
Abstract
Methionyl-tRNA synthetase (MARS) catalyzes the ligation of methionine to tRNA. Heterozygous MARS mutations have been reported to cause Charcot-Marie-Tooth disease, axonal, type 2U (CMT2U). Homozygous or compound heterozygous mutations in MARS gene would cause interstitial lung and liver disease (ILLD), a severe disease onset in infancy or early childhood. Here we report a Chinese ILLD family with two affected boys diagnosed by exome sequencing. They carry novel compound heterozygous MARS mutations (p.Asp145Asn and p.Phe802Ser). Their phenotype is concordant with ILLD description. As ILLD patients were only reported by two studies, we summarized all the reported patients and characterized the principle clinical features as interstitial lung disease, developmental delay, postnatal growth failure, non-life-threatening liver dysfunction and anemia. Genotype–phenotype correlation analysis suggests most of the ILLD mutations locate in the catalytic domain of MARS. ILLD and CMT2U might have different disease mechanism.
Similar content being viewed by others


Introduction
MARS (OMIM 156560) gene encodes cytoplasmic Methionyl-tRNA synthetase, an enzyme that charges Methionyl to the cognate tRNA. Compound heterozygous or homozygous mutations in MARS could cause a severe disease onset in infancy or early childhood, interstitial lung and liver disease (ILLD, OMIM 615486). ILLD was reported in two studies. One patient presented with a multi-organ dysfunction,1 while the major clinical features are lung disease and liver disorder in ILLD patients from Réunion or nearby islands.2 Here we report the genetic and clinical findings in a Chinese family that suffered from ILLD.
Clinical presentation
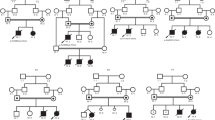
The non-consanguineous parents are healthy with no known family history of genetic disorders. The proband (II.1, Figure 1a) was admitted to clinic at 41 days because of microcytic anemia (pale face). Bone marrow biopsy suggested erythroid hyperplasia with maturation arrest. Sideroblastic anemia was excluded by Prussian blue staining. Head MRI showed arachnoid cyst. Chest radiograph revealed increased shadow of lung. Gradually he developed cough complicated with severe pulmonary infection and suffered from malnutrition secondary to feeding difficulty. At 6 months, liver function test showed liver damages (Supplementary Table 1). The boy had developmental delay and could not lift his head at 8 months. Digital clubbing and hepatomegaly were present. He died from respiration failure at the age of 9 months.

The family pedigree and the clinical feature of the Chinese ILLD family. (a) The family pedigree. The proband II.1 is marked with arrow. II.2, II.3 and II.5 were aborted; (b) clubbed fingers of II.4; (c) clubbed toes of II.4; (d) photo of II.4 taken at 39 months. A full color version of this figure is available at the Journal of Human Genetics journal online.
II.4 (Figure 1a) was referred to the hospital for feeding difficulty at 23 days. He had microcytic anemia and liver dysfunction (Supplementary Table 1) and was diagnosed with pneumonia, acidosis and lung fibrosis. Head MRI showed bilateral ventricular enlargement. At 2 months, intermittent lactic acidosis occurred. Atrial septal defect and pulmonary artery dysplasia were detected by ultrasonic cardiogram. Magnetic Resonance Cholangiopancreatography illustrated partially narrowed proximal choledochal duct. Bone marrow biopsy showed bone marrow cell active proliferation. CT scan suggested interstitial lung alteration. Digital clubbing could be noticed (Figures 1b and c). Cholestasis relieved at age of 39 months (Supplementary Table 1). However, the protein level stayed low. At 51 months, he could not sit without support. He could speak ’papa/mama’. He was malnourished with extremely thin subcutaneous fat tissue (Figure 1d).
The two boys’ growth curves showed postnatal growth failure (Supplementary Figure 1). Both their birth weights were 3 kg (within −1 s.d.). They however showed failure to thrive immediately (<−3 s.d. at 1 month). II.4’s weight stopped to increase since 25 months. The detailed clinical information of this family was summarized in Table 1. Common clinical features are interstitial lung disease, microcytic anemia, developmental delay, postnatal growth failure and liver dysfunction.
For II.5, no abnormality was observed in the second trimester by ultrasound. All family members in this study provided informed consent for DNA study and publication.
Results
Exome sequencing was performed on I.2 and II.4 to detect potential disease causing variants in this family (Supplementary Methods). Two heterozygous variants in MARS gene (NM_004990.3:c.433G>A, p.Asp145Asn (Figure 2a); NM_004990.3:c.2405 T>C, p.Phe802Ser (Figure 2b)) were considered to be the top candidate variants from the candidate variant list (Supplementary Table 2). They cosegregate within the family (Figure 1a). The two variants are not presented in any cohort variant databases and an inhouse database containing 150 exomes. They are predicted to be deleterious by in silico tools (Supplementary Table 3). The encoded amino acids are highly conserved (Figure 2c).

The MARS mutations found in this family and literatures. The IGV illustration of the exome data and Sanger sequencing for (a) c.433G>A and (b) c.2405 T>C; (c) amino acid sequence alignment of MARS orthologs. Asp145Asn and Phe802Ser locate in the conserved region (Screenshot from UCSC genome browser); (d) the MARS mutation distribution in protein domain structure. Mutations reported in this manuscript is marked with grey box. The MARS domain structure was published by Hadchouel et al.2 The Rossmann fold domain and the CP domain compose the catalysis center. Anti-codon binding domain forms the α-helix bundle. A full color version of this figure is available at the Journal of Human Genetics journal online.
MARS gene encodes cytoplasmic methionyl-tRNA synthetase.3 Mutations in MARS gene will cause ILLD, an autosomal recessive entity characterized by respiratory insufficiency and liver disease in infancy or early childhood. The phenotype of II.1 and II.4 are compatible to the disease description of ILLD. On basis of the ACMG guideline the two mutations can be classified as ’likely pathogenic’.4
Discussion
Exome sequencing of II.4 and I.2 revealed the family was suffering with ILLD by identification of the compound heterozygous mutations in MARS gene. In 2013, van Meel et al.1 described a patient (P1) with multi-organ dysfunction. Hadchouel et al.2 reported patients from Réunion islands, Tunisia and France. The main clinical features are pulmonary alveolar proteinosis and liver dysfunction. To further delineate the clinical features of ILLD, we summarized the phenotypes of reported patients together with the two Chinese siblings in Table 1. Lung is the most affected organ. All patients developed interstitial lung disease since infancy. Majority of them showed failure to thrive and developmental delay. Liver disease was noticed in most patients except the ones from Tunisia and France. However, liver dysfunction is not life threatening. Liver function panel of II.4 (Supplementary Table 1) demonstrated his liver dysfunction alleviated at 39 months. Total bilirubin (TBIL) was in the normal range, and direct bilirubin (DBIL) was slightly below the lower limit, indicating his cholestasis relieved. Anemia was presented in P1 and the Chinese boys. In the manuscript that reported the clinical features of the Réunion patients,5 we noticed more than half of them also had anemia. Therefore, main ILLD features include interstitial lung disease, developmental delay, postnatal growth failure, non-life-threatening liver dysfunction and anemia.
It is noteworthy that anemia could also be observed in patients harboring mutations in other aminoacyl-tRNA synthetase (ARS) genes. Patients with homozygous LARS mutations presented microcytic anemia.6 Mitochondrial ARS genes, YARS27, 8, 9 and LARS210 have been reported to cause sideroblastic anemia. This suggests ARS plays an essential role in hematologic system.
Eight ILLD mutations have been reported to our knowledge (Supplementary Table 3). Although some patients carry the same MARS genotypes, clinical severity is different from each other. This indicates phenotype of MARS mutations is highly heterogeneous.
Heterozygous MARS mutations were reported to cause Charcot–Marie–Tooth disease, axonal, type 2U (CMT2U, OMIM 616280) in two cases.11, 12 CMT2U is a late-adult onset neurologic disorder. In one family, compound heterozygous MARS mutations were linked to spastic paraplegia-70 (SPG70).13 However, more evidence is required to establish the pathogenicity (Supplementary Table 3). All the mutations and the carrier phenotype were submitted to the online MARS database.14
MARS protein3 is illustrated in Figure 2d. The catalytic domain is composed of two Rossmann fold domains and one ’connective polypeptide’ (CP) insertion.15 Most of the ILLD mutations locate in the catalytic domain (Figure 2d). The two CMT2U mutations locate in the stem-contact fold domain and the α-helix bundle domain. By searching the family history, heterozygous ILLD mutation carriers do not develop CMT2U in their elder age. This might imply different mechanism of ILLD and CMT2U.
In conclusion, we describe the genetic findings and clinical features of the first Chinese ILLD family. ILLD clinical features can include interstitial lung disease, developmental delay, postnatal growth failure, non-life-threatening liver dysfunction and anemia. To date, most of the ILLD mutations locate in the catalytic domain of MARS.
References
van Meel, E., Wegner, D. J., Cliften, P., Willing, M. C., White, F. V., Kornfeld, S. et al. Rare recessive loss-of-function methionyl-tRNA synthetase mutations presenting as a multi-organ phenotype. BMC Med. Genet. 14, 106 (2013).
Hadchouel, A., Wieland, T., Griese, M., Baruffini, E., Lorenz-Depiereux, B., Enaud, L. et al. Biallelic mutations of methionyl-tRNA synthetase cause a specific type of pulmonary alveolar proteinosis prevalent on Reunion Island. Am. J. Hum. Genet. 96, 826–831 (2015).
Deniziak, M. A. & Barciszewski, J. Methionyl-tRNA synthetase. Acta. Biochim. Pol. 48, 337–350 (2001).
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the association for molecular pathology. Genet. Med. 17, 405–424 (2015).
Enaud, L., Hadchouel, A., Coulomb, A., Berteloot, L., Lacaille, F., Boccon-Gibod, L. et al. Pulmonary alveolar proteinosis in children on La Reunion Island: a new inherited disorder? Orphanet. J. Rare Dis. 9, 85 (2014).
Casey, J., McGettigan, P., Lynam-Lennon, N., McDermott, N., Regan, M., Conroy, R. et al. Identification of a mutation in LARS as a novel cause of infantile hepatopathy. Mol. Genet. Metab. 106, 351–358 (2012).
Ardissone, A., Lamantea, E., Quartararo, J., Dallabona, C., Carrara, F., Moroni, I. et al. A novel homozygous YARS2 mutation in two Italian siblings and a review of literature. JIMD. Rep. 20, 95–101 (2015).
Nakajima, J., Eminoglu, T. F., Vatansever, G., Nakashima, M., Tsurusaki, Y., Saitsu, H. et al. A novel homozygous YARS2 mutation causes severe myopathy, lactic acidosis, and sideroblastic anemia 2. J. Hum. Genet. 59, 229–232 (2014).
Riley, L. G., Cooper, S., Hickey, P., Rudinger-Thirion, J., McKenzie, M., Compton, A. et al. Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia—MLASA syndrome. Am. J. Hum. Genet. 87, 52–59 (2010).
Riley, L. G., Rudinger-Thirion, J., Schmitz-Abe, K., Thorburn, D. R., Davis, R. L., Teo, J. et al. LARS2 variants associated with hydrops, lactic acidosis, sideroblastic anemia, and multisystem failure. JIMD. Rep. 28, 49–57 (2016).
Hyun, Y. S., Park, H. J., Heo, S. H., Yoon, B. R., Nam, S. H., Kim, S. B. et al. Rare variants in methionyl- and tyrosyl-tRNA synthetase genes in late-onset autosomal dominant Charcot-Marie-Tooth neuropathy. Clin. Genet. 86, 592–594 (2014).
Gonzalez, M., McLaughlin, H., Houlden, H., Guo, M., Yo-Tsen, L., Hadjivassilious, M. et al. Exome sequencing identifies a significant variant in methionyl-tRNA synthetase (MARS) in a family with late-onset CMT2. J Neurol. Neurosurg. Psychiatry. 84, 1247–1249 (2013).
Novarino, G., Fenstermaker, A. G., Zaki, M. S., Hofree, M., Silhavy, J. L., Heiberg, A. D. et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science 343, 506–511 (2014).
MARS variant database. http://databases.lovd.nl/shared/genes/MARS.
Casina, V. C., Lobashevsky, A. A., McKinney, W. E., Brown, C. L. & Alexander, R. W. Role for a conserved structural motif in assembly of a class I aminoacyl-tRNA synthetase active site. Biochemistry. 50, 763–769 (2011).
Acknowledgements
We thank the patients and their family members for their participation. This work was funded by National Natural Science Foundation of China (81400872 to YS, 81170811, 30973216 to WJQ), and Shanghai Health Bureau (20134005 to WJQ).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Sun, Y., Hu, G., Luo, J. et al. Mutations in methionyl-tRNA synthetase gene in a Chinese family with interstitial lung and liver disease, postnatal growth failure and anemia. J Hum Genet 62, 647–651 (2017). https://doi.org/10.1038/jhg.2017.10
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2017.10
This article is cited by
-
Fatal systemic disorder caused by biallelic variants in FARSA
Orphanet Journal of Rare Diseases (2022)
-
The recurrent missense mutation p.(Arg367Trp) in YARS1 causes a distinct neurodevelopmental phenotype
Journal of Molecular Medicine (2021)
-
Charcot–Marie–Tooth disease type 2A with an autosomal-recessive inheritance: the first report of an adult-onset disease
Journal of Human Genetics (2018)

{kind=link}