
Abstract
We have recently described a family with a condition (Santos syndrome (SS; MIM 613005)) characterized by fibular agenesis/hypoplasia, hypoplastic femora and grossly malformed/deformed clubfeet with severe oligodactyly, ungual hypoplasia/anonychia, sometimes associated with mild brachydactyly and occasional pre-axial polydactyly. Autosomal dominant inheritance with incomplete penetrance was suggested, but autosomal recessive inheritance could not be ruled out, due to the high frequency of consanguineous matings in the region where the family lived. This report deals with linkage studies and exome sequencing, disclosing a novel variant in WNT7A, c.934G>A (p.Gly312Ser), as the cause of this syndrome. This variant was present in homozygous state in five individuals typically affected by the SS syndrome, and in heterozygous state in the son of one affected homozygous individual. The heterozygous boy presented only unilateral complex polysyndactyly and we hypothesize that he either presents a distinct defect or that his phenotype results from a rare, mild clinical manifestation of the variant in heterozygous state. Variants in WNT7A are known to cause at least two other limb defect disorders, the syndromes of Fuhrmann and Al-Awadi/Raas-Rothschild. Despite their variable degree of expressivity and overlap, the three related conditions can be differentiated phenotypically in most instances.
Similar content being viewed by others


Introduction
In 2008, we described a new hereditary condition, presently known as ‘Santos syndrome’ (SS; MIM 613005), comprising asymmetric lower limb defects with hypoplastic femora, fibular agenesis/hypoplasia, grossly malformed/deformed clubfeet with severe oligodactyly; upper limbs with acromial dimples, limited extension, flexion, pronation, and supination of forearm and hands, severe ungual hypoplasia/anonychia, sometimes associated with mild brachydactyly, and occasionally pre-axial polydactyly.1
The condition was compared with two syndromes presenting fibular agenesis associated with other defects/deformities: brachydactyly–ectrodactyly with fibular ‘aplasia’ or hypoplasia (MIM 113310)2, 3, 4 and Fuhrmann syndrome (FS; MIM 228930).5, 6, 7, 8 Its clinical presentation did not perfectly fit the reported phenotypes of either conditions or those of other phenotypically related syndromes.
Pedigree analysis suggested autosomal dominant inheritance with a low penetrance, but the possibility of autosomal recessive inheritance could not be discarded because of the high regional population inbreeding rate, as already stated clearly on the 2008 paper. The frequencies of consanguineous matings in the region range from 9 to 33%, with corresponding average inbreeding coefficients (F) varying from 0.3 to 1.0%.9, 10
Below we report on the identification of the genetic variant responsible for the syndrome.
Materials and methods
The research was approved by the Ethics Committee of the Institute of Biosciences at the University of São Paulo (CAAE: 37297114.7.0000.5464).
Patients
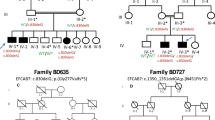
This study covers 12 individuals from the family with SS, originally described by Santos et al.:1 five individuals (IV:1, IV:16; IV:19; IV:20 and IV:31) with the complete SS phenotype, one individual (V:12) with a milder and atypical phenotype (unilateral polysyndactyly without lower limb defects) and six unaffected individuals (IV:17, IV:29, IV:32, V:13, V:14 and V:15). Genomic DNA was extracted from blood samples. Pedigree is shown in Figure 1 and clinical findings are summarized in Table 1.

Pedigree of family. The black symbols represent individuals with the complete phenotype of Santos syndrome (upper and lower limb malformations with anonychia/ungual hypoplasia); the half-filled symbol represents the individual with polysyndactyly of upper limb without anonychia/ungual hypoplasia or lower limb malformations (originally depicted in Santos et al.1). Below the symbols are shown the genotypes for the variant c.934G>A in gene WNT7A.
Array-based comparative genomic hybridization
Investigation of copy number changes was performed by array-based comparative genomic hybridization (array-CGH), using a 180 K chip (OGT, Oxford, UK) with genomic DNA samples from the proband IV:31 and his mildly affected son V:12. Cytosure Genomic DNA Labelling kit (OGT) and Agilent Oligo a-CGH Hybridization kit (Agilent Technologies, Santa Clara, CA, USA) were used, according to the manufacturers’ protocol. Gains and losses of genomic sequences were called using the aberration detection statistical algorithm ADM-2, with a sensitivity threshold of 6.0.
SNP-array and microsatellite markers
Genomic DNA from affected individuals (IV:1, IV:16; IV:19; IV:20; IV:31 and V:12) was submitted to SNP-Array (50K) assays (Affymetrix GeneChip Human Mapping 50 K Array, Affymetrix, Santa Clara, CA, USA), using the manufacturer's reagents (XbaI) and following the GeneChip Mapping 10K 2.0 Assay Manual. Scanning was performed in a GenechipScanner 3000 and interpreted with Affymetrix Genotyping Console software (Affymetrix). Multipoint logarithm of odds (LOD) score values were calculated, for each autosome, by the Merlin program11 under dominant and recessive inheritance models, assuming a rare allele (frequency of 0.001). The penetrance rate under the dominant inheritance model was estimated as 0.324 (95% confidence limits: 0.139–0.585).1 Twenty-two polymorphic microsatellite markers mapped to chromosome 3 (ABI Prism Linkage Mapping Sets v2.5) were genotyped in the 12 samples. Multipoint LOD score values were calculated using the Merlin program.
Whole-exome sequencing and segregation of candidate variants
DNA samples from one affected individual with SS phenotype (IV:16) and from the individual with the atypical clinical presentation (V:12) were submitted to whole-exome sequencing. The library was prepared with Nextera rapid capture kit (Illumina, San Diego, CA, USA), sequence capture was performed with Illumina Exome enrichment kit (~62 Mb target size) and sequencing was performed using HiSeq 2500. Fastq files were aligned against reference GRCh37 with Burrows-Wheeler Aligner (BWA);12 realignment of indel regions, discovery of variants and recalibration of base qualities were performed using GATK software13 for the production of VCF files; the VCF was annotated by ANNOVAR software.14
Variant frequencies were compared with public variant databases: 1000 Genomes (http://www.internationalgenome.org/), NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), Exome Aggregation Consortium (http://exac.broadinstitute.org/) and Online Archive of Brazilian Mutations (http://abraom.ib.usp.br/). Polyphen-2,15 SIFT,16 Provean17 and MutationTaster218 were used for in silico damage prediction to the protein. Protein sequence alignment near the best candidate variant (WNT7A) was performed by Universal Protein Resource (Uniprot) catalog (http://www.uniprot.org/).
Sanger sequencing
To investigate the segregation of the variants in WNT7A and TBX5 genes, the coding sequences were amplified by PCR. The products were bi-directionally Sanger sequenced with the BigDye Terminator v3.1 Cycle Sequencing Kit method in ABI 3730 DNA Analyzer (Applied Biosystems, Foster City, CA, USA). The results were analyzed by the Bioedit program (Ibis Biosciences, Carlsbad, CA, USA).
Results
CNV and linkage analysis
No pathogenic copy number variation was detected by array-CGH.
LOD score values, using SNP-array and microsatellite data, were calculated by three different strategies: (1) under recessive model considering individual V:12 as unaffected by SS; (2) under recessive model considering individual V:12 as affected by SS; and (3) under dominant (k=0.324) model considering individual V:12 as affected by SS.
In strategy (1), by SNP-array data analysis, positive LOD score values were found in two regions on chromosome 3: maximum positive values of 1.406 and 1.735 were observed, respectively, in the regions between 15 and 20 cM and between 87 and 123 cM (Supplementary Figure S1). As the result indicated chromosome 3 as candidate, we performed additional LOD score calculations using data from 22 microsatellite markers along the chromosome. The results were consistent with those obtained with SNP-array data: maximum positive values of 2.098 and 2 were observed, respectively, in the regions between pTer and 42 cM, and between 80 and 139 cM (Supplementary Figure S1). According to family reports, individuals III:5 and III:6 might be remotely related. Such possibility led us to perform additional LOD score calculations, under an autosomal recessive model, but considering individuals I:1 and I:6 as sibs. The results pointed to significant positive LOD score values at the same chromosome 3 locations. The maximum values, when SNPs were used, for the first and second regions were 2.876 and 3.235, respectively; after microsatellite data analysis, the maximum values for the first and second regions were 2.098 and 2.0, respectively (Supplementary Figure S1).
In strategies (2) and (3), with SNP-array and microsatellite data, positive LOD score values were achieved only for chromosome 3, but the maximum LOD score values were slightly lower in strategy (2), and markedly lower in strategy (3) (data not shown), when compared with results from strategy (1). In strategy (2) the maximum LOD score was 1.786 (with SNP-array) and 2.047 (with microsatellites). In strategy (3) the maximum LOD score was 0.897 (with SNP-array) and 1.053 (with microsatellites).
In conclusion, linkage data pointed only to chromosome 3 as harboring the pathogenic mutation that causes SS. Additionally, we present the transmission of the microsatellite haplotypes near the WNT7A gene region (Supplementary Figure S2), the best candidate gene, according to linkage data and sequencing results presented in the next section.
Whole-exome sequencing and segregation of candidate variants
We obtained, by sample, approximately 70M reads, average read length of 99 bp, average coverage of 120X and 98% of target bases with more than 20 reads.
The variants were filtered by two different strategies: (1) IV:16 and V:12 separately, under the assumption that IV:16 and V:12 were affected by two distinct genetic diseases; and (2) IV:16 and V:12 together, considering that both phenotypes were caused by the same genetic alteration. In both strategies, we screened exonic and non-synonymous variants for quality, and checked against public variant databases and 60 samples sequenced simultaneously as controls. We selected variants occurring in homozygous state with frequencies lower than 0.01, and variants in heterozygous state with frequencies lower than 0.001. The remaining variants were analyzed considering association with (a) limb development and its disorders (OMIM (https://www.omim.org/) and literature), (b) limb defects, after mouse knocking out (MGI (http://www.informatics.jax.org/)) and/or (c) WNT and SHH signaling pathways (Kegg (http://www.genome.jp/kegg/pathway.html)).
Following strategy (1), for individual IV:16, we selected only autosomal variants in homozygosis, and obtained nine variants in nine genes: ANKRD53, GCFC2, SMN, NCF1, APBB1IP, GLB1L2, GPRC5B, OR5K1 and WNT7A. Among these variants, only two are located in the candidate chromosome 3: the missense variants c.G592A in OR5K1 and c.934G>A in WNT7A gene. After analysis of these nine genes according to the criteria (a), (b) and/or (c), described above, only the variant c.934G>A in the WNT7A gene remained.
Following strategy (1), for individual V:12, we obtained 179 variants in heterozygous state, one in homozygous state (in FAM21A gene) and three in hemizygous state in chromosome X (in COL4A6, PLXNB3 and OPN1MW genes). After selecting genes according to associations (a), (b) and (c) described above, only six heterozygous variants were obtained: c.G934A in WNT7A (Fuhrmann syndrome MIM 228930), c.A3382G in FREM2 (Fraser syndrome MIM 219000), c.C113A in CEP152 (Seckel syndrome MIM 210600), c.G1220A in IFT122 (cranioectodermal dysplasia MIM 218330), c.G496A in TRPV3 (Olmsted syndrome MIM 614594) and c.C698A in TBX5 (Holt-Oram syndrome MIM 142900). Fuhrmann, Fraser and Seckel syndromes, and cranioectodermal dysplasia are autosomal recessive disorders and could not be explained by a variant in heterozygous state. The Olmsted and Holt-Oram syndromes are autosomal dominant syndromes, but Olmsted syndrome does not include the clinical findings seen in individual V:12. This led us to investigate the segregation of the variant in the TBX5 gene, which had been previously associated with radial limb defects, and might explain the clinical features of V:12. However, Sanger sequencing revealed that the TBX5 variant was present in his non-affected mother (IV:32) and in one of his non-affected sisters (V:15) who, as V:12 subject, were heterozygous for both variants, c.C848A in TBX5 and c.934G>A in WNT7A. This excluded TBX5 gene as the cause of the phenotype of V:12.
According to the strategy (2), we selected variants shared by the individuals V:12 and IV:16; 14 variants were found, 11 in heterozygous state in both patients and three homozygous variants in IV:16 and in heterozygous state in V:12. After selecting genes associated with (a), (b) and (c), only the variant c.934G>A (p.Gly312Ser) in WNT7A, on chromosome 3, remained as candidate.
Summing up, linkage studies, associated with all strategies of filtering variants after exome sequencing, indicated the WNT7A variant in homozygosis as the best explanation for the characteristic clinical features of SS: it is located on chromosome 3, and WNT7A variants have been related to limb development and clinical features similar to the SS phenotype. The other variant located in the candidate chromosome 3, c.G592A (p.Val198Ile) in OR5K1 gene, was excluded as candidate because OR5K1 protein is an olfactory receptor. There are no variants in this gene previously associated with clinical presentations in humans and pathogenicity predictions by Polyphen-2, SIFT, Provean and MutationTaster indicated that this variant is benign.
Sanger sequencing revealed that the candidate variant c.934G>A in WNT7A was present in homozygosis in five affected individuals with the complete phenotype (IV:1, IV:16, IV:19, IV:20 and IV:32); it was found in heterozygous state in the individual with the milder distinct phenotype (V:12), as well as in four unaffected individuals (V:31, V:13, V:14 and V:15). The genotypes are shown in Figure 1.
The c.934G>A variant in WNT7A has never been reported in patients with limb defects, nor in sequencing databases. It is predicted to result in a p.Gly312Ser amino acid substitution, and the glycine at position 312 lies in a highly conserved region across different species (Figure 2). PolyPhen-2 and SIFT predicted that this variant is probably damaging (scores of 1.0 and 0.001, respectively); MutationTaster2 predicted that it was disease causing, and Provean, that it was deleterious (score of 5.42).

Alignment of the amino acid sequence near the p.Gly312Ser WNT7A mutation, across different species. In gray, the conserved amino acid glycine (at position 312 in humans).
Discussion
Limb development in human embryos begins in the fourth week after fertilization. Limb buds originate from the lateral plate mesoderm, which is in close contact with the ectoderm.19 The ectodermal bulge of limb buds (apical ectodermal ridge) is a region that controls, through the action of the fibroblast growth factor proteins FGF4 and FGF8, limb development in the proximal–distal axis,20, 21 while the anterior–posterior axis determines the ulnar/radial and fibular/tibial growth width.22 SHH protein, produced in the zone of polarizing activity of posterior (ulnar and fibular) mesoderm, is responsible for the development of posterior structures,23 in addition to inducing the expression of FGF4 in the apical ectodermal ridge.24 In the dorsal (nails and thin hairy skin) and ventral (palm and sole) axes, the WNT7A protein, produced from the dorsal ectoderm, induces the local expression of LMX-1B, which is responsible for the development of dorsal structures.25 WNT7A also plays a role in the maintenance of SHH expression in zone of polarizing activity.8, 26 Ungual hypoplasia (sometimes anonychia), marked fibular hypoplasia (sometimes agenesis) and femoral hypoplasia in affected individuals are clinical features present in patients previously reported with WNT7A variants.8, 27, 28, 29, 30, 31 Anonychia and ungual hypoplasia probably arise from the disruption of the dorsal–ventral axis by lack of induction of LMX-1B by WNT7A, while fibular hypoplasia/aplasia would result from disturbances (due to decreased SHH expression in zone of polarizing activity) in the development of the antero-posterior axis. Finally, developmental disruption of the proximal–distal axis (possibly due to SHH-FGF4 feedback deficiency) may lead to brachydactyly.32, 33
All WNT7A variants in humans have so far been reported in FS (MIM 228930) and Al-Awadi/Raas-Rothschild syndrome (AARRS, MIM 276820), both rare autosomal recessive conditions. WNT7A variants in homozygosis were reported most often in inbred Muslim populations from the Middle East. The literature records six different pathogenic WNT7A variants: p.Glu72Lys (FS);27, 33 p.Ala109Thr (FS);8 p.Arg222Trp (FS);28, 33 p.Glu272X (AARRS);29 p.Arg292Cys (AARRS)8, 29 and p.Gly204Ser (AARRS and FS).30, 31 The variant p.Gly312Ser in WNT7A is described in this paper for the first time (Figure 3).

Scheme of WNT7A gene. The light gray boxes indicate all known mutations associated with Fuhrmann syndrome and the dark gray ones indicate all known mutations associated with AARR syndrome (mutation c.610G>A is associated with both AARR and Fuhrmann syndromes). An ellipse indicates the location of the novel mutation (this report) related to Santos syndrome.
Woods et al.8 demonstrated, in vivo and in vitro, that in FS there is a partial loss of function of the WNT7A protein, whereas in AARRS there is a complete loss of WNT7A function. This might account for the clinical features shared by these syndromes, and the more severe AARRS phenotype. Both AARRS and FS are characterized by anonychia/hypoplastic nails, variable degrees of ulnar ray deficiency, pelvic dysgenesis and severe lower limb hypoplasia (sometimes aplasia). Usually FS patients present a less severe phenotype, with 4–5 digits per hand and rarely phocomelia, while AARRS affected individuals have 1–3 digits per hand and, frequently, phocomelia.33 However, it is striking that variant p.Gly204Ser was found in patients with FS and AARRS (Figure 3), showing that this correlation is not absolute.
Accordingly, both SS and FS might result from partial loss of function of the WNT7A protein. Similarities and differences between these and other syndromes have been discussed in our 2008 paper,1 which pointed out that, despite the variable degree of overlap and expressivity of the features between the FS and SS, (1) severe distal ulnar defects might be present only in FS, (2) femur and pelvis defects seemed to be much more severe and frequent in FS, while (3) foot malformation was usually more severe and conspicuously typical in SS. In spite of the occasional overlap of some signs in the three conditions (FS, AARRS and SS), it is clear that they can be differentiated in most instances. Since all three syndromes represent the phenotype of pathogenetic variants in the WNT7A gene, they could be certainly referred to as a group of ‘WNT7A-related syndromes with ungueal and fibular defects,’ but the nomenclature SS, AARRS and FS could be retained to describe the three subgroups of clinical phenotypes.
Although variant c.934G>A in homozygosis explains perfectly the phenotype of Santos syndrome, intriguingly the son (V:12) of the proband presents the WNT7A variant in heterozygous state, and his clinical presentation is quite different from that of other affected family members (Table 1), without the main features expected to result from variants in WNT7A. So, additionally, his exome sequencing was individually filtered, in an attempt to identify variants in other genes that could explain his clinical presentation. The best candidate found was the heterozygous variant c.C848A in TBX5. Heterozygous variants in TBX5 cause Holt-Oram syndrome, which is characterized by radial anomalies, often including thriphalangeal thumb, associated with atrial septal defects.34 The complex limb malformation of individual V:12 in his left hand includes a thriphalangeal thumb. However, segregation analysis ruled out this variant as a candidate because it was found in samples from two unaffected individuals (his mother and his sister). No other candidate variant was found that could explain why he was affected. Three alternative hypotheses might explain the phenotype of V-12: (1) the milder phenotypic expression of the WNT7A variant in heterozygous state; (2) unidentified environmental factors; (3) an additional undetected distinct causative genetic variant, his phenotype not being related with the WNT7A variant. If this is the case, the defect is not due to a coding region alteration or the variant was not identified by our approach of filtering variants detected by the exome sequencing, which was based on information about protein function related to limb development.
In conclusion, the cause of SS is convincingly explained by the homozygosis of a WNT7A gene variant. The variant we found is allelic to those responsible for FS and AARRS. The proband’s son presents distinct limb defects, possibly without any relationship with Santos syndrome. Our findings contribute to broaden the spectrum of clinical presentation of WNT7A variants.
References
Santos, S. C., Pardono, E., Ferreira da Costa, M. I., de Melo, A. N., Graciani, Z., de Albuquerque e Souza, A. C. et al. A previously undescribed syndrome combining fibular agenesis/hypoplasia, oligodactylous clubfeet, anonychia/ungual hypoplasia, and other defects. Am. J. Med. Genet. A 146A, 3126–3131 (2008).
Lewin, S. O. & Opitz, J. M. Fibular a/hypoplasia: review and documentation of the fibular developmental field. Am. J. Med. Genet. Suppl. 2, 215–238 (1986).
Genuardi, M., Zollino, M., Bellussi, A., Fuhrmann, W. & Neri, G. Brachy/ectrodactyly and absence or hypoplasia of the fibula: an autosomal dominant condition with low penetrance and variable expressivity. Clin. Genet. 38, 321–326 (1990).
Evans, J. A., Reed, M. H. & Greenberg, C. R. Fibular aplasia with ectrodactyly. Am. J. Med. Genet. 113, 52–58 (2002).
Fuhrmann, W., Fuhrmann-Rieger, A. & de Sousa, F. Poly-, syn- and oligodactyly, aplasia or hypoplasia of fibula, hypoplasia of pelvis and bowing of femora in three sibs—a new autosomal recessive syndrome. Eur. J. Pediatr. 133, 123–129 (1980).
Fuhrmann, W., Fuhrmann-Rieger, A., Jovanović, V. & Rehder, H. A new autosomal recessive skeletal dysplasia syndrome—prenatal diagnosis and histopathology. Prog. Clin. Biol. Res. 104, 519–524 (1982).
Pfeiffer, R. A., Stöss, H., Voight, H. J. & Wündisch, G. F. Absence of fibula and ulna with oligodactyly, contractures, right-angle bowing of femora, abnormal facial morphology, cleft lip/palate and brain malformation in two sibs: a possibly new lethal syndrome. Am. J. Med. Genet. 29, 901–908 (1988).
Woods, C. G., Stricker, S., Seemann, P., Stern, R., Cox, J., Sherridan, E. et al. Mutations in WNT7A cause a range of limb malformations, including Fuhrmann syndrome and Al-Awadi/Raas-Rothschild/Schinzel phocomelia syndrome. Am. J. Hum. Genet. 79, 402–408 (2006).
Santos, S., Kok, F., Weller, M., de Paiva, F. R. & Otto, P. A. Inbreeding levels in Northeast Brazil: strategies for the prospecting of new genetic disorders. Genet. Mol. Biol. 33, 220–223 (2010).
Weller, M., Tanieri, M., Pereira, J. C., Almeida Edos, S., Kok, F. & Santos, S. Consanguineous unions and the burden of disability: a population-based study in communities of Northeastern Brazil. Am. J. Hum. Biol. 24, 835–840 (2012).
Abecasis, G. R., Cherny, S. S., Cookson, W. O. & Cardon, L. R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 30, 97–101 (2002).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P. et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Ng, P. C. & Henikoff, S. Predicting the effects of amino acid substitutions on protein function. Annu. Rev. Genomics Hum. Genet. 7, 61–80 (2006).
Choi, Y., Sims, G. E., Murphy, S., Miller, J. R. & Chan, A. P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 7, e46688 (2012).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362 (2014).
Niswander, L. Pattern formation: old models out on a limb. Nat. Rev. Genet. 4, 133–143 (2003).
Niswander, L., Jeffrey, S., Martin, G. R. & Tickle, C. A positive feedback loop coordinates growth and patterning in the vertebrate limb. Nature 371, 609–612 (1994).
Sun, X., Mariani, F. V. & Martin, G. R. Functions of FGF signalling from the apical ectodermal ridge in limb development. Nature 418, 501–508 (2002).
Tickle, C. The number of polarizing region cells required to specify additional digits in the developing chick wing. Nature 289, 295–298 (1981).
Riddle, R. D., Johnson, R. L., Laufer, E. & Tabin, C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell 75, 1401–1416 (1993).
Zúñiga, A., Haramis, A. P., McMahon, A. P. & Zeller, R. Signal relay by BMP antagonism controls the SHH/FGF4 feedback loop in vertebrate limb buds. Nature 401, 598–602 (1999).
Riddle, R. D., Ensini, M., Nelson, C., Tsuchida, T., Jessell, T. M. & Tabin, C. Induction of the LIM homeobox gene Lmx1 by WNT7a establishes dorsoventral pattern in the vertebrate limb. Cell 83, 631–640 (1995).
Parr, B. A. & McMahon, A. P. Dorsalizing signal Wnt-7a required for normal polarity of D-V and A-P axes of mouse limb. Nature 374, 350–353 (1995).
Garavelli, L., Wischmeijer, A., Rosato, S., Gelmini, C., Reverberi, S., Sassi, S. et al. Al-Awadi-Raas-Rothschild (limb/pelvis/uterus-hypoplasia/aplasia) syndrome and WNT7A mutations: genetic homogeneity and nosological delineation. Am. J. Med. Genet. A 155A, 332–336 (2011).
Kantaputra, P. N., Mundlos, S. & Sripathomsawat, W. A novel homozygous Arg222Trp missense mutation in WNT7A in two sisters with severe Al-Awadi/Raas-Rothschild/Schinzel phocomelia syndrome. Am. J. Med. Genet. A 152A, 2832–2837 (2010).
Al-Qattan, M. M., Al-Abdulkareem, I., Ballow, M. & Al Balwi, M. A report of two cases of Al-Awadi Raas-Rothschild syndrome (AARRS) supporting that ‘apparent’ phocomelia differentiates AARRS from Schinzel phocomelia syndrome (SPS). Gene 527, 371–375 (2013).
Eyaid, W., Al-Qattan, M. M., Al Abdulkareem, I., Fetaini, N. & Al Balwi, M. A novel homozygous missense mutation (c.610G>A, p.Gly204Ser) in the WNT7A gene causes tetra-amelia in two Saudi families. Am. J. Med. Genet. A 155A, 599–604 (2011).
Al-Qattan, M. M., Shamseldin, H. E. & Alkuraya, F. S. The WNT7A G204S mutation is associated with both Al-Awadi-Raas Rothschild syndrome and Fuhrmann syndrome phenotypes. Gene 516, 168–170 (2013).
Wang, B., Sinha, T., Jiao, K., Serra, R. & Wang, J. Disruption of PCP signaling causes limb morphogenesis and skeletal defects and may underlie Robinow syndrome and brachydactyly type B. Hum. Mol. Genet. 20, 271–285 (2011).
Al-Qattan, M. M. Molecular basis of the clinical features of Al-Awadi-Raas-Rothschild (limb/pelvis/uterus-hypoplasia/aplasia) syndrome (AARRS) and Fuhrmann syndrome. Am. J. Med. Genet. A 161A, 2274–2280 (2013).
Temtamy, S. A. & McKusick, V. A. The genetics of hand malformations. Birth Defects Orig. Artic. Ser. 14: i-xviii, 1–619 (1978).
Acknowledgements
We thank Mr Guilherme Yamamoto for assistance in bioinformatics; Dr Carla Rosenberg and Silvia S Costa for array-CGH experiments and exome library preparation; Dr Angela M Vianna-Morgante and Dr Bernd Wollnik (from Universitätsmedizin Göttingen, Göttingen, Germany) for helpful discussions. We also thank CEPID-FAPESP (Centro de Estudos do Genoma humano e Células-tronco, 2013/08028-1) and CAPES for the financial support, and the patients for their cooperation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Alves, L., Santos, S., Musso, C. et al. Santos syndrome is caused by mutation in the WNT7A gene. J Hum Genet 62, 1073–1078 (2017). https://doi.org/10.1038/jhg.2017.86
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2017.86

{kind=link}
{kind=link}