligand-assisted reprecipitation approaches
Nano Research (2023)
-
Lattice distortion inducing exciton splitting and coherent quantum beating in CsPbI3 perovskite quantum dots
Nature Materials (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
