
Abstract
Swine influenza A viruses (SwIV) cause significant economic losses in animal husbandry as well as instances of human disease1 and occasionally give rise to human pandemics2, including that caused by the H1N1/2009 virus3,4. The lack of systematic and longitudinal influenza surveillance in pigs has hampered attempts to reconstruct the origins of this pandemic4. Most existing swine data were derived from opportunistic samples collected from diseased pigs in disparate geographical regions, not from prospective studies in defined locations, hence the evolutionary and transmission dynamics of SwIV are poorly understood. Here we quantify the epidemiological, genetic and antigenic dynamics of SwIV in Hong Kong using a data set of more than 650 SwIV isolates and more than 800 swine sera from 12 years of systematic surveillance in this region, supplemented with data stretching back 34 years. Intercontinental virus movement has led to reassortment and lineage replacement, creating an antigenically and genetically diverse virus population whose dynamics are quantitatively different from those previously observed for human influenza viruses. Our findings indicate that increased antigenic drift is associated with reassortment events and offer insights into the emergence of influenza viruses with epidemic potential in swine and humans.
Similar content being viewed by others

Main
All major SwIV lineages of North American or European origin—classical swine (CS), European or Eurasian avian-like swine (EA) and triple-reassortant swine (TRIG) (see Supplementary Information for an overview)—co-circulate in southern China4,5, and both human (H3N2) and avian (H5N1 and H9N2) viruses have been isolated from swine in the region6,7,8,9. To address the critical lack of structured swine influenza data, we undertook virological and serological analysis of Hong Kong SwIV surveillance samples. Most (80–95%) of the swine slaughtered in Hong Kong originate from provinces in mainland China (Supplementary Fig. 1 and Supplementary Table 1), the region with the world’s largest swine population10,11,12.
We isolated and subtyped 573 H1N1 and H1N2, 97 H3 and 2 H9N2 viruses from fortnightly sampling of swine slaughtered between May 1998 and January 2010 (Fig. 1a, b). We found no H5N1 viruses. From August 1998 to December 2002, the isolates were mostly CS H1N1 viruses. EA H1N1 viruses were detected only from 2001 onwards and TRIG H1N2 from 2002 onwards. During 2002–05, viruses classified as CS, EA, TRIG and H3N2 co-circulated and fluctuated in relative prevalence (Fig. 1b). After 2005, EA H1N1 viruses became dominant and H3N2 viruses disappeared, although CS H1N2 and TRIG H1N2 viruses continued to be isolated sporadically (Fig. 1b). All three SwIV H1 lineages (CS, EA and TRIG) have co-circulated with H1N1/2009 after the introduction of the latter virus into pigs5.

a, b, Percentage prevalence (a) and year-averaged relative frequency (b) of the major HA variants of SwIV. Colour codes and numbers of isolates (n) of H1 and H3 subtype viruses detected from swine in Hong Kong between 1998 and 2010 are shown. The viruses detected include CS, EA, TRIG, H1N1/2009 and human seasonal H1N1 and H3N2 viruses. The orange line indicates the number of pigs sampled during the surveillance period.
Comprehensive phylogenetic analyses of the genes encoding surface antigens haemagglutinin (HA) and neuraminidase (NA) in all H1N1 and H1N2 isolates (including 93 H1N1 viruses isolated during 1976–79 and 1993–94) confirmed that most isolates belonged to the CS or EA lineages (Fig. 2 and Supplementary Fig. 2a–c). All pre-1998 viruses were CS except two ‘pure avian’ viruses from 1993 (ref. 13); much greater HA diversity was observed after 1998 (Fig. 2a). Notably, our CS isolates do not form a single monophyletic group; rather, they are interspersed with North American CS viruses, indicating multiple introductions of CS into the study area. In contrast, EA viruses are monophyletic, indicating a single introduction. All Hong Kong TRIG viruses form a single group except for isolate Sw/HK/78/2003, indicating that this virus was introduced from North America separately. The Hong Kong TRIG viruses diverge from the North American TRIG and H1N1/2009 HA lineages soon after the emergence of TRIG H1N2 viruses in North America and thus constitute a third distinct TRIG HA gene lineage (Fig. 2a)14.

a–c, Haemagglutinin H1 (a), neuraminidase N1 (b), neuraminidase N2 (c). Representative avian, swine and human influenza A viruses obtained from GenBank are represented by black, dark grey and light grey, respectively. Colour codes of H1N1 and H1N2 subtype viruses detected from swine during 1977–2010 in Hong Kong are shown in the key. Arrow A indicates the antigenically divergent EA-reassortant viruses discussed in the text (for example, Sw/HK/72/2007, Fig. 4). Scale bars represent substitutions per site. Fully detailed phylogenies including sequence names are provided in Supplementary Fig. 2a–c.
Molecular clock phylogenies of 221 whole genomes (33.2% of isolates) revealed that CS viruses isolated from 1976 to 1994 contained only CS genome segments: no reassortment was detected during this period (Fig. 3a and Supplementary Fig. 3). However, several reassortant SwIV were detected between 1998 and 2010. In addition to the CS, EA, TRIG and H1N1/2009 viruses, we detected 14 ‘genotypes’ generated by reassortment between circulating swine and human/avian lineages (Supplementary Figs 3 and 4). Most of the newly identified reassortants were observed only transiently and usually contained genome segments from viruses that were dominant at that time (Supplementary Fig. 4). Excepting the CS H1N2 virus, which acquired the human H3N2 neuraminidase gene repeatedly, we detected no preferential direction of horizontal gene transfer among SwIV strains.

a, CS; b, EA. Coloured boxes adjacent to tips show the lineage classification of each gene segment of SwIV isolated in this study. Arrows indicate the long branches that lead to newly detected reassortant SwIV. Purple node bars represent 95% credible intervals of lineage divergence times. A fully detailed HA phylogeny including sequence names is shown in Supplementary Fig. 3a.
Three of the 14 reassortant genotypes were isolated repeatedly (Supplementary Fig. 4); specifically, (1) CS H1N2 viruses, (2) novel H1N2 reassortants and (3) Sw/HK/72/2007-like (EA-reassortant) strains, which have acquired an NS gene from TRIG viruses and which belong to a divergent EA lineage (Fig. 2). Since their initial detection in 2007, EA-reassortant viruses have become the dominant EA lineage, constituting 12.5% of all EA viruses in 2007, 15.4% in 2008 and 41.4% in 2009. Because most reassortant ‘genotypes’ were isolated only once, we hypothesize that few are adapted for continuous circulation (although we cannot exclude stochastic demographic effects or sampling bias as alternative explanations). SwIV diversity in our population is probably increased by pig movements: breeding pigs constitute the bulk of live pigs imported into China and data indicate that imports have increased since 1990 (refs 11,12).
For all genome segments, molecular clock phylogenies exhibited long branches leading to several reassortant lineages (Fig. 3). This was also observed for the H1N1/2009 virus4 and indicates a long period of unsampled diversity. Upon first detection, these reassortant lineages tend to be more closely related to viruses circulating in our population five to eleven years previously, rather than to co-circulating strains (that is, they do not arise from the contemporaneous part of the phylogenetic ‘backbone’).
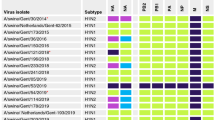
We found extensive antigenic crossreaction among CS, TRIG and H1N1/2009 viruses (Supplementary Table 2 and Supplementary Fig. 5). Ferret antisera to these viruses also crossreacted with early EA viruses (2001–03) but reacted poorly with more recent EA-reassortant strains (Fig. 4). Interestingly, the six novel EA-reassortant viruses tested (sampled between 2007 and 2009) crossreacted weakly with all ferret antisera used, including the antiserum to Sw/HK/NS29/2009. This group thus represents an antigenically distinct SwIV lineage. Excepting the earliest EA reassortant (Sw/HK/72/2007), all remaining EA reassortants reacted well against antisera raised to the EA-reassortant virus Sw/HK/1559/2009 (Fig. 4), indicating progressive antigenic change of EA-reassortant viruses during our study.

Titres are shaded according to their respective major SwIV HA lineages (see Figs 1–3); low (1:20, 1:40) and non-reactive titres (<1:10) are shaded in lighter colours. Underlined values represent homologous antibody titres. EA-reassortant viruses (indicated by asterisks) showed poor crossreactivity against antisera raised towards CS, TRIG, H1N1/2009 and late (2006–2009) ‘pure’ EA viruses. Excepting the earliest EA-reassortant virus (Sw/HK/72/2007), all remaining EA-reassortants reacted well against antisera raised towards the EA-reassortant virus Sw/HK/1559/2009, indicating progressive antigenic change of this novel reassortant.
The earliest of the above-mentioned EA reassortants (Sw/HK/72/2007) had acquired two amino acid changes in HA antigenic sites and later EA reassortants (for example, Sw/HK/1559/2008 and Sw/HK/1532/2009) had a further five changes at antigenic sites (Supplementary Figs 3a and 6). These findings support the hypothesis that EA-reassortant viruses have antigenically drifted away from crossreacting antibodies arising from CS, TRIG and early-EA virus infection. The observation that antigenic change occurred in the reassortant EA virus lineage rather than in the parental lineage indicates that reassortment may facilitate the generation of SwIV antigenic diversity.
Although SwIV isolation rates declined after EA viruses became predominant (Fig. 1), serological data indicate that overall SwIV seroprevalence has not declined (Supplementary Tables 3 and 4). To test whether EA viruses have a competitive advantage over CS strains, we intranasally infected five-week-old, previously influenza-naive pigs with SwIV representative of the lineages isolated here (Methods and Supplementary Fig. 7). EA viruses showed the highest and most prolonged virus shedding, closely followed by TRIG viruses; CS viruses showed lower peak viral titres. Thus, the replicative advantage of EA viruses, together with the low prevalence of crossreactive antibodies to EA in swine (15% in 2000, 26% in 2004; Supplementary Table 4), may help to explain the replacement of other SwIV lineages with EA viruses.
We tracked the evolution in our EA viruses of amino acids previously associated with adaptation of avian influenza to other species15,16,17. Purported avian residues were maintained at most of these sites (Supplementary Table 5) despite the circulation of these viruses in swine for more than 30 years18,19. However, the PDZ- (post-synaptic density protein, Drosophila disc large tumor suppressor and zonula occludens-1 protein) ligand at the 3′ end of EA virus non-structural (NS) 1 genes showed significant host-specific evolution: early European EA viruses had the avian ESEV motif, with a change to GSEV/GPEV motifs observed in several hosts. By 1999, most viruses sampled had the GPKV motif previously described from pigs16. CS and TRIG viruses that contributed the NS gene to H1N1/2009 have a truncated NS gene, as do the antigenically variant Sw/HK/72/2007-like viruses. The role of the truncated NS gene in inter-species transmission clearly merits further study. Furthermore, a modest but significant (P < 0.01) change in selection pressure was observed between European EA viruses isolated shortly after cross-species transmission (non-synonymous to synonymous (dN/dS) substitution rate ratio of 0.24; 95% confidence interval = 0.22–0.27) and those isolated later (dN/dS = 0.17; 95% confidence interval = 0.14–0.20), consistent with the hypothesis that host-specific selection increased viral adaptation after the introduction of EA viruses into swine (Supplementary Table 6).
Our unique longitudinal study reveals a genetically and antigenically dynamic SwIV population within a single region and provides a baseline for future studies of the virus elsewhere. The epidemiology and evolution of SwIV seem to be strongly shaped by gene flow among continents and species, facilitating the reassortment of diverse lineages and occasionally resulting in antigenic change. Although we confirm that the H1N1/2009 virus was not generated within our study’s catchment, the processes of lineage emergence, importation, reassortment and replacement described here are probably representative of the H1N1/2009 source population. We show that reassortments between EA and TRIG viruses do occur, generating reassortants that establish themselves as stable lineages in swine. SwIV reassortants containing H1N1/2009-like genome segments have also been transiently detected5,20.
Despite clear evidence of inter-continental SwIV movement, gene flow is not so frequent that the global SwIV population acts as a single gene pool (as observed for human influenza A21,22); instead a higher diversity of mammalian-adapted viruses in global swine populations is supported. Crucially, the co-circulation of multiple SwIV lineages facilitates the production of new genomic combinations. The evolutionary consequences of increased SwIV movement are hard to predict but require consideration given an increasingly globalized future.
Our study reveals a frequent generation of new reassortants but the survival and persistence of only a few, a process we term ‘recombinant chatter’23. Our data also indicate that reassortment and antigenic change are linked. This phenomenon was described in North America CS viruses after the events that generated the TRIG viruses24; it has also been observed in human influenza21. After reassortment, evolution in HA antigenic domains may arise for several reasons: (1) because of herd-immune selection pressure; (2) because those residues are under weak selective constraint; or (3) to compensate for fitness costs of mutations accruing elsewhere in the genome. The role of reassortment in driving genome-wide evolution requires detailed investigation.
We found that the quantitative dynamics of SwIV genomic diversity and lineage turnover (Supplementary Fig. 8) are slower, less periodic and less predictable than the repeated annual replacements typically seen for human influenza A. The reasons for SwIV lineage change are unclear: previously, selection arising from herd immunity was considered less important for pigs than for humans because the short lifespan of farmed swine (∼150 days) lowers the chance of re-infection, reducing the cross-protection that probably drives antigenic drift. Furthermore, maternally acquired swine immunity does not seem to interrupt SwIV infection or transmission, despite masking clinical illness25. Our surveillance data, animal infection experiments and serological data show that one reason for lineage change may be a competitive advantage of EA over CS and TRIG viruses.
The hypothesis that pigs are important in pandemic emergence, as facilitators of reassortment among influenza viruses26, has regained favour after the emergence of H1N1/2009. This virus represented a subtype already endemic in humans, implying that other H1 and H3 viruses prevalent in swine are credible pandemic candidates, especially when corresponding immunity in humans is absent. Indeed, we found that there are other swine viruses (for example, Sw/HK/NS29/09) to which humans lack herd immunity (Supplementary Table 7). Hence, future assessment of zoonotic potential must combine the evaluation of crossreactive immunity in humans, the assessment of transmissibility in animal models and ongoing surveillance of SwIV genetic diversity. The H1N1/2009 virus has already infected swine and reassorted with other SwIV, indicating that circulating SwIV will continue to acquire novel non-SwIV genes5 (notably, avian viruses such as H9N2 and H5N1 are occasionally detected in swine in Asia6,8,9,27). Avian-to-swine and swine-to-human host adaptation of influenza viruses are both poorly understood in comparison to avian-to-human adaptation and are a priority for future research.
Methods Summary
Systematic swine influenza A virus surveillance was initiated in May 1998 at a central slaughterhouse in Hong Kong. During 1999–2007, about 15–20% of the pigs were farmed locally in Hong Kong and the remainder were imported from several provinces in China; however, since 2008 the proportion of locally produced pigs fell to 5% (see Supporting Information). About 128 nasal and tracheal swabs were collected twice monthly from August 1998 to April 2009; since May 2009, sample numbers were doubled (Fig. 1a). Genes encoding surface proteins (HA and NA) were sequenced for all 573 H1N1 viruses isolated from 1998 to 2010 and for 93 swine H1 viruses from our repository, isolated during the periods 1976–1978 and 1993–1994. Full genome sequencing was carried out for 221 representative viruses. To estimate the genetic diversity and the level of gene reassortment, phylogenetic trees were constructed for each genomic segment independently (Supplementary Fig. 2). On the basis of the phylogenetic relationships of each gene segment, major swine virus lineages circulating in Hong Kong were identified and a more detailed Bayesian phylogenetic analysis for each lineage was conducted, thereby estimating rates of viral evolution and dates of divergence (Supplementary Fig. 3).
Online Methods
Surveillance
Systematic influenza surveillance was conducted from May 1998 until January 2010 in swine at an abattoir in Hong Kong, where tracheal or nasal swabs were collected fortnightly from slaughtered swine. During 1999–2007, about 15–20% of the pigs were farmed locally in Hong Kong and the remainder were imported from several provinces in China; however, since 2008 the proportion of locally produced pigs fell to 5% (see Supporting Information). Serum samples were collected from 50 slaughtered pigs each month. Routine virological surveys were also conducted in Hong Kong in 1977–79 and 1993–94. Swab materials were inoculated into nine-to-ten-day-old embryonated chicken eggs and Madin Darby canine kidney (MDCK) cells; virus isolates were identified and subtyped by haemagglutination inhibition assays as previously described24.
Virus isolation and sequencing
Viral RNA extraction, complementary DNA synthesis, PCR and sequencing were carried out as described5,28. Viral RNA was extracted directly from infected allantoic fluid or cell culture using the QIAamp viral RNA minikit (Qiagen). cDNA was synthesized by reverse transcription; gene amplification by PCR was performed using specific primers for each gene segment. PCR products were purified with the QIAquick PCR purification kit (Qiagen) and sequenced using synthetic oligonucleotides. Reactions were performed using the Big Dye-Terminator v3.1 Cycle Sequencing Reaction Kit on an ABI PRISM 3700 DNA Analyser (Applied Biosystems) following the manufacturer’s instructions. All sequences were assembled and edited with Lasergene version 6.1 (DNASTAR). The HA and NA genes were sequenced for all viruses collected in this study and full genome sequencing was conducted for representative viruses, selected on the basis of HA and NA gene diversity and including representative viruses sampled on each positive sampling occasion. All novel reassortants detected on the basis of full genome sequencing were subjected to plaque cloning and full genome sequencing (of at least six randomly selected clones per virus) to confirm that the reassortant was not an artefact of mixed infection.
Antigenic analyses
The antigenic characteristics of SwIV were compared using a haemagglutination inhibition assay with ferret antisera raised against representative influenza A viruses. Ferret antisera raised against Sw/HK/4167/1999 (CS H1N1), Sw/HK/1110/2006 (TRIG H1N2), Sw/HK/NS29/2009 (EA H1N1) and A/California/4/2009 were produced at the Department of Infectious Diseases at St Jude Children’s Research Hospital, Memphis, Tennessee and the Department of Microbiology, The University of Hong Kong. The haemagglutination inhibition assay started at 1:40 dilutions for ferret antisera.
To detect antibody prevalence towards major SwIV lineages, we used the haemagglutination inhibition assay with five representative viruses including the antigenically divergent Sw/HK/72/2007-like EA viruses. This allowed us to quantify changes in seroprevalence in serum collected from swine during 2000, 2004, 2009 and 2010.
Experimental infection of pigs
To characterize in vivo replicative behaviour of viruses from the major SwIV lineages, we experimentally infected local domestic hybrid (Putian white and Nianbian variant) pigs (Sus scrofa domesticus) obtained from a commercial herd and confirmed to be sero-negative and free of influenza virus by HI assays and virus isolation in MDCK cells. Pigs were infected with representative strains belonging to the CS (Sw/HK/4167/1999, Sw/HK/1304/2003), TRIG (Sw/HK/1110/2006), EA (Sw/HK/NS29/2009) and novel EA-reassortant (Sw/HK/72/2007) lineages isolated in this study. Two five-week-old pigs (one male and one female) were intranasally infected with 1 ml of Eagle’s minimal essential medium (MEM) containing 106 50% tissue culture infectious doses (TCID50) of a virus strain. Nasal swabs were collected for 14 d after inoculation from each piglet and placed in 0.6 ml of virus transport medium. Virus shedding in the nasal swabs of pigs was calculated in MDCK by the 50% end-point method29 and was expressed as TCID50 ml−1 of swab. Animal experiments were carried out in biosafety level three containment facilities at 20–21 °C and 76.5 ± 2.1% relative humidity. Experiments were approved by the Shantou University Medical College and conducted in compliance with university guidelines on animal ethics and welfare.
Molecular evolution and adaptation
Global dN/dS rate ratios for each Hong Kong swine lineage and the haemagglutinin gene of European EA viruses were estimated using the codon-based single likelihood ancestor counting method30. To determine whether selection was acting differentially on major lineages, the dN/dS rate ratio estimate for a lineage was enforced to other co-circulating lineages. A likelihood ratio test was conducted to evaluate whether this fit was significantly worse than unconstrained analysis (and vice versa), with a critical P value of 0.01. This test was repeated using the upper and lower limits of the confidence interval.
Phylogenetic analyses
Phylogenetic trees were inferred using the neighbour-joining method, using genetic distances calculated by maximum likelihood under the Hasegawa, Kishino and Yano (HKY) model with gamma-distributed among-site rate variation (HKY+Γ). The parameters of this model were estimated using maximum likelihood on an initial tree. Temporal phylogenies and rates of evolution were inferred using a ‘relaxed molecular clock’ model that allows evolutionary rates to vary among lineages in a Bayesian Markov chain Monte Carlo (MCMC) framework31 This was used to sample phylogenies and dates of divergence while constraining each sequence to its known date of sampling. A model comprising codon-position-specific HKY+Γ substitution models was used. For all analyses employing Bayesian MCMC sampling, a chain length of at least 50 million steps was used with a 10% ‘burn-in’ removed. At least two independent runs of each chain were performed and compared to ensure adequate sampling. To estimate changes in genetic diversity during our sampling period we used a coalescent-based flexible demographic model32 to the above MCMC approach. An estimate of the relative genetic diversity (Net, where Ne is the effective population size and t is the generation time) is obtained by integrating uncertainty across the tree topologies.
Accession codes
Primary accessions
GenBank/EMBL/DDBJ
Data deposits
Sequences generated in this study have been deposited with GenBank under the accession numbers CY084470–CY085121, CY085301–CY086876 and CY087041–CY087142.
References
Shinde, V. et al. Triple-reassortant swine influenza A (H1) in humans in the United States, 2005–2009. N. Engl. J. Med. 360, 2616–2625 (2009)
Smith, G. J. D. et al. Dating the emergence of pandemic influenza A viruses. Proc. Natl Acad. Sci. USA 106, 11709–11712 (2009)
Garten, R. J. et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 325, 197–201 (2009)
Smith, G. J. D. et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 459, 1122–1125 (2009)
Vijaykrishna, D. et al. Reassortment of pandemic H1N1 viruses in swine. Science 328, 1529 (2010)
Peiris, J. S. M. et al. Cocirculation of avian H9N2 and contemporary “human” H3N2 influenza A viruses in pigs in southeastern China: potential for genetic reassortment? J. Virol. 75, 9679–9686 (2001)
Yu, H. et al. Genetic evolution of swine influenza A (H3N2) viruses in China from 1970–2006. J. Virol. 46, 1067–1075 (2008)
Cong, Y. L. et al. Antigenic and genetic characterization of H9N2 swine influenza viruses in China. J. Gen. Virol. 88, 2035–2041 (2007)
Li, H. Y. et al. Isolation and characterization of H5N1 and H9N2 influenza viruses from pigs in China. Chinese J. Vet. Prev. Med. 26, 1–6 (2004)
Clements, A. C. A., Pfeiffer, D. U., Otte, M. J., Morteo, K. & Chen, L. A global livestock production and health atlas (GLiPHA) for interactive presentation, integration and analysis of livestock data. Prev. Vet. Med. 56, 19–32 (2002)
Zhang, J. & Beckman, C. People’s Republic of China: Agricultural situation: Livestock and Products 2008. (USDA Foreign Agriculture Service, 2008)
Wang, R. China – pork powerhouse of the world. Advances Pork Prod. 17, 33–46 (2006)
Guan, Y. et al. Emergence of avian H1N1 influenza viruses in pigs in China. J. Virol. 70, 8041–8046 (1996)
Lorusso, A. et al. Genetic and antigenic characterization of H1 influenza viruses from United States swine from 2008. J. Gen. Virol. 92, 919–930 (2011)
Finkelstein, D. B. et al. Persistent host markers in pandemic and H5N1 influenza viruses. J. Virol. 81, 10292–10299 (2007)
Obenauer, J. C. et al. Large-scale sequence analysis of avian influenza isolates. Science 311, 1576–1580 (2006)
Taubenberger, J. K. et al. Characterization of the 1918 influenza virus polymerase genes. Nature 437, 889–893 (2005)
Pensaert, M., Ottis, K., Vanderputte, J., Kaplan, M. M. & Buchmann, P. A. Evidence for the natural transmission of influenza A virus from wild ducks to swine and its potential for man. Bull. World Health Organ. 59, 75–78 (1981)
Dunham, E. et al. Different evolutionary trajectories of European avian-like and classical swine H1N1 influenza A viruses. J. Virol. 83, 5485–5494 (2009)
Moreno, A. et al. Novel H1N2 swine influenza reassortant strain in pigs derived from the pandemic H1N1/2009 virus. Vet. Microbiol. 149, 472–477 (2011).
Rambaut, A. et al. The genomic and epidemiological dynamics of human influenza A virus. Nature 453, 615–619 (2008)
Russell, C. A. et al. The global circulation of seasonal influenza A (H3N2) viruses. Science 320, 340–346 (2008)
Wolfe, N. D., Dunavan, C. P. & Diamond, J. Origins of major human infectious diseases. Nature 447, 279–283 (2007)
Webby, R. J. et al. Evolution of swine H3N2 influenza viruses in the United States. J. Virol. 74, 8243–8251 (2000)
Loeffen, W. L. A., Heinen, P. P., Bianchi, A. T. J., Hunneman, W. A. & Verheijden, J. H. M. Effect of maternally derived antibodies on the clinical signs and immune response in pigs after primary and secondary infection with an influenza H1N1 virus. Vet. Immunol. Immunopathol. 92, 23–35 (2003)
Scholtissek, C., Hinshaw, V. S. & Olsen, C. W. Influenza in pigs and their role as the intermediate host. In Textbook of Influenza (eds Nicholson, K. G., Webster, R. G. & Hay, A. J. ) 137–145 (Blackwell Scientific, 1998)
Nidom, C. A. et al. Influenza A (H5N1) viruses from pigs, Indonesia. Emerg. Infect. Dis. 16, 1515–1523 (2010)
Poon, L. L. M. et al. Rapid detection of reassortment of pandemic influenza H1N1. Clin. Chem. 56, 1340–1344 (2010)
Reed, L. J. & Muench, H. A. Simple method of estimating fifty percent endpoints. Am. J. Hyg. 27, 493–497 (1938)
Kosakovsky Pond, S. L. & Frost, S. D. W. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 22, 1208–1222 (2005)
Drummond, A. J., Ho, S. Y., Phillips, M. J. & Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 4, e88 (2006)
Drummond, A. J., Rambaut, A., Shapiro, B. & Pybus, O. G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22, 1185–1192 (2005)
Acknowledgements
This research was supported in part by the National Institute of Allergy and Infectious Diseases (NIAID) contract HHSN26600700005C and the Area of Excellence Scheme of the University Grants Commission (grant AoE/M-12/06) of the Hong Kong SAR Government. We acknowledge the Food and Environmental Hygiene Department of Hong Kong for facilitating the study. We acknowledge support from The Royal Society of London (O.G.P.), UK COSI (S.B.), NIAID (G.J.D.S.), the Agency for Science, Technology and Research and the Ministry of Health, Singapore (D.V., G.J.D.S and J.B.). We thank C. Y. H. Leung for producing some of the ferret antisera used in this study.
Author information
Authors and Affiliations
Contributions
J.S.M.P. and Y.G. conceived the study, conducted surveillance, performed analyses and co-wrote the paper. D.V., G.J.D.S. and O.G.P. conceived the study, performed analyses, co-wrote the paper and contributed equally to this work. H.Z., S.B., L.L.M.P., S.R., J.B., R.A.P.M.P. and H.C. performed analyses, S.K.M. conducted surveillance, C.L.C. conducted sequencing, K.F.S. and R.G.W. initiated surveillance in 1976 and provided viruses and R.J.W. provided viruses and reagents. All authors commented on and edited the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
This file contains Supplementary Figures 1-8 with legends, Supplementary Tables 1-8, Supplementary Notes and additional references. (PDF 14791 kb)
Rights and permissions
About this article
Cite this article
Vijaykrishna, D., Smith, G., Pybus, O. et al. Long-term evolution and transmission dynamics of swine influenza A virus. Nature 473, 519–522 (2011). https://doi.org/10.1038/nature10004
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature10004
This article is cited by
-
Quasispecies and viral gene expression analysis of the influenza A virus H1N1 strains isolated from human, mallard duck and pig
Biologia (2023)
-
Evidence for the circulation of genotype 4 Eurasian avian-like lineage swine H1N1 influenza A viruses on Korean swine farms, obtained using a newly developed one-step multiplex RT-qPCR assay
Archives of Virology (2023)
-
Are pigs overestimated as a source of zoonotic influenza viruses?
Porcine Health Management (2022)
-
Genetic characterization and pathogenicity of a Eurasian avian-like H1N1 swine influenza reassortant virus
Virology Journal (2022)
-
Rapid detection of influenza A (H1N1) virus by conductive polymer-based nanoparticle via optical response to virus-specific binding
Nano Research (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
