
Abstract
De novo dominant mutations in the aldehyde dehydrogenase 18 family member A1 (ALDH18A1) gene have recently been shown to cause autosomal dominant cutis laxa with progeroid features (MIM 616603). To date, all de novo dominant mutations have been found in a single highly conserved amino acid residue at position p.Arg138. We report an 8-year-old male with a clinical diagnosis of autosomal dominant cutis laxa (ADCL) with progeroid features and a novel de novo missense mutation in ALDH18A1 (NM_002860.3: c.377G>A (p.Arg126His)). This is the first report of an individual with ALDH18A1-ADCL due to a substitution at a residue other than p.Arg138. Knowledge of the complete spectrum of dominant-acting mutations that cause this rare syndrome will have implications for molecular diagnosis and genetic counselling of these families.
Similar content being viewed by others

Introduction
De Barsy syndrome is a rare genetic disease characterized by cutis laxa, a progeroid appearance, ophthalmological abnormalities and intellectual disability.1, 2, 3 Biallelic mutations in either PYCR14 or ALDH18A15, 6 have been identified as responsible for De Barsy syndrome (MIM 219150 and 614438). These genes encode mitochondrial enzymes (PYCR1 and P5CS, respectively) that are involved in the biosynthesis of proline, ornithine and arginine.
A dominant form of De Barsy syndrome (MIM 616603) that is caused by de novo mutations in ALDH18A1 was recently recognized and is referred to as ALDH18A1-autosomal dominant cutis laxa with progeroid features (ALDH18A1-ADCL).7, 8 The individuals described to date share many of the features characteristic of the recessive form of the disease. All reported de novo mutations have involved a recurrent substitution of arginine at position 138 for glutamine, leucine or tryptophan. Here we report a novel de novo heterozygous substitution in ALDH18A1, p.Arg126His in an individual affected with ALDH18A1-ADCL.
Case report
The child was born at term to healthy consanguineous parents (fourth cousins) of Somali descent. He had short long bones antenatally and presented at birth with small stature, cutis laxa, truncal hypotonia, inguinal hernias, bilateral congenital hip dislocations and congenital cataracts. He was later diagnosed with nystagmus and central corneal opacities.
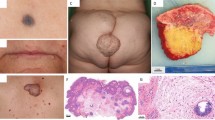
At age 8 years, his height was 96 cm (Z=−5.8) and weight was 11.6 kg (Z=−7.26). His head circumference was 45.4 cm, (Z=−5.0) which is the 50th centile for a 10-month-old boy. The trajectories of his growth parameters were consistent over time. He had global developmental delay and truncal hypotonia; he walked with support and used 3–5 word sentences. Dysmorphic features included a broad forehead with a triangular face, down-slanted palpebral fissures, a small mouth, soft lax skin and bilaterally adducted thumbs (Figures 1a and c–e).

(a) The patient at 8 months with cutis laxa, a broad forehead, triangular facies and adducted thumbs. (b) T2-weighted coronal brain MRI at age 3 years showing no structural malformations, with tortuosity of the vertebral and carotid system, and their proximal branches. (c) The patient’s feet and (d) right hand at age 8 demonstrating cutis laxa. (e) The patient at age 8, with broad forehead and cutis laxa again noted. A full color version of this figure is available at the Journal of Human Genetics journal online.
A brain MRI performed at 15 months of age and at 3 years of age revealed prominent ventricles and marked tortuosity of the intracranial arteries (Figure 1b). He had an older sister and younger brother who were developmentally appropriate and well grown.
A clinical diagnosis of De Barsy syndrome was given at 2 years of age. Sanger sequencing of ALDH18A1 and PYCR1 revealed a single variant at ALDH18A1: c.377G>A. Chromosomal SNP microarray showed no regions of homozygosity encompassing PYCR1 and ALDH18A1, or other chromosomal deletions or duplications. Given the lack of homozygosity at these loci, and the known consanguinity in the parents, a novel form of autosomal recessive cutis laxa was suspected. The family was enrolled in the Care4Rare Canada research project. Research ethics board approval was obtained as was free and informed consent from the family. Whole-exome sequencing (WES) of genomic DNA from the proband was performed and analyzed as previously described.9 The single heterozygous missense variant in ALDH18A1 was observed: NM_002860.3: c.377G>A, (p.Arg126His).
In order to identify rare copy-number variants, two tools were used: FishingCNV,10 which compared the read depth of the variants on the WES data against the distribution of reads in more than 48 control individuals with unrelated genetic disorders, and eXome-Hidden Markov Model,11 which used principal component analysis to normalize read depth, filtering out extremely variable targets not derived from copy-number changes. These analyses did not identify any significant copy-number variation in the WES data, including the genes ALDH18A1 and PYCR1.
At the time of interpretation, only the recessive form of ALDH18A1-ADCL had been described and the p.Arg126His variant was not pursued further. No other compelling candidates were identified. Upon publication of Fischer-Zirnsak et al.’s7 report of de novo mutations in ALDH18A1 causing ALDH18A1-ADCL, the p.Arg126His variant was reviewed. Subsequent sequencing in unaffected parents demonstrated that it was a de novo variant (Figure 2), which had not been reported in presumed healthy control databases, including EVS,12 1000 Genomes13 and the ExAC browser.14

Sanger sequencing traces showing the identified mutation in ALDH18A1 (c.377G>A(p.Arg126His)) in the affected patient and its absence in the patient’s parents. A full color version of this figure is available at the Journal of Human Genetics journal online.
The arginine at codon 126 is a moderately conserved residue (to Tetraodon nigroviridis) in the gamma-glutamyl kinase domain of P5CS. Its conversion to a histidine is predicted to be deleterious by several in silico programs, including SIFT15 and Mutation Taster.16 PolyPhen-217 predicts the variant to be possibly damaging.
Discussion
We report a novel dominant mutation in a patient with ALDH18A1-ADCL. Evidence that the c.377G>A variant is responsible for the disease includes: (1) its de novo status, (2) the absence of the variant in control databases, (3) the in silico predicted deleterious effect, (4) the clinical similarity between our patient and the others reported, (5) p.Arg126His is in proximity to the known p.Arg138 substitution and located in the same domain of P5CS, suggesting a similar functional effect and (6) that no other candidates were identified by WES. Our patient’s features are similar to those previously described, including growth restriction, microcephaly, cranial vessel tortuosity, cataracts, dysmorphic features and cutis laxa (Figure 1).
The identification of a de novo dominant mutation responsible for ALDH18A1-ADCL had implications for genetic counselling. A sibling recurrence risk was considered low (<1%), although the possibility of gonadal mosaicism could potentially increase this risk. This was markedly different than the recessive form hypothesized to be present in this family prior to recognition of the dominant form.
The mechanism by which dominant mutations in ALDH18A1 cause a similar phenotype to that of recessive mutations is still unclear. Studies on p.Arg138 patient cell lines have demonstrated altered mitochondrial localization.7 These cell lines also have decreased proline levels, which has also been seen in cell lines with biallelic mutations.7 Fischer-Zirnsak et al.7 speculated that the mutant P5CS protein may have a dominant-negative effect on the wild-type P5CS protein.
In conclusion, we report the first individual with ALDH18A1-ADCL due to a substitution other than p.Arg138. Further study of ALDH18A1-ADCL and associated mutations in ALDH18A1 will enhance our understanding of this disease and its pathogenesis, and will have implications for diagnosis and genetic counselling.
References
De Barsy, A. M., Moens, E. & Dierckx, L. Dwarfism, oligophrenia, and elastic-tissue hypoplasia: a new syndrome? Lancet 2, 47 (1967).
Guerra, D., Fornieri, C., Bacchelli, B., Lugli, L., Torelli, P., Balli, F. et al. The De Barsy syndrome. J. Cutan. Pathol. 31, 616–624 (2004).
Kivuva, E. C., Parker, M. J., Cohen, M. C., Wagner, B. E. & Sobey, G. De Barsy syndrome: a review of the phenotype. Clin Dysmorphol. 17, 99–107 (2008).
Reversade, B., Escande-Beillard, N., Dimopoulou, A., Fischer, B., Chng, S. C., Li, Y. et al. Mutations in PYCR1 cause cutis laxa with progeroid features. Nat. Genet. 41, 1016–1021 (2009).
Bicknell, L. S., Pitt, J., Aftimos, S., Ramadas, R., Maw, M. A. & Robertson, S. P. A missense mutation in ALDH18A1, encoding Δ1 -pyrroline-5-carboxylate synthase (P5CS), causes an autosomal recessive neurocutaneous syndrome. Eur. J. Hum. Genet. 1691, 1176–1186 (2008).
Skidmore, D. L., Chitayat, D., Morgan, T., Hinek, A., Fischer, B., Dimopoulou, A. et al. Further expansion of the phenotypic spectrum associated with mutations in ALDH18A1, encodingΔ1-pyrroline-5-carboxylate synthase (P5CS). Am. J. Med. Genet. A 155, 1848–1856 (2011).
Fischer-Zirnsak, B., Escande-Beillard, N., Ganesh, J., Tan, Y. X., Al Bughaili, M., Lin, A. E. et al. Recurrent de novo mutations affecting residue Arg138 of pyrroline-5-carboxylate synthase cause a progeroid form of autosomal-dominant cutis laxa. Am. J. Hum. Genet. 97, 483–492 (2015).
Nozaki, F., Kusunoki, T., Okamoto, N., Yamamoto, Y., Miya, F., Tsunoda, T. et al. ALDH18A1-related cutis laxa syndrome with cyclic vomiting. Brain Dev. 38, 678–684 (2016).
Beaulieu, C. L., Majewski, J., Schwartzentruber, J., Samuels, M. E., Fernandez, B. A., Bernier, F. P. et al. FORGE Canada Consortium: Outcomes of a 2-year national rare-disease project. Am. J. Hum. Genet. 94, 809–817 (2014).
Shi, Y. & Majewski, J. FishingCNV: a graphical software package for detecting rare copy number variations in exome-sequencing data. Bioinformatics 29, 1461–1462 (2013).
Fromer, M., Moran, J. L., Chambert, K., Banks, E., Bergen, S. E., Ruderfer Douglas, M. et al. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am. J. Hum. Genet. 91, 597–607 (2012).
Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA, USA. Available at http://evs.gs.washington.edu/EVS/ (accessed on September 2016).
1000 Genomes Project Consortium 1000 Genomes Project Consortium, Auton, A. 1000 Genomes Project Consortium, Brooks, L. D. 1000 Genomes Project Consortium, Durbin, R. M. 1000 Genomes Project Consortium, Garrison, E. P. 1000 Genomes Project Consortium, Kang, H. M. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Fennell, T., O'Donnell-Luria, A. H. et al. Analysis of protein-coding genetic variation in 60 706 humans. Nature 536, 285–291 (2016).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects on coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 8, 1073–1082 (2009).
Schwarz, J. M., Rödelsperger, C., Schuelke, M. & Seelow, D. Mutationtaster evaluates disease- causing potential of sequence alterations. Nat. Methods 7, 575–576 (2010).
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P. et al. A. method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010).
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Care4Rare Canada Consortium Gene Discovery Steering Committee K.M. Boycott (lead; University of Ottawa), A.E. MacKenzie (co-lead; University of Ottawa), J. Majewski (McGill University), M. Brudno (University of Toronto), D.E. Bulman (University of Ottawa) and D.A. Dyment (University of Ottawa).
Rights and permissions
About this article

Cite this article
Bhola, P., Hartley, T., Bareke, E. et al. Autosomal dominant cutis laxa with progeroid features due to a novel, de novo mutation in ALDH18A1. J Hum Genet 62, 661–663 (2017). https://doi.org/10.1038/jhg.2017.18
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2017.18
This article is cited by
-
Novel mutations in the ALDH18A1 gene in complicated hereditary spastic paraplegia with cerebellar ataxia and cognitive impairment
Journal of Human Genetics (2018)
