
Abstract
Although carbon tetrachloride (CCl4)-induced acute and chronic hepatotoxicity have been extensively studied, little is known about the very early in vivo effects of this organic solvent on oxidative stress and mitochondrial function. In this study, mice were treated with CCl4 (1.5 ml/kg ie 2.38 g/kg) and parameters related to liver damage, lipid peroxidation, stress/defense and mitochondria were studied 3 h later. Some CCl4-intoxicated mice were also pretreated with the cytochrome P450 2E1 inhibitor diethyldithiocarbamate or the antioxidants Trolox C and dehydroepiandrosterone. CCl4 induced a moderate elevation of aminotransferases, swelling of centrilobular hepatocytes, lipid peroxidation, reduction of cytochrome P4502E1 mRNA levels and a massive increase in mRNA expression of heme oxygenase-1 and heat shock protein 70. Moreover, CCl4 intoxication induced a severe decrease of mitochondrial respiratory chain complex IV activity, mitochondrial DNA depletion and damage as well as ultrastructural alterations. Whereas DDTC totally or partially prevented all these hepatic toxic events, both antioxidants protected only against liver lipid peroxidation and mitochondrial damage. Taken together, our results suggest that lipid peroxidation is primarily implicated in CCl4-induced early mitochondrial injury. However, lipid peroxidation-independent mechanisms seem to be involved in CCl4-induced early hepatocyte swelling and changes in expression of stress/defense-related genes. Antioxidant therapy may not be an efficient strategy to block early liver damage after CCl4 intoxication.
Similar content being viewed by others


Main
The mechanisms of carbon tetrachloride (CCl4) hepatotoxicity have been thoroughly studied since the seventies using in vivo models of acute and chronic CCl4 poisoning as well as perfused livers and isolated or cultured hepatocytes.1 A consensus has emerged that CCl4 toxicity is a mutifactorial process involving the generation of CCl4-derived free radicals, lipid peroxidation, covalent binding to macromolecules, loss of calcium homeostasis, nucleic acid hypomethylation and inflammatory cytokines.1, 2, 3 Most of the studies related to the very early events of CCl4 toxicity (ie minutes to few hours) have been performed in vitro on isolated hepatocytes, showing early findings such as swelling of hepatocytes, disorganization of endoplasmic reticulum, mitochondrial morphological injury and increased levels of free cytoplasmic calcium.4, 5, 6 Consequently, little is known about the early effects of this organic solvent in vivo, particularly on mitochondrial function. It has been shown recently in a murine model of liver fibrosis that chronic administration of CCl4 for 6 weeks led to mitochondrial DNA (mtDNA) alterations, reduced glutathione (GSH) depletion and decreased aconitase activity.7 In that study, overexpression of Bcl-2 reduced liver fibrosis for the first 3 weeks of treatment by protecting hepatocytes against mitochondrial damage, but subsequently failed to prevent fibrosis with the persistence of the aggression. CCl4 is activated by cytochrome P450 (CYP)2E1, and very marginally by other CYPs (CYP2B and CYP3A), to form the trichloromethyl (CCl3.) free radical, which can react with oxygen to produce the trichloromethyl peroxy radical (CCl3OO.). Both radicals are highly reactive species that may covalently bind to macromolecules to form nucleic acid, protein and lipid adducts.1, 2, 3 However, the evidence for such interactions with liver DNA in vivo is limited.2 Indeed, several studies using radiolabeled CCl4 reported a modest binding to hepatocyte DNA,8, 9, 10 which was not confirmed by the identification of adducts by mass spectrometry.2 As regards lipids, CCl3OO. radical initiates lipid peroxidation by pulling out a hydrogen atom in the vicinity of a polyunsaturated fatty acid double bond. After propagation of the peroxidation process, lipids are finally degraded in small molecules such as malondialdehyde (MDA) or 4-hydroxynonenal (HNE), which are highly reactive aldehydes that can form protein and DNA adducts.10, 11 In this study, we used an in vivo model to explore the very early toxic events, particularly regarding mitochondria, occurring after CCl4 administration. Inhibition of CCl4 activation by the CYP2E1 inhibitor diethyldithiocarbamate (DDTC) and impairment of CCl4-induced lipid peroxidation by antioxidants allowed us to establish a direct link between lipid peroxidation and mitochondrial alterations.
MATERIALS AND METHODS
Animals and Treatments
Wild-type 7-week-old male mice Swiss CD1 weighing 28 to 30 g were purchased from Charles River (L’arbresle, France). Mice were accommodated in an animal care facility accredited by the French veterinary authorities and fed ad libitum on a normal diet (A04 biscuits, UAR, Villemoison sur Orge, France). After 1 week of acclimatization, animals received a single intraperitoneal injection of CCl4 (1 or 1.5 ml/kg, corresponding to 1.58 and 2.38 g/kg) and were killed 3 or 24 h later by cervical dislocation. In other sets of experiments, mice were pretreated intraperitoneally once a day for 4 days either with the CYP2E1 inhibitor DDTC (750 mg/kg dissolved in phosphate buffered saline) or with the vitamin E analog Trolox C (100 mg/kg dissolved in corn oil) or dehydroepiandrosterone (DHEA), a hormone with antioxidant properties (30 mg/kg dissolved in corn oil). The dosage of each compound has been chosen based on previous investigations.12, 13, 14, 15, 16 On the fourth day, 3 h after the last injection, mice received one injection of 1.5 ml/kg of CCl4 and were killed 3 or 24 h later. Control mice were injected with the same volume of each vehicle. Four independent series of experiments have been conducted with at least six mice per group for the 3 h timepoint and two separate series including at least six mice per group for the 24 h time-point. Blood was collected from the retro-orbitary sinus and the liver was rapidly removed, frozen in liquid nitrogen and kept at −80 °C or immediately processed for histology, electron microscopy and immuohistochemistry. All experiments were performed according to national guidelines for the use of animals in biomedical research and approved by the local Ethics Committee in Animal Experiment of Rennes 1 University.
Plasma Studies
Plasma alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities were measured on an automatic analyser AU2700 (Olympus Diagnostics, Rungis, France) with Olympus commercial Kits OSR6107 and OSR6109, respectively. Plasma interleukin-6 (Il-6) and tumor necrosis factor-alpha (TNF-α) levels were measured with the corresponding Mouse Duoset ELISA kits from R&D Systems Europe (Lille, France).
Liver Histology and Immunohistochemistry
For liver histology, samples were fixed with 10% buffered formalin, pH 7.4, and embedded in paraffin. Sections were stained with hematoxylin and eosin (H&E). For immunohistochemistry, paraffin-embedded tissue was cut at 4 μm, mounted on positively charged slides and dried at 58 °C for 60 min. Immunohistochemical staining was performed on the Discovery Automated IHC stainer using the Ventana DABMap detection kit (Ventana Medical Systems, Tucson, AZ). Briefly, endogen peroxidase was blocked with Inhibitor-D 3% H2O2 (Ventana) for 4 min at 37 °C. After rinsing, slides were incubated at 37 °C for 60 min with a 1:100 dilution of rabbit polyclonal anti-MDA (Alexis, Lausen, Switzerland) antibody. Signal enhancement was performed using a biotinylated goat anti-rabbit immunoglobin IgG (H+L) secondary antibody (Vector laboratory, Burlingame, CA). Slides were then counterstained for 4 min with hematoxylin and rinsed. After removal from the instrument, slides were manually dehydrated and coverslips were placed on the slides. To quantify swelling of hepatocytes, five areas next to the central vein have been selected in three different slides of control, CCl4-, DDTC- and DDTC+ CCl4-treated mice. The size of centrilobular hepatocytes for each group was calculated by dividing the surface area by the number of hepatocytes counted with the imaging software ‘NIS elements’. The ratio obtained for control mice was set at 100%.
Transmission Electron Microscopy
Mice liver was cut into 2 mm sections that were fixed in 2.5% glutaraldehyde for 1 h, extensively washed with 0.2 M cacodylate buffer and post fixed in 0.2% osmium tetroxide. After further rinses with cacodylate buffer, the samples were dehydrated with increasing concentrations of acetone and infiltrated in a mixture of acetone-Eponate (50/50) for 2 h and in pure Eponate for 3 h. Finally, the samples were embedded in DMP30-Eponate for 24 h at 60 °C. Sections (0.5 μm) were cut on a LEICA IC7 microtome and stained with toluidine blue. Ultrathin sections (90 nm) were obtained, placed on grids, stained with uranyl acetate and lead citrate and examined in a JEOL 1400 transmission electron microscope at 120 KV.
Lipid Peroxidation, Hepatic Glutathione and Enzymatic Activities
Lipid peroxidation was assessed by measuring thiobarbituric acid reactants (TBARs).7 GSH levels and aniline hydroxylase activity were determined in liver homogenates prepared in 0.1 M phosphate buffer, pH 7.4, as described before.17 Aconitase activity, which can be impaired by superoxide anion, was assessed in homogenates as previously described.7 To evaluate whether CCl4 treatment could induce early apoptosis, the activity of caspases 2, 3, 8 and 9 was measured using the fluorescent ApoAlert Caspase Profiling Assay Plate (Clontech, Mountain View, CA) as described before.18 A positive control (rat liver extract after ischemia reperfusion) was included in the assay and a specific inhibitor of each caspase was incubated with the protein extract to demonstrate the specificity of the signal.
Western Blot Analyses
The protein expression of CYP2E1, manganese superoxide dismutase (MnSOD), heme oxygenase 1 (HO-1), heat shock protein (Hsp) 70 and heat shock cognate protein (Hsc) 70 was studied by western blot. Liver homogenates were prepared from frozen fragments in NaCl/Tris-base buffer containing 0.1% SDS, 1% Nonidet P40 and protease inhibitors. After protein determination with the Lowry method, 30 μg protein were subjected to SDS 12% polyacrylamide gel electrophoresis. After transfer to nitrocellulose membrane, immunoblotting was carried out with antibodies against CYP2E1 (Euromedex, Souffelweyersheim, France), MnSOD (Tebu-bio, Le Perray en Yvelines, France), Hsp70 (Tebu-bio, Le Perray en Yvelines, France), HO-1 (Gentaur, Kampenhout, Belgium) and Hsc70 (Tebu-bio). Blots were then incubated with appropriate secondary antibodies and proteins were detected by enhanced chemiluminescence in a Chemi-smart imager (Fisher Scientific SAS, Illkirch, France). Quantification was performed with the BIO-1D software and protein expressions were normalized with Hsc70.
DNA Isolation and Long PCR Experiments
Total hepatic DNA was extracted with the Nucleospin Tissue Kit (Macherey Nagel, Hoerdt, France) and samples from treated and untreated mice were subjected to 0.7% agarose gel electrophoresis to detect a smear (reflecting necrosis) or internucleosomal fragmentation (traducing apoptosis).
Long PCR experiments allowed the detection of mtDNA lesions (adducts, abasic sites and DNA strand breaks) that hamper the progression of polymerases.19, 20 Indeed, this technique is based on the rationale that randomly distributed blocking lesions are more likely to hamper amplification of a long stretch of mtDNA than a very short fragment. The selected primers (Table 1) allowed the amplification of a 317-bp mtDNA short fragment and a 6365-bp mtDNA long fragment. PCR reactions were performed with the Expand Long PCR System (Roche Diagnostics, Indianapolis, IN), as recommended by the manufacturer. The thermocycler profile included initial denaturation at 94 °C for 2 min, 25 cycles of 94 °C for 45 s, 56 °C for 1 min and 68 °C for 10 min, and final extension at 68 °C for 7 min. PCR products were subjected to electrophoresis on ethidium bromide-containing agarose gels and the intensity of each PCR fragment was determined using a transilluminator (Fisher Scientific SAS) and BIO-1D software. The long/short mtDNA intensity ratio was thus calculated for each mouse.
RNA Isolation and Reverse Transcription
Total RNA was extracted with the SV total RNA isolation system (Promega, Madison, WI) that includes a direct DNAse treatment step. RNA quantity and purity were assessed with a Nanodrop ND-1000 spectrophotometer (Nyxor Biotech, Paris, France) and RNA quality was checked with a Bioanalyzer 2100 (Agilent Technologies, Massy, France). Total RNA (1 μg) was reversed transcribed into cDNA using the high capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA).
Real time Quantitative PCR (RT-qPCR)
RT-qPCR was performed on cDNA using the SYBR Green PCR Master Mix (Applied Biosystems) and the Step One Plus qPCR apparatus (Applied Biosystems). Primer pairs selected for each transcript are indicated in Table 1. Amplification of specific transcripts was confirmed by the melting curve profiles generated at the end of each run. Moreover, PCR specificity was further ascertained with an agarose gel electrophoresis by checking the length of the amplicons. Expression of the mouse ribosomal protein S6 (S6) was used as reference and the 2−ΔΔCt method was used to express the relative expression of each selected gene.
RT-qPCR was also carried out to determine mtDNA levels. To this end, mtDNA/nuclear (n)DNA ratio was assessed by performing the RT-qPCR directly on hepatic DNA (25 ng) with specific primers of mtDNA and nDNA. Primers were designed to amplify cytochrome c oxidase 2 (COX2) for mtDNA and S6 for nDNA (Table 1).
Citrate Synthase, Complex I and Complex IV Activities
Activity of citrate synthase and mitochondrial chain complexes I and IV were measured on liver homogenates at 30 °C using a Specord 205 Analytikjena spectrophotometer. Briefly, mouse livers were homogenized in a buffer containing 50 mM Tris HCl, 150 mM KCl, pH 7.5. After centrifugation for 20 min at 1000 g at 4 °C, complex IV, complex I and citrate synthase activities were measured in the supernatant. Complex IV activity was measured in 10 mM KH2PO4 buffer, pH 7.0, using 75 μM reduced cytochrome c. Complex I activity was measured in 100 mM KH2PO4 buffer, pH 7.5, containing 100 mM MgCl2, 50 mg/ml BSA, 10 mM KCN, 2.5 mg/ml antimycin, 25 mM decylubiquinone and with or without 0.6 mg/ml rotenone for the measurement of rotenone complex I sensitivity. After 4 min incubation, the reaction was started by adding 2 mM NADH and the rate of NADH disappearance was recorded at 340 nm. Citrate synthase was assayed by using 1 mM 5-5′-dithiobis(2-nitrobenzoic acid), 10 mM oxaloacetate, 10 mM acetyl-CoA and triton X-10. Complex IV activity was expressed in nmol of cytochrome c/min/mg protein, complex I activity in nmol of NADH/min/mg protein and citrate synthase activity in nmol of dithiobis(2-nitrobenzoic acid)/min/mg protein.
Statistical Analysis
Results are obtained from two or four separate experiments with at least six mice per group and data are means±s.e.m. of 12 to 28 mice in each group. Statistical analysis was performed by one-way ANOVA followed by a Fisher's test, using StatView 4.5.
RESULTS
Dose of CCl4 Inducing Moderate Hepatic Toxicity 3 h after Treatment
Hepatocyte cytolysis and liver damage were evaluated by ALT and AST measurement in plasma and by H&E staining of liver sections 3 and 24 h after treatment with 1 or 1. 5 ml/kg of CCl4. Plasma ALT levels were increased by 2 and 5-fold 3 h after the injection of 1 and 1.5 ml/kg of CCl4, respectively, and by 260 and 240-fold 24 h later (Figure 1a). Likewise, AST levels were augmented by 1.2 and 8-fold after 3 h with 1 and 1.5 ml/kg of CCl4 and by 90 and 100-fold 24 h later (Figure 1b). With both doses of CCl4, H&E staining showed swelled centrilobular hepatocytes 3 h after the treatment and large necrotic areas with few infiltrating inflammatory cells (as shown by the arrows in Figure 1c) after 24 h. Considering the moderate elevation of transaminases and pathological changes of the liver 3 h after the injection of 1.5 ml/kg of CCl4, this dose was selected for further experiments. Inflammatory mediators TNF-α (Figure 1d) and Il-6 (Figure 1e) were already increased in plasma 3 h after CCl4 administration by 3 and 3.5-fold respectively. TNF-α further increased (60-fold) whereas Il-6 leveled off (2.5-fold) 24 h later. CCl4 did not activate any of the tested caspases (ie caspase 2, 3, 8, 9) showing that no significant apoptosis could be detected in our model until 24 h (Table 2). Migration of total hepatic DNA on agarose gel showed an intact DNA 3 h after CCl4 treatment. In contrast, a smear of DNA was observed 24 h after CCl4 injection suggesting necrosis (Figure 1f). To further characterize alterations of liver cells, transmission electron microscopy was performed (Figure 2). At 3 h after CCl4 administration, important ultrastructural changes could already be observed consisting in the appearance of lipid droplets and lysosomes vacuolization, degranulation of components of the endoplasmic reticulum and clarification of the mitochondrial matrix. Moreover, some mitochondria have fused, some have lost their crests and the membrane of few of them has been disrupted as indicated by the black arrows in Figure 2d. Twenty four h later, the cells showed a markedly more altered morphology with numerous lipid droplets in the cytoplasm, tangled webs of endoplasmic reticulum and mitochondrial lysis. Most of the remaining mitochondria presented altered structure with internal vesicles and calcium crystalloids as previously described in rats (indicated by the white arrows in Figure 2f).5, 21

CCl4-induced liver toxicity 3 or 24 h after administration assessed by (a) ALT and (b) AST levels, (c) H&E staining, (d) TNF-α and (e) Il-6 plasma levels and (f) hepatic DNA integrity on agarose gel. Data are means±s.e.m. for 10–14 mice in each group.*Significantly different from control mice (P<0.05). H&E staining was performed on liver slices and representative photographs of the liver of mice treated for 3 or 24 h with 1 or 1.5 ml/kg CCl4 are shown at × 250 magnification. Arrows indicate inflammatory cell infiltrates. CV, centrilobular vein.

Liver ultrastructural changes assessed by transmission electron microscopy 3 or 24 h after administration of 1.5 ml/kg CCl4. (a, b) control mice liver; (c, d) CCl4-treated mice liver for 3 h and (e, f) CCl4-treated mice liver for 24 h. Magnification × 8000 for a, c and e and × 15 000 for b, d and f. Black arrows show the disruption of the mitochondrial membrane. White arrows indicate the presence of calcium crystalloids. lip, lipid droplet; ly, lysosome; mt, mitochondria; rer, rough endoplasmic reticulum.
Inhibition of CYP2E1 by DDTC
To determine the role of CCl3. free radical in CCl4 early toxicity, we inhibited its formation with DDTC, the primary metabolite of disulfiram and a well-known inhibitor of CYP2E1 in vitro and in vivo.22 As expected, CCl4 by itself reduced CYP2E1-supported aniline hydroxylase activity and protein expression by 85 and 50%, respectively, 3 h only after treatment (Figure 3a and b). Indeed, CCl4 is a CYP2E1 suicide substrate and CCl3. binding to the active site leads to its inactivation and degradation.23, 24 Moreover, CYP2E1 mRNA was decreased by 50% by CCl4 (Figures 3c). DDTC reduced CYP2E1 mRNA and protein by 30% and aniline hydroxylase activity by 55%. Pretreatment of mice with DDTC prevented CCl4-induced diminution of CYP2E1 mRNA, but not that of protein expression and activity (Figure 3). However, at the 24 h time point, DDTC completely prevented the major decrease in CYP2E1 levels and activity induced by CCl4 (Figure 3).

CYP2E1 inhibition by DDTC. CYP2E1 (a) mRNA expression, (b) protein expression and (c) activity were measured 3 and 24 h after CCl4 treatment (1.5 ml/kg), with or without pretreatment with DDTC. Transcript levels of CYP2E1 were measured by RT-qPCR. Expression of S6 was used to normalize mRNA levels. CYP2E1 protein expression was assessed by western blotting and normalized with Hsc70. Data are expressed relatively to the ratio found in control mice arbitrarily set at 1. Aniline hydroxylase activity supported by CYP2E1 was measured in liver homogenates and results are expressed as nmol/min/mg protein. Data are mean±s.e.m. for 18–28 mice (18 mice in DDTC and DDTC+CCl4 groups and 28 mice in control and CCl4 groups). *Significantly different from control mice (P<0.05). #Significantly different from CCl4-treated mice (P<0.05).
Prevention of CCl4-Induced Early Toxicity with DDTC
In order to evaluate early toxicity, plasma levels of aminotransferases were first measured 3 h after CCl4 administration. DDTC by itself did not change ALT or AST levels (Figure 4a). Pretreatment of mice with DDTC partially prevented the elevation of ALT (3-fold instead of 5-fold increase) and AST (3.5-fold instead of 8-fold). Of note, 24 h after CCl4 administration, DDTC totally prevented transaminase augmentations (Figure 4b). Swelling of centrilobular hepatocytes observed 3 h after CCl4 treatment was prevented by DDTC (Figure 4c). Indeed, quantitative image analysis showed that CCl4 treatment increased the size of centrilobular hepatocytes by 39% (139.2±3.3% vs 100.0±4.5% for control mice) and that pretreatment with DDTC significantly prevented this augmentation (105.6±7.1%).

Prevention by DDTC of CCl4-induced (a) ALT and (b) AST elevation and (c) histological modifications. H&E staining was performed on liver slices from eight mice per group. ALT and AST data are mean±s.e.m. for 18–28 mice (3 h time-point) or 9 mice (24 h time-point) per group. *Significantly different from control mice (P<0.05). #Significantly different from CCl4-treated mice (P<0.05). Representative pictures (magnification × 250) of the liver of control mice or mice treated for 3 h with 1.5 ml CCl4, with or without pretreatment with DDTC, as well as inlays of centrilobular hepatocytes at higher magnification ( × 500) are shown. CV, centrilobular vein.
We also assessed lipid peroxidation by TBARs measurement and histoimmunochemistry using an anti-MDA antibody. CCl4 is a prototypical lipid peroxidative agent that indeed induced early lipid peroxidation in liver. In CCl4-treated mice, TBARs were increased by 23% (Figures 5a) and anti-MDA antibody staining revealed that lipid peroxidation was predominant in centri- and mediolobular hepatocytes (Figure 5b). Pretreatment with DDTC completely prevented lipid peroxidation induced by CCl4 (Figures 5a and b). Noticeably, DDTC decreased TBARs levels by 26% below control values. Besides being a CYP2E1 inhibitor, DDTC is a copper chelator and possesses antioxidant properties,25, 26 which can explain the decrease of basal lipid peroxidation. As a consequence, inhibition of CCl4-induced lipid peroxidation by 37% may not only result from CYP2E1 inhibition but also from the other properties of DDTC.

Prevention by DDTC of CCl4-induced lipid peroxidation and GSH moderate depletion. Mice were treated or not treated for 3 h with 1.5 ml CCl4, with or without pretreatment with DDTC. Lipid peroxidation was assessed by (a) TBARs measurement and (b) immunohistochemistry using an anti-MDA antibody. (c) GSH was measured in liver homogenates and results are expressed as nmol/mg protein. Data are mean±s.e.m. for 18–28 mice per group. *Significantly different from control mice (P<0.05). #Significantly different from CCl4-treated mice (P<0.05).
We then measured total hepatic GSH and aconitase activity. CCl4 induced a minor GSH depletion (30.9±1.4 and 26.9±1.0* nmol/mg protein in control and CCl4-treated mice, respectively (*, significantly different from control mice, P<0.05%)), indicating that defense mechanisms were still effective after such a short treatment, whereas DDTC increased significantly GSH levels up to 37.3±1.8 nmol/mg protein. When CCl4-injected mice were pretreated with DDTC, GSH remained at the same level than with DDTC alone, ie 36.3±1.8 nmol/mg protein (Figure 5c). No modification of aconitase activity was found after CCl4 or DDTC administration (not shown).
To further assess oxidative stress, we measured mRNA and protein expression of several stress/defense-related genes, namely HO-1, Hsp70 and MnSOD. HO-1 and Hsp70 mRNA expression was highly induced after 3 h of CCl4 treatment probably to counteract CCl3. formation, but was almost normalized after 24 h. This transient induction was prevented by DDTC at both time points (Figures 6a and c). Regarding protein expression, inducible HO-1 was weakly expressed and almost unchanged 3 h after CCl4 treatment, but was strongly induced afterwards. DDTC by itself also induced HO-1 protein at the 24 h timepoint, but to a lower extent, and was able to counteract CCl4 action (Figure 6b). In our model, Hsp70 protein was constitutively expressed and was slightly increased by 20% 24 h after CCl4 treatment (Figure 6d). Concerning MnSOD, mRNA expression decreased in a time-dependent manner. This diminution, which was prevented by DDTC, had no impact on protein expression after 3 and 24 h of treatment (Figures 6e and f).

Effect of DDTC on CCl4-induced modifications of stress and defense-related proteins. Mice were treated or not for 3 and 24 h with 1.5 ml CCl4, with or without pretreatment with DDTC. Transcript levels of (a) HO-1, (c) Hsp70 and (e) MnSOD were measured by RT-qPCR. Expression of S6 was used to normalize mRNA levels. (b) HO-1, (d) Hsp70 and (f) MnSOD protein expression levels were assessed by western blotting and normalized with Hsc70. Data are expressed relatively to the ratio found in control mice arbitrarily set at 1 and are means±s.e.m. for 18 different mice in each group at the 3 h timepoint (three series with six mice per group) and for six mice per group at the 24 h timepoint. *Significantly different from control mice (P<0.05). #Significantly different from CCl4-treated mice (P<0.05).
Next, several mitochondrial parameters were investigated. MtDNA levels were assessed by measuring mtDNA/nDNA ratio by RT-qPCR whereas qualitative alterations were determined using long PCR amplification. A 30% depletion of mtDNA, prevented by DDTC, was observed 3 h after CCl4 administration (Figure 7a). Noticeably, the hepatic mtDNA pool was totally restored 24 h after CCl4 treatment (mtDNA/nDNA ratio was 1.04). This could result from reparation processes in the less injured mitochondria of centrilobular hepatocytes but also from compensation mechanisms in adjacent intact cells. Moreover, the long/short mtDNA fragment ratio was halved 3 h after CCl4 administration and this reduction was totally avoided by DDTC pretreatment (Figures 7b and c).

Prevention by DDTC of CCl4-induced mitochondrial dysfunction. Mice were treated or not for 3 h with 1.5 ml CCl4, with or without pretreatment with DDTC. MtDNA (a) depletion and (b) damage were assessed by RT-qPCR and long PCR, respectively. (c) A representative picture of an agarose gel with bands corresponding to the amplifications of the short (317 bp) and the long fragment (6365 bp) obtained by long PCR is shown. The activity of mitochondrial respiratory chain (d) complex I and (e) complex IV were measured by spectrophotometry. Data are mean±s.e.m. for 18–28 mice per group. *Significantly different from control mice (P<0.05). #Significantly different from CCl4-treated mice (P<0.05).
To further examine the effects of DDTC on CCl4-induced mitochondrial dysfunction, we analyzed the activity of three mitochondrial enzymes. We first assessed citrate synthase activity, which was not significantly modified by CCl4 or DDTC treatments indicating that mitochondrial mass remained unchanged during the experiment (not shown). We then measured the activity of complex I and IV of the mitochondrial respiratory chain. We previously found an inhibition of both complexes after chronic CCl4 intoxication.7 However, in the present model, complex I activity was not modified by CCl4 treatment whereas it was decreased by 30% by DDTC (Figure 7d). Disulfiram and DDTC have been reported to have deleterious effect on the mitochondrial electron transport system, and particularly on the respiration supported by complexes I and II.27, 28 In contrast, complex IV activity was severely reduced after only 3 h of CCl4 exposure, whereas DDTC pretreatment partially restored this activity from 15% to 60% of control values (Figure 7e). Interestingly, CCl4 treatment had no effect on mtDNA-encoded COX2 mRNA (not shown), thus indicating that decreased complex IV activity was not due to reduced protein synthesis.
Limited Protection Against CCl4-Induced Alterations with Antioxidants
DDTC blocks the CYP2E1-mediated generation of the CCl3. radical and subsequently lipid peroxidation and formation of protein, lipid or DNA adducts. To investigate the specific role of lipid peroxidation in the development of CCl4-induced early hepatotoxicity (ie 3 h after CCl4 administration), we tested the effects of two antioxidants on several parameters that were corrected by DDTC pretreatment. As expected, DHEA and Trolox C, inhibited lipid peroxidation induced by CCl4 (Table 3). However, both compounds failed to prevent CCl4-induced morphological changes in liver (Figure 8) and plasma ALT and AST elevation (Table 3). Interestingly, 24 h later, levels of ALT and AST in CCl4 mice pretreated with DHEA were still similar to those of CCl4-treated mice (12 450±2865 vs 17 818±4234 IU/l for ALT and 9253±1250 vs 10 385±834 IU/l for AST). Parameters reflecting CCl4-induced mitochondrial dysfunction such as complex IV inhibition, mtDNA depletion and mtDNA oxidative damage, were corrected by both antioxidants. In contrast, CCl4-induced early changes in CYP2E1, HO-1, Hsp70 and MnSOD mRNA expression were not prevented by DHEA and Trolox C (Table 3).

Effect of antioxidants on CCl4-induced histological modifications. H&E staining was performed on liver slices from eight mice per group. Representative pictures of the liver of control mice or mice treated for 3 h with 1.5 ml CCl4, with or without pretreatment with Trolox or DHEA are shown at × 250 magnification. CV, centrilobular vein.
DISCUSSION
In this study, we showed that CCl4 administration induced very early signs of hepatotoxicity in mice that were mediated through lipid peroxidation-dependent and independent mechanisms. In particular, we demonstrated that several mitochondrial alterations were directly linked to lipid peroxidation (Figure 9). A single dose of CCl4 induced swelling of hepatocytes, mild cytolysis and lipid peroxidation 3 h only after its administration. These events were accompanied by mtDNA depletion and damage, mitochondria ultrastructural alterations, inhibition of mitochondrial respiratory chain complex IV (also referred to as cytochrome c oxidase) activity as well as changes in the expression of different stress/defense-related proteins such as CYP2E1, HO-1, Hsp70 and MnSOD. However, our data suggested that CCl4-induced early toxicity was not associated with a major oxidative stress. Indeed, GSH levels were marginally decreased, aconitase activity was not affected and MnSOD protein expression was not enhanced, suggesting the absence of superoxide anion overproduction. Nevertheless, the increase in HO-1 and Hsp70 mRNAs 3 h after CCl4 administration indicated that CCl3. radical formation rapidly induced the release of stress and defense signals.

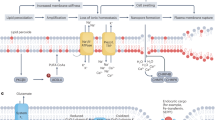
Scheme showing lipid peroxidation-dependent and -independent early toxicity induced by CCl4. Mitochondrial alterations including mtDNA damage and respiratory chain complex IV inhibition are prevented by both DDTC and antioxidants whereas mild cytolysis, swelling of centrilobular hepatocytes and increase of defense/stress-related enzymes are prevented only by DDTC. This suggests that mitochondrial alterations seen 3 h after the treatment result from lipid peroxidation induced by CCl4 metabolic activation. Other modifications seem to be directly linked to the formation of the free radicals CCl3. and CCl3OO. and do not depend on lipid peroxidation.
Pretreatment with the CYP2E1 inhibitor DDTC significantly alleviated CCl4-induced early hepatocyte morphological changes and concomitant transaminitis. Moreover, DDTC completely prevented liver lipid peroxidation, mtDNA alterations, cytochrome c oxidase inactivation as well as early induction of HO-1 and Hsp70 transcripts. On the other hand, although DHEA and Trolox C inhibited CCl4-induced lipid peroxidation, mtDNA alterations and mitochondrial dysfunction, these antioxidants failed to prevent early swelling of centrilobular hepatocytes, transaminitis as well as induction of HO-1 and Hsp70. Thus, although lipid peroxidation appeared to be involved in CCl4-induced early mtDNA damage and dysfunction, it might not be responsible for other hepatocytic events such as swelling and induction of stress/defense-related genes (Figure 9). Other investigations will be required to determine the role of CCl4-derived radical(s) in these lipid peroxidation-independent events.
In the current study, the preventive effect of the antioxidants DHEA and Trolox C against CCl4-induced lipid peroxidation was associated with a preservation of mtDNA integrity and complex IV activity, thus suggesting a direct relationship between these events. Indeed, previous data have shown that some lipid peroxidation end products such as MDA or 4-HNE are able to damage key mitochondrial constituents including mtDNA and some enzymes involved in mitochondrial respiration such as complex IV and α-ketoglutarate dehydrogenase.29, 30, 31, 32, 33, 34 Interestingly, Humphries et al showed that 4-HNE was unable to alter complex I activity,34 which may explain why this activity was not modified after CCl4 administration in our study. The mitochondrial genome is more susceptible to oxidative damage than nDNA due to the absence of histone, incomplete DNA repair capacity and proximity to the mitochondrial respiratory chain, which is the main intracellular source of reactive oxygen species.20, 35, 36 Previous studies on acute ethanol intoxication in mice reported a rapid fall of hepatic mtDNA levels which could be, at least in part, related to lipid peroxidation.37, 38, 39 Thus both ethanol and CCl4 induce lipid peroxidation and mtDNA depletion 2 or 3 h following their administration. The mechanisms by which mitochondrial genome lesions induced by lipid peroxidation (adducts, oxidized bases or single strand breaks) could cause mtDNA degradation and depletion are not well understood. However, as mitochondria do not possess the nucleotide excision repair pathway, damaged mtDNA molecules harboring numerous strand breaks and/or bulky adducts may be preferentially degraded by mitochondrial endonucleases thus leading to mtDNA depletion.20, 36, 40 Noteworthy, mtDNA pools were restored 24 h after CCl4 or ethanol administration,38 suggesting the presence of compensation and reparation processes in the liver. This rapid adaptive response could restrain the toxic consequences of mtDNA lesions after a single administration of CCl4 or ethanol whereas a repeat exposure could induce the accumulation of damaged mtDNA molecules, inhibiting the resynthesis and inducing prolonged mtDNA depletion.7, 20 In the present study, CCl4-induced early mtDNA depletion and damage were not associated with nDNA fragmentation, thus suggesting that mtDNA was specifically affected by lipid peroxidation products as previously described in vitro in isolated rat mitochondria.29 A direct role of CCl4-derived radicals cannot be completely excluded. Indeed, CYP2E1-mediated metabolism of CCl4 leads to the formation of CCl3. and CCl3OO. radicals, which are able to alkylate proteins and nucleic acids to generate CCl4-derived adducts.1, 2Anterior data from in vitro experiments with isolated hepatocytes indicated that mitochondrial related events such as reduction of intracellular ATP or perturbation of calcium homeostasis appeared as a result of attack by free radical metabolites rather than a consequence of lipid peroxidation.41 However, CCl4-derived radicals binding to mtDNA has been documented in vitro,10 but not demonstrated, or to a little extent, in vivo.2, 8 Thus, our study sustains the major role of lipid peroxidation in mitochondrial events following CCl4 administration in vivo. As the presence of CYP2E1 in liver mitochondria is now well established,17, 42, 43 it is conceivable that metabolic activation of CCl4 in situ may lead to lipid peroxidation initiation in the very close vicinity of mtDNA, thus favoring its degradation.
An interesting observation in our study was CCl4-induced early reduction of CYP2E1 mRNA levels. Moreover, decreased CYP2E1 mRNA expression was almost totally prevented by DDTC pretreatment but not by the antioxidants DHEA and Trolox C, thus suggesting that CYP2E1 transcripts could be specifically degraded by CCl4-derived radical(s). Previous data suggested that CYP2E1 mRNA is one of the most abundant transcript among CYPs (56% of total CYP mRNA), whereas the protein is relatively poorly expressed (7% of total CYP proteins).44, 45 Although this could reflect a particular ability of CYP2E1 transcripts to undergo significant degradation in normal circumstances,46 acute CCl4 intoxication could accelerate this putative CYP2E1 mRNA degradative pathway. Interestingly, current investigations in our laboratory also showed acetaminophen-induced early reduction of CYP2E1 mRNA levels (unpublished data). These data suggest that different types of reactive metabolites could trigger CYP2E1 transcript degradation. However, other mechanisms such as an inhibition of CYP2E1 transcription consequent to inflammatory stimulation could also have an important role in CYP2E1 mRNA decrease.47
In conclusion, our data suggest that CCl4-induced early signs of hepatotoxicity could be mediated through two different mechanisms involving or not lipid peroxidation (Figure 9). Lipid peroxidation triggered mtDNA degradation and mitochondrial dysfunction but not other CCl4-induced deleterious events such as hepatocyte swelling, abnormal expression of HO-1 and Hsp70, and reduction of CYP2E1 mRNA levels. Antioxidants such as Trolox and other vitamin E analogs have been shown to be hepatoprotective molecules in animal models of chronic toxicity where inflammation and fibrosis are primarily involved,48 but might not be as efficient on early signs of toxicity. In studies performed with rats, Trolox and other vitamin E analogs (α- and γ-tocopherol) partially or did not protect against CCl4-induced elevation of ALT 6 h after its administration.49, 50 In another work where the effect of vitamin E and IRFI 042, an analogous compound, were evaluated 48 h after CCl4 administration to rats, a partial reduction of ALT was observed with IRFI 042, but none with vitamin E.51 Thus, the present study and previous data52 clearly indicate that reducing CYP2E1 activity could be a better strategy than antioxidants to prevent CCl4-induced early hepatotoxicity.
References
Weber LW, Boll M, Stampfl A . Hepatotoxicity and mechanism of action of haloalkanes: carbon tetrachloride as a toxicological model. Crit Rev Toxicol 2003;33:105–136.
Manibusan MK, Odin M, Eastmond DA . Postulated carbon tetrachloride mode of action: a review. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev 2007;25:185–209.
Recknagel RO, Glendee Jr EA, Dolak JA, et al. Mechanisms of carbon tetrachloride toxicity. Pharmacol Ther 1989;43:139–154.
Berger ML, Reynolds RC, Combes B . Carbon tetrachloride-induced morphologic alterations in isolated rat hepatocytes. Exp Mol Pathol 1987;46:245–257.
Tyson CA, Story DL, Stephens RJ . Ultrastructural changes in isolated rat hepatocytes exposed to different CCl4 concentrations. Biochem Biophys Res Commun 1983;114:511–517.
Brattin WJ, Pencil SD, Waller RL, et al. Assessment of the role of calcium ion in halocarbon hepatotoxicity. Environ Health Perspect 1984;57:321–323.
Mitchell C, Robin MA, Mayeuf A, et al. Protection against hepatocyte mitochondrial dysfunction delays fibrosis progression in mice. Am J Pathol 2009;17:1929–1937.
Rocchi P, Prodi G, Grilli S, et al. In vivo and in vitro binding of carbon tetrachloride with nucleic acids and proteins in rat and mouse liver. Int J Cancer 1973;11:419–425.
Diaz Gomez MI, Castro JA . Covalent binding of carbon tetrachloride metabolites to liver nuclear DNA, proteins, and lipids. Toxicol Appl Pharmacol 1980;56:199–206.
Levy GN, Brabec MJ . Binding of carbon tetrachloride metabolites to rat hepatic mitochondrial DNA. Toxicol lett 1984;22:229–234.
Kadiiska MB, Gladen BC, Baird DD, et al. Biomarkers of oxidative stress study II: are oxidation products of lipids, proteins, and DNA markers of CCl4 poisoning? Free Radic Biol Med 2005;38:698–710 .
Deamer NJ, Genter MB . Olfactory toxicity of diethyldithiocarbamate (DDTC) and disulfiram and the protective effect of DDTC against the olfactory toxicity of dichlobenil. Chem Biol Interact 1995;95:215–226.
Li L, Xu B, Zhu Y, et al. DHEA prevents Abeta(25-35)-impaired survival of newborn neurons in the dentate gyrus through a modulation of PI3K-Akt-mTOR signaling. Neuropharmacology 2010;59:323–333.
Ben-Nathan D, Lachmi B, Lustig S, et al. Protection by dehydroepiandrosterone in mice infected with viral encephalitis. Arch Virol 1991;120:263–271.
Diaz Z, Laurenzana A, Mann KK, et al. Trolox enhances the anti-lymphoma effects of arsenic trioxide, while protecting against liver toxicity. Leukemia 2007;21:2117–2127.
Silver PJ, Gordon RJ, Horan PJ, et al. Low molecular weight analogs of trolox with potent antioxidant activity in vitro and in vivo. Drug Dev Res 1992;27:45–52.
Robin MA, Sauvage I, Grandperret T, et al. Ethanol increases mitochondrial cytochrome P450 2E1 in mouse liver and rat hepatocytes. FEBS Lett 2005;579:6895–6902.
Fromenty B, Vadrot N, Massart J, et al. Chronic ethanol consumption lessens the gain of body weight, liver triglycerides, and diabetes in obese ob/ob mice. J Pharmacol Exp Ther 2009;331:23–34.
Fromenty B, Demeilliers C, Mansouri A, et al. Escherichia coli exonuclease III enhances long PCR amplification of damaged DNA templates. Nucleic Acids Res 2000;28:E50.
Demeilliers C, Maisonneuve C, Grodet A, et al. Impaired adaptive resynthesis and prolonged depletion of hepatic mitochondrial DNA after repeated alcohol binges in mice. Gastroenterology 2002;123:1278–1290.
Smuckler EA . Structural and functional chages in acute liver injury. Environ Health Perspect 1976;15:13–25.
Brady JF, Xiao F, Wang MH, et al. Effects of disulfiram on hepatic P450IIE1, other microsomal enzymes, and hepatotoxicity in rats. Toxicol Appl Pharmacol 1991;108:366–373.
Dai Y, Cederbaum AI . Inactivation and degradation of human cytochrome P4502E1 by CCl4 in a transfected HepG2 cell line. J Pharmacol Exp Ther 1995;275:1614–1622.
Tierney DJ, Haas AL, Koop DR . Degradation of cytochrome P450 2E1: selective loss after labilization of the enzyme. Arch Biochem Biophys 1992;14:9–16.
Susa N, Ueno S, Furukawa Y . Protective effect of diethyldithiocarbamate pretreatment on chromium (VI)-induced cytotoxicity and lipid peroxidation in primary cultures of rat hepatocytes. J Vet Med Sci 1997;60:71–76.
Liu J, Shigenaga MK, Yan LJ, et al. Antioxidant activity of diehtyldithiocarbamate. Free Radic Res 1996;24:461–472.
Kuroda MA, Cuellar A . Deleterious effects of disulfiram on the respiratory electron transport system of liver mitochondria. Int J Biochem 1993;25:87–91.
Bachurin SO, Shevtzova EP, Lermontova NN, et al. The effects of dithiocarbamates on neurotoxic action of 1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine (MPTP) and on mitochondrial respiratory chain. Neurotoxicology 1996;17:897–903.
Hruszkewycz AM . Evidence for mitochondrial DNA damage by lipid peroxidation. Biochem. Biophys Res Commun 1988;153:191–197.
Picklo MJ, Montine TJ . Acrolein inhibits respiration in isolated brain mitochondria. Biochim Biophys Acta 2001;1535:145–152.
Ikeda K, Toda M, Tanaka K, et al. Increase of lipid hydroperoxides in liver mitochondria and inhibition of cytochrome oxidase by carbon tetrachloride intoxication in rats. Free Radic Res 1998;28:403–410.
Chen J, Petersen DR, Schenker S, et al. Formation of malondialdehyde adducts in livers of rats exposed to ethanol: role in ethanol-mediated inhibition of cytochrome c oxidase. Alcohol Clin Exp Res 2000;24:544–552.
Chen J, Schenker S, Frosto TA, et al. Inhibition of cytochrome c oxidase activity by 4-hydroxynonenal (HNE). Role of HNE adduct formation with the enzyme subunits. Biochim Biophys Acta 1998;1380:336–344.
Humphries KM, Yoo Y, Szweda LI . Inhibition of NADH-linked mitochondrial respiration by 4-hydroxy-2-nonenal. Biochemistry 1998;37:552–557.
Croteau DL, Stierum RH, Bohr VA . Mitochondrial DNA repair pathways. Mutat Res 1999;434:137–148.
Begriche K, Massart J, Robin MA, et al. Drug-induced toxicity on mitochondria and lipid metabolism: Mechanistic diversity and deleterious consequences for the liver. J Hepatol 2011;54:773–794.
Mansouri A, Demeilliers C, Amsellem S, et al. Acute ethanol administration oxidatively damages and depletes mitochondrial dna in mouse liver, brain, heart, and skeletal muscles: protective effects of antioxidants. J Pharmaco Exp Ther 2001;298:737–743.
Mansouri A, Gaou I, De Kerguenec C, et al. An alcoholic binge causes massive degradation of hepatic mitochondrial DNA in mice. Gastroenterology 1999;117:181–190.
Larosche I, Lettéron P, Berson A, et al. Hepatic mitochondrial DNA depletion after an alcohol binge in mice: probable role of peroxynitrite and modulation by manganese superoxide dismutase. J Pharmacol Exp Ther 2010;332:886–897.
Rothfuss O, Gasser T, Patenge N . Analysis of differential DNA damage in the mitochondrial genome employing a semi-long run real-time PCR approach. Nucleic Acids Res 2010;38:E24.
Albano E, Bellomo G, Carini R, et al. Mechanisms responsible for carbon tetrachloride-induced pertubation of mitochondrial calcium homeostasis. FEBS Lett 1985;192:184–188.
Robin MA, Anandatheerthavarada HK, Fang JK, et al. Mitochondrial targeted cytochrome P450 2E1 (P450 MT5) contains an intact N terminus and requires mitochondrial specific electron transfer proteins for activity. J Biol Chem 2001;276:24680–24689.
Knockaert L, Fromenty B, Robin MA . Mechanisms of Mitochondrial Targeting of Cytochrome P450 2E1: physiopathological role in liver injury and obesity. FEBS J 2011;278:4252–4260.
Shimada T, Yamazaki H, Mimura M, et al. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther 1994;270:414–423.
Bièche I, Narjoz C, Asselah T, et al. Reverse transcriptase-PCR quantification of mRNA levels from cytochrome (CYP)1, CYP2 and CYP3 families in 22 different human tissues. Pharmacogenet Genomics 2007;17:731–742.
Mohri T, Nakajima M, Fukami T, et al. Human CYP2E1 is regulated by miR-378. Biochem Pharmacol 2010;79:1045–1052.
Riddick DS, Lee C, Bhathena A, et al. Transcriptional suppression of cytochrome P450 genes by endogenous and exogenous chemicals. Drug Metab Dispos 2004;32:367–375.
Galicia-Moreno M, Rodríguez-Rivera A, Reyes-Gordillo K, et al. Trolox down-regulates transforming growth factor-beta and prevents experimental cirrhosis. Basic Clin Pharmacol Toxicol 2008;103:476–481.
Yachi R, Igarashi O, Kiyose C . Protective effects of vitamin E analogs against carbon tetrachloride-induced fatty liver in rats. J Clin Biochem Nutr 2010;47:148–154.
Rudnicki M, Silveira MM, Pereira TV, et al. Protective effects of Passiflora alata extract pretreatment on carbon tetrachloride induced oxidative damage in rats. Food Chem Toxicol 2007;45:656–661.
Campo GM, Squadrito F, Ceccarelli S, et al. Reduction of carbon tetrachloride-induced rat liver injury by IRFI 042, a novel dual vitamin E-like antioxidant. Free Radic Res 2001;34:379–393.
Wong FW, Chan WY, Lee SS . Resistance to carbon tetrachloride-induced hepatotoxicity in mice which lack CYP2E1 expression. Toxicol Appl Pharmacol 1998;153:109–118.
Acknowledgements
We are grateful to Pascale Bellaud from the Histopathology platform for her excellent technical support and to Bruno Turlin for the expertise in histopathology. We also acknowledge Agnès Burel for technical expertise in transmission electronic microscopy (IFR140, Microscopy Rennes Imaging Center). This work was supported by INSERM (Institut Nationale de la Santé et de la Recherche Médicale). Laetitia Knockaert was a recipient of a co-fellowship from INSERM and Région Bretagne.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Although lipid peroxidation is implicated in early mitochondrial injury associated with liver fibrosis, lipid peroxidation-independent mechanisms are responsible for early hepatocyte swelling and changes in expression of stress/defense related genes. Therefore, antioxidant therapy may not be an efficient strategy to block early liver damage.
Rights and permissions
About this article
Cite this article
Knockaert, L., Berson, A., Ribault, C. et al. Carbon tetrachloride-mediated lipid peroxidation induces early mitochondrial alterations in mouse liver. Lab Invest 92, 396–410 (2012). https://doi.org/10.1038/labinvest.2011.193
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2011.193
Keywords
This article is cited by
-
Mitochondrial stress activates YAP/TAZ through RhoA oxidation to promote liver injury
Cell Death & Disease (2024)
-
Neuroblastoma RAS viral oncogene homolog (N-RAS) deficiency aggravates liver injury and fibrosis
Cell Death & Disease (2023)
-
Lepidium meyenii Walp (red maca) Supplementation Prevents Acrylamide-Induced Oxidative Stress and Liver Toxicity in Rats: Phytochemical Composition by UHPLC–ESI–MS/MS
Plant Foods for Human Nutrition (2022)
-
Period1 mediates rhythmic metabolism of toxins by interacting with CYP2E1
Cell Death & Disease (2021)
-
Hepatospecific ablation of p38α MAPK governs liver regeneration through modulation of inflammatory response to CCl4-induced acute injury
Scientific Reports (2019)
