
Abstract
Genomic imprinting is an epigenetic phenomenon that leads to parent-specific differential expression of a subset of genes. Most imprinted genes form clusters, or imprinting domains, and are regulated by imprinting control regions. As imprinted genes have an important role in growth and development, aberrant expression of imprinted genes due to genetic or epigenetic abnormalities is involved in the pathogenesis of human disorders, or imprinting disorders. Beckwith–Wiedemann syndrome (BWS) is a representative imprinting disorder characterized by macrosomia, macroglossia and abdominal wall defects, and exhibits a predisposition to tumorigenesis. The relevant imprinted chromosomal region in BWS is 11p15.5, which consists of two imprinting domains, IGF2/H19 and CDKN1C/KCNQ1OT1. BWS has five known causative epigenetic and genetic alterations: loss of methylation (LOM) at KvDMR1, gain of methylation (GOM) at H19DMR, paternal uniparental disomy, CDKN1C mutations and chromosomal rearrangements. Opposite methylation defects, GOM and LOM, at H19DMR are known to cause clinically opposite disorders: BWS and Silver–Russell syndrome, respectively. Interestingly, a recent study discovered that loss of function or gain of function of CDKN1C also causes clinically opposite disorders, BWS and IMAGe (intrauterine growth restriction, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital anomalies) syndrome, respectively. Furthermore, several clinical studies have suggested a relationship between assisted reproductive technology (ART) and the risk of imprinting disorders, along with the existence of trans-acting factors that regulate multiple imprinted differentially methylated regions. In this review, we describe the latest knowledge surrounding the imprinting mechanism of 11p15.5, in addition to epigenetic and genetic etiologies of BWS, associated childhood tumors, the effects of ART and multilocus hypomethylation disorders.
Similar content being viewed by others

Introduction
Genomic imprinting is an epigenetic phenomenon that leads to parent-specific differential expression of a subset of mammalian genes. So far, >100 imprinted genes have been identified in humans and mice, and most imprinted genes often form clusters, or imprinting domains. The expression of imprinted genes within these domains is regulated by imprinting control regions (ICRs).1, 2 ICRs are identical to differentially methylated regions (DMRs), which are characterized by DNA methylation on one of the two parental alleles, or maternally methylated DMRs and paternally methylated DMRs. In addition, there are two classes of imprinted DMRs, gametic DMRs and somatic DMRs. Gametic DMRs acquire DNA methylation during gametogenesis, and the methylation is maintained from zygote to somatic cells during all developmental stages. Most gametic DMRs are identical to ICRs. Methylations of somatic DMRs are established during early embryogenesis after fertilization under the control of nearby ICRs.1, 2
As most imprinted genes have an important role in the growth and development of embryos, placental formation, and metabolism, aberrant expression of imprinted genes due to epigenetic or genetic abnormalities is often implicated in the pathogenesis of human disorders such as congenital anomalies and tumors.1, 2 Epigenetic abnormality leading to aberrant expression of imprinted genes mostly includes aberrant hypomethylation or hypermethylation at ICRs. Genetic abnormalities include uniparental disomies, chromosomal deletions, duplications, translocations, inversions of imprinting domains, and point mutations of imprinted genes. Representative imprinting disorders and their corresponding imprinted loci are as follows: Beckwith–Wiedemann syndrome (BWS) at 11p15.5, Prader-Willi/Angelman syndromes at 15q11-q13, pseudoparahypothyroidism type 1b at 20q13.3, Silver–Russell syndrome (SRS) at 11p15.5 and chromosome 7, and transient neonatal diabetes mellitus type 1 at 6q24.
Here, we review BWS, focusing especially on imprinting mechanisms of 11p15.5, epigenetic and genetic etiologies leading to aberrant expression of corresponding imprinted genes, relationships between epigenetic/genetic alterations and clinical features, and associated childhood tumors. We also describe the relationship between assisted reproductive technology (ART) and imprinting disorders and explore multilocus hypomethylation disorders (MHDs).
Clinical features and causative alterations of BWS
BWS (OMIM #130650) is a pediatric overgrowth disorder that is characterized by the peculiar traits of prenatal and postnatal macrosomia, macroglossia, abdominal wall defects as originally described by Beckwith and Wiedemann.3, 4 The incidence has been reported to be 1 in 13 700,5 and the male-to-female ratio is ∼1:1. BWS also shows other variable features, including anterior ear lobe creases and/or posterior helical pits, neonatal hypoglycemia, intra-abdominal visceromegaly, cytomegaly of adrenal fetal cortex, renal abnormalities, hemihyperplasia and cleft palate. The development of embryonal tumors (for example, Wilms’ tumor, hepatoblastoma and rhabdomyosarcoma) is an important feature of BWS, and the overall tumor risk has been estimated at 7.5% with a range of 4–21%.6, 7 Although several clinical criteria have been proposed so far,8, 9, 10 there is no single unified criterion. However, a criteria scheme proposed by Weksberg et al.11 is generally accepted for clinical diagnosis: the presence of at least three major findings, or two major findings and one minor finding, from those reported in Table 1. Simpson–Golabi–Behmel syndrome, Costello syndrome, Perlman syndrome, Sotos syndrome and mucopolysaccharidosis VI (Maroteaux–Lamy syndrome) are considered as differential diagnoses.
Approximately 85% of BWS cases are sporadic; the other 15% are familial showing autosomal dominant inheritance. The relevant imprinted chromosomal region in BWS, 11p15.5, consists of two independent imprinting domains, IGF2/H19 and CDKN1C/KCNQ1OT1. Several causative alterations have been identified for sporadic cases of BWS: loss of methylation (LOM) at KvDMR1 (∼50%), gain of methylation (GOM) at H19DMR (∼5%), paternal uniparental disomy (patUPD; ∼20%), CDKN1C mutations (∼5%), duplications of 11p15 (<1%) and translocations or inversions involving 11p15 (<1%) (Table 2).11, 12, 13 However, no alteration of 11p15.5 can be found for ∼20% of BWS cases. Interestingly, among these causative alterations, methylation abnormalities, such as KvDMR1-LOM and H19DMR-GOM, and patUPD are mosaic in the patients; however, other genetic alterations including CDKN1C mutation are essentially not mosaic.
Imprinting mechanisms of 11p15.5 and etiologies of BWS
The IGF2/H19 domain
The important genes in this domain are insulin-like growth factor 2 (IGF2) and H19. IGF2 is expressed from the paternal allele, and the gene product has an important role in development and growth, whereas H19 is a maternally-expressed, non-coding RNA, which may function as a tumor suppressor, but whose precise biological role remains unresolved.14, 15 One study reported that H19 is a miRNA precursor expressed in human keratinocytes and neonatal mice, suggesting its involvement during development.16 The ICR of this domain is H19DMR, which is located 2 kb upstream of H19 and is methylated on the paternal but not the maternal allele (Figure 1). The methylation of H19DMR is established during spermatogenesis.17, 18 This ICR, which contains seven CCCTC-binding factor (CTCF) binding sites in human and four in mouse, regulates the reciprocal expression of IGF2 and H19 by functioning as a chromatin insulator. On the maternal allele, CTCF binding at the insulator elements within unmethylated H19DMR blocks enhancers downstream of H19 from accessing IGF2 promoters. On the paternal allele, as the methylation of H19DMR prevents CTCF binding, the enhancers can access IGF2 promoters.19, 20 Thus, these mechanisms lead to paternal expression of IGF2 and maternal expression of H19. Recent chromatin conformation studies showed that CTCF binding at regulatory regions other than H19DMR and the enhancers surrounding the domain formed allele-specific chromatin loops, depending on the methylation of H19DMR, in order to regulate the expression of IGF2 and H19. For these CTCF-dependent chromatin loop formations, the recruitment of cohesin to CTCF-binding sites is required and cohesin stabilizes the chromatin conformations.21, 22

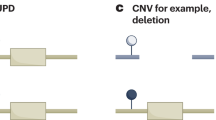
Imprinting domains at 11p15.5. Upper panel indicates the imprinting mechanisms in normal individuals. As for the IGF2/H19 domain, the insulator model is shown. On the maternal chromosome, the binding of CTCF to unmethylated H19DMR blocks enhancers from accessing IGF2 promoters. In contrast, on the paternal allele, as the methylation of H19DMR prevents CTCF binding, the enhancers can access IGF2 promoters. Thus, these mechanisms lead to paternal expression of IGF2 and maternal expression of H19. Please refer to the text for the chromatin loop model. As for the CDKN1C/KCNQ1OT1 domain, on the paternal chromosome, it has been proposed that CDKN1C is repressed by KCNQ1OT1 RNA coating and by a silencer and an insulator near the KvDMR1, which is likely regulated by CTCF. A putative enhancer within the KCNQ1 locus acts on maternal expression of CDKN1C. The lower panel displays causative alterations of BWS. Vertical arrows with maternal translocations and inversions indicate chromosomal break points. Blue: paternal expressed genes; red: maternal expressed genes; green diamond: enhancers (putative enhancer in CDKN1C/KCNQ1OT1 domain); wavy line: non-coding RNA transcribed from the paternal KCNQ1OT1 gene.
In ∼5% of BWS patients, gain of DNA methylation occurs on the normally unmethylated maternal H19DMR (H19DMR-GOM) (Figure 1, Table 2). Aberrant DNA methylation at maternal H19DMR is accompanied by a change of histone modification from accessible H3K9ac and bivalent H3K4me2/H3K27me3 to repressive H3K9me3 and H4K20me3.22 The aberrant DNA methylation prevents CTCF binding to maternal H19DMR, and the chromatin loop formation changes from maternal-type to paternal-type due to aberrant DNA methylation and histone modification change. The chromatin conformation change drags the enhancers into the vicinity of IGF2, leading to biallelic expression and loss of imprinting of IGF2 and reduced expression of H19. Overexpression of IGF2 and reduced expression of H19 induce the BWS phenotype. One representative phenotype of H19DMR-GOM is hemihyperplasia (Table 2).12
The majority of GOM cases show an isolated epigenetic alteration; however, ∼20% of GOM cases are associated with genetic alterations, which are variable length microdeletions including CTCF-binding sites and point mutations and a deletion at the octamer-binding protein (OCT) binding site.23 These genetic alterations lead to maternal H19DMR not being able to maintain an unmethylated status.24, 25 However, the mechanism by which isolated H19DMR-GOM occurs is still unknown. As a certain number of cases with isolated H19DMR-GOM show variable hypermethylation, patients have an epigenetic mosaic of normal cells and aberrantly methylated cells, indicating that GOM occurs in the post-fertilization stage, especially after implantation.26, 27, 28
Epimutation of H19DMR is also a cause of SRS (OMIM #180860), which is characterized by opposite clinical phenotypes such as growth restriction.29 In ∼40% of SRS patients, methylated paternal H19DMR becomes hypomethylated (H19DMR-LOM), leading to increased H19 expression and decreased IGF2 expression.30 In contrast to BWS, essentially no mutations of H19DMR have been found in SRS patients with H19DMR-LOM. One SRS patient did exhibit a de novo mutation in H19DMR; however, as the mutation did not involve any putative protein-binding sites, it remains unknown if the mutation affected the methylation status of H19DMR.23 As a majority of cases with H19DMR-LOM show variable hypermethylation, LOM also occurs in the post-fertilization stage.29, 31
The CDKN1C/KCNQ1OT1 domain
The important genes in this domain are CDKN1C and KCNQ1OT1. CDKN1C encodes cyclin-dependent kinase inhibitor and shows preferential maternal expression. KCNQ1OT1 is a paternally-expressed, long non-coding RNA. The ICR of this domain is KvDMR1, located in intron 10 of the KCNQ1 gene, and it is methylated on the maternal but not the paternal allele (Figure 1). The methylation of KvDMR1 is established during oogenesis.17, 18 As KvDMR1 overlaps with the promoter of KCNQ1OT1, the paternal KCNQ1OT1 is expressed from unmethylated paternal KvDMR1 in the opposite direction of KCNQ1, and it functions to silence genes in the domain in cis.32 In mice, Kcnq1ot1 RNA interacts with G9a and the PRC2 complex, which mediates repressive histone modifications such as H3K9me3 and H3K27me3, and forms a repressive nuclear compartment that leads to gene silencing within the domain, including of Cdkn1c. However, this mechanism is specific to the placenta.33, 34 In mouse liver, Kcnq1ot1 RNA interacts with Dnmt1 to mediate maintenance of somatic DMRs, some of which overlap the Cdkn1c promoter, and silences genes within the domain.35 In addition, the identification of paternal allele-specific CTCF binding to KvDMR1 suggests that a repressive element within KvDMR1 likely regulated by CTCF acts to silence paternal Cdkn1c specifically and without promoter methylation in a subset of tissues (for example, kidney, liver and lung).36, 37 In humans, although KCNQ1OT1 coats the neighboring regions of chromatin-containing CDKN1C, the CDKN1C promoter does not show DMR, and H3K9me may not be involved in CDKN1C repression.38, 39 In two BWS families with significantly reduced expression of CDKN1C, maternal microdeletions for most parts of the KCNQ1 gene impact KvDMR1 and the following KCNQ1OT1 gene, but not CDKN1C, suggesting the presence of an enhancer element within the KCNQ1 locus for maternal expression of CDKN1C.40, 41 In addition, the DNA fragment containing KvDMR1 has been shown to have both silencer and insulator activities with CTCF binding.42 Therefore, researchers have proposed that CDKN1C is repressed on the paternal chromosome by KCNQ1OT1 RNA coating and by both a silencer and an insulator near KvDMR1, which is likely regulated by CTCF binding that prevents the CDKN1C promoter from accessing the enhancer downstream of KvDMR1.41
Loss of DNA methylation on the normally methylated maternal KvDMR1 (KvDMR1-LOM) accounts for ∼50% of BWS patients (Figure 1, Table 2). KvDMR1-LOM is accompanied by loss of H3K9me2, and this leads to expression of KCNQ1OT1 RNA, which in turn results in repression of CDKN1C expression on the maternal chromosome with the mechanism as proposed above.39, 41, 43, 44 In addition, only three families have been reported to have maternal transmission of the microdeletions containing KvDMR1, leading to reduced expression of CDKN1C.40, 41, 45 Such reduced expression induces the BWS phenotype.
Representative phenotypes of KvDMR1-LOM include omphalocele and hemihyperplasia (Table 2).12 As certain cases with isolated KvDMR1-LOM also display variable hypomethylation, patients are epigenetic mosaic, which indicates that LOM occurs in the post-fertilization stage.46, 47, 48, 49 Interestingly, monozygotic twins discordant for BWS are found predominantly for females. This could be in part explained by reduction of the amount of DNMT1 to maintain KvDMR1 methylation during the overlap in timing shared by X-inactivation and twinning.46
Paternal uniparental disomy
patUPD of 11p is found in ∼20% of patients (Figure 1, Table 2). All patients with patUPD are mosaic for patUPD cells and normal biparental cells, indicating occurrence of somatic recombination at the post-fertilization stage. Thus, UPD is always paternal isodisomy. Romanelli et al.50 analyzed nine patients with patUPD using SNP arrays, and found that the minimal patUPD size was ∼2.7 Mb from telomere to the centromeric side of KvDMR1 (Figure 1). As the minimal region includes both ICRs, H19DMR and KvDMR1, both H19DMR hypermethylation and KvDMR1 hypomethylation occur depending on the percentage of mosaicism and IGF2 overexpression; reduced expression of CDKN1C must be induced. Meanwhile, Romanelli et al.50 could not find hot-spots of mitotic recombination break points. One representative phenotypes of patUPD is hemihyperplasia, which can affect various regions of the body (Table 2).12
The largest patUPD size is the whole genome, denoted as genome-wide patUPD. Non-mosaic genome-wide patUPD results in hydatidiform mole formation. In contrast, individuals with mosaic genome-wide patUPD are born alive. To date, 11 patients with genome-wide patUPD have been reported.51, 52, 53, 54, 55, 56, 57, 58 Among these, half of the patients were diagnosed as BWS and only two displayed phenotypes associated with transient neonatal diabetes mellitus type 1 and upd(14)pat syndrome.51, 57 In addition, one patient with parthenogenic chimerism/mosaicism showed a SRS-like phenotype.59 These findings suggested an epi-dominant effect of aberrant methylation of 11p15 on clinical features. However, genome-wide patUPD patients with BWS phenotypes display atypical and varied phenotypes. This would be attributable to a paternal epigenotype for all ICRs and being homozygous for mutations of autosomal recessive genes. In addition, patients exhibit a significantly increased predisposition for tumor development. This also would be attributable to inactivation of tumor suppressor genes, or activation of oncogenes.
CDKN1C mutation
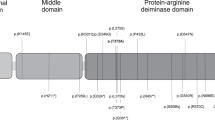
As mentioned before, CDKN1C is a gene responsible for the pathogenesis of BWS within the CDKN1C/KCNQ1OT1 domain, and it exhibits maternal preferential expression. This gene contains three exons divided by two introns encoding a 316 amino-acid protein, which is a strong inhibitor of several G1 cyclin/Cdk complexes and a negative regulator of cell proliferation.60, 61 The CDKN1C (p57KIP2) protein consists of three distinct domains: a cyclin-dependent kinase inhibitory domain, a proline and alanine repeat domain, and a QT domain (Figure 2). The cyclin-dependent kinase inhibitory domain contains a cyclin-binding region, a cyclin-dependent kinase-binding region and a 310 helix, which are both necessary and sufficient to bind and inhibit cyclin-dependent kinase activity.60, 61, 62 Proline and alanine repeats interact with the LIM domain kinase 1 and regulate actin dynamics.62, 63, 64 The QT domain contains a proliferating cell nuclear antigen (PCNA) binding domain, which can prevent DNA replication in vitro and S-phase entry in vivo, and a nuclear localization signal.60, 62, 65

Mutations of CDKN1C in BWS and IMAGe syndrome.67, 68, 69 The mutations in BWS are loss-of-function mutations, which are either amino-acid substitution mutations localized to the cyclin-dependent kinase inhibitory domain or truncating mutations. The mutations in IMAGe syndrome that lead to growth restriction are missense mutations specific to the PCNA-binding domain, considered a gain-of-function mutation. Blue: amino-acid substitution mutations; red: truncating mutations.
The mutations are found in ∼5% of sporadic cases, whereas dominant maternal transmission of germline mutations are found in 40% of familial BWS cases.11, 12 The mutations in sporadic cases should occur on the maternal allele because of maternal expression of CDKN1C. Approximately 30 mutations have been reported since the initial report by Hatada et al.66, 67, 68 These mutations are either missense mutations localized to the cyclin-dependent kinase inhibitory domain or nonsense mutations, both of which result in loss of function and lead to the BWS phenotype (Figure 2). Representative phenotypes of CDKN1C mutations include omphalocele and cleft palate (Table 2).12
Recently, missense mutations in the PCNA binding domain have reported in the undergrowth developmental disorder IMAGe syndrome (OMIM #614732), which is characterized by intrauterine growth restriction, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital anomalies (Figure 2).69 Only maternal transmission of the mutation results in IMAGe syndrome, consistent with imprinting inheritance. Targeted expression of patient-associated mutations in Drosophila caused restricted eye and wing growth, suggesting a gain-of-function mechanism. The gain of function might be due to abolishment of PCNA dependent CDKN1C monoubiquitination.69 It is intriguing that two opposite phenotypes, BWS and IMAGe syndrome, occur because of the mutations of the same CDKN1C gene. The biological role and molecular mechanism of the monoubiquitination should be elucidated to understand how the two disorders differ.
Chromosomal rearrangements
Chromosomal rearrangements involving 11p—including duplications, balanced translocations and inversions—occur in <2% of BWS patients (Figure 1, Table 2). Paternal duplications of 11p15 result in BWS due to overexpression of IGF2,70 whereas maternal duplications of 11p15 result in SRS.71 SRS and BWS phenotypes associated with 11p duplications in a single family have been reported.72 In this family, a SRS child was born from a mother with BWS phenotypes due to paternal duplication. Representative phenotypes of BWS due to duplication causes developmental delay (Table 2).12
So far at least 12 cases harboring translocations or inversions have been reported, with most break points of the translocations and inversions falling in the KCNQ1 locus.73, 74, 75, 76, 77 BWS develops when these are transmitted maternally. Three cases harboring inv(11)(p13;p15.5), inv(11)(p11.2;p15.5) and t(11;17)(p15.5;q21.3), respectively, have been seen to exhibit KvDMR1-LOM. However, a fibroblast with inv(11)(p15.5;q13) and a rhabdoid tumor line with t(11;22) have shown signs of reduced expression of CDKN1C with normal methylation at KvDMR1. These are consistent with the enhancer blocking insulator model mentioned before.75, 76, 77 However, the remaining cases showed neither KvDMR1-LOM nor reduced expression of CDKN1C. Therefore, the developmental mechanism for BWS harboring translocations and inversions is largely unknown.
Different risks for childhood tumors in each alteration type
Embryonal malignancies are the tumors most commonly associated with BWS—for example, Wilms’ tumor, hepatoblastoma, adrenocortical carcinoma, rhabdomyosarcoma and neuroblastoma—but other malignant or benign tumors are occasionally observed.6, 7 Although overall tumor risk is ∼7.5%, it is different for each causative alteration (Table 2). H19DMR-GOM and patUPD show the highest tumor risk, at >25%, especially for Wilms’ tumor and hepatoblastoma. KvDMR1-LOM has a rate of developing hepatoblastoma, rhabdomyosarcoma and gonadoblastoma other than Wilms’ tumors of ∼5%.10 The lowest risk is found in CDKN1C mutations with <5% of cases affected. Only neuroblastomas have been found in patients with CDKN1C mutations.78, 79 Wilms’ tumors are frequently seen in patients with H19DMR-GOM or patUPD, but never seen in patients with KvDMR1-LOM or CDKN1C mutations, suggesting a critical role of IGF2 overexpression in Wilms’ tumor development. In fact, IGF2 loss of imprinting is found in 60–70% of sporadic Wilms’ tumors without 11p LOH.80, 81 Furthermore, IGF2 loss of imprinting was also observed in ∼21% of sporadic hepatoblastomas without 11p LOH, and aberrant methylations at H19DMR, H19 promoter, IGF2-DMR0 or IGF2-DMR2 were observed in ∼55% of sporadic hepatoblastomas without 11p LOH, suggesting the importance of IGF2 overexpression for hepatoblastoma development as well (Rumbajan JM et al., submitted).82 In addition, although many kinds of adult tumors display reduced CDKN1C expression, of which certain cases show KvDMR1-LOM, the risk of embryonal tumorigenesis is low in BWS patients with KvDMR1-LOM or CDKN1C mutations, suggesting different contributions of CDKN1C to tumor development between adulthood and childhood.
ART and BWS
The worldwide usage of ART has increased. Several reports have raised concerns that the risk of imprinting disorders, such as BWS and Angelman syndrome, are increased in children conceived by ART, especially through in vitro fertilization and intracytoplasmic sperm injection, as the first reported associations in 2002 and 2003 between Angelman syndrome and BWS, respectively, with ART.83, 84, 85 The risk of BWS is estimated to be six to nine times higher in children conceived by ART than in children conceived naturally.86 The causative alteration for most of ART-related BWS is KvDMR1-LOM. The cause of Angelman syndrome is also LOM at SNRPN.
Animal studies have suggested that ovarian stimulation and culture medium for the embryo can affect DNA methylation and the expression of several imprinted genes.87, 88, 89, 90 In fact, ‘large offspring syndrome’ has been described as caused by LOM of the maternal Igf2r after sheep embryo culture.91 However, in humans, although ovarian stimulation may predispose to aberrant methylation at imprinted loci,92 it is still unclear whether the procedure of ART affects methylation at imprinted loci because ART populations are different from naturally conceived populations having low fertility rates, increased frequency of reproductive loss and advanced age.93 Indeed, male infertility is strongly associated with aberrant methylation at both maternal and paternal alleles.94, 95 It has been reported that there are no phenotypic differences between ART-related BWS and naturally conceived BWS.96 However, Lim et al.97 provided evidence that ART-related BWS had a significantly lower frequency of exomphalos and higher risk of tumor development than Wilms’ tumor. Larger size studies are needed to better understand the correlation between ART and BWS.
Multilocus hypomethylation disorders
Hypomethylations at several other imprinted loci have been reported to occur in BWS patients with KvDMR1-LOM.47, 48, 49, 97 As this phenomenon was also seen in patients with transient neonatal diabetes mellitus type 1 and SRS, a new entity of imprinting disorders such as MHD has been proposed.49, 98, 99, 100, 101 The literature indicates an overall frequency of multilocus hypomethylation in BWS patients with KvDMR1-LOM of 20% (49/244).49, 98, 99, 100, 101 IGF2R-DMR2, GNAS, NESPAS, PEG1 and PLAGL1 are frequently hypomethylated DMRs. In BWS patients, only maternally methylated DMRs displayed hypomethylation; however, several SRS patients with H19DMR-LOM showed hypomethylation at DLK1/GTL2 IG-DMR, another paternally methylated DMR, indicating involvement of both maternally and paternally methylated DMRs. In addition, a certain SRS showed hypomethylation at both H19DMR and KvDMR1.48, 100 As these hypomethylations were mosaic, they were presumed to be due to a post-fertilization event.
Lim et al.97 reported that ART-related BWS show multilocus hypomethylation more frequently than naturally conceived BWS; however, no such difference was observed by Rossignol et al.47 One study reported that BWS with multilocus hypomethylation displayed characteristics not usually associated with BWS, such as speech retardation, peri/postnatal apnea, feeding difficulties and hearing problems; additionally, nevus flammeus and hemihypertrophy were significantly lower in patients with multilocus hypomethylation.49 However, three other studies reported no difference in clinical features between MHDs and monolocus hypomethylation disorders.47, 48, 97 As the studies so far have analyzed only limited numbers of DMRs, further investigation of all known DMRs are needed.
The involvement of trans-acting factors in these MHD has been suggested. In fact, in one study, homozygous and compound heterozygous mutations of ZFP57, which encodes a KRAB zinc-finger protein and is required for the post-fertilization maintenance of maternal and paternal methylation imprinting at multiple loci, were found in transient neonatal diabetes mellitus type 1 patients with multilocus hypomethylation.102 However, no mutations were found in 27 BWS patients with KvDMR1-LOM probably without multilocus hypomethylation.103 KAP1, a protein associated with ZFP57, interacts with DNMT1 and binds to many ICRs in embryonic stem cells to maintain DNA and histone methylation.104, 105 Mice with maternal deletions of Trim28, a homolog of human KAP1, show aberrant DNA demethylation at a few ICRs.106 Mutation searches of KAP1 in MHD patients have not been reported to date.
Other candidates for trans-acting factors are NLRP2 and NLRP7, which are members of the Nod-like receptor protein (NLRP) family. Some NLRPs are components of the inflammasome, an assembly that is implicated in the sensing of, and inflammatory reaction to, extracellular pathogens and intracellular noxious compounds.107 Mutations of NLRP2 were identified in a familial case of BWS with KvDMR1-LOM and PEG1-LOM, suggesting a role of NLRP2 in the establishment or maintenance of ICRs.108 However, the mutation has not been corroborated by other studies yet. Mutations of NLRP7 and C6ORF221 account for familial biparental hydatidiform mole, which is a maternal effect recessive disorder resulting from failure of maternal imprints.109, 110 Mutation searches of NLRP7 were performed on the mother of a patient showing both transient neonatal diabetes mellitus type 1 and BWS features with multilocus hypomethylation, but they were unsuccessful.99 In addition, DNMT3L, which is required for establishing maternal imprints, was not mutated in two BWS patients with severe multilocus hypomethylation.49 Mutation searching of all candidate trans-acting factors should be performed over a large number of MHD patients to explore this matter further.
In addition, one circular chromosome conformation capture (4C) study revealed that maternal H19DMR interacts with the autosomal region, and imprinting domains were strongly overrepresented in the 4C library, suggesting the involvement of higher order chromatin interaction in the regulation of imprinting.111 The involvement of physical chromosome interactions in MHD should also be further elucidated.
Conclusions
Although H19DMR-GOM, KvDMR1-LOM, patUPD and CDKN1C mutations, and chromosomal rearrangements account for ∼80% of BWS phenotypes, several questions about these alterations still remain to be clarified. In addition, at least 20% of patients do not have these associated alterations, suggesting the existence of other, unknown epigenetic/genetic defects. Furthermore, other issues, such as the effect of ART on imprinting disorders and the mechanism of multilocus imprinting establishment/maintenance, should be clarified. Further investigations of all of these issues must be elucidated in order to understand the molecular basis of BWS and related imprinting disorders.
References
Abramowitz, L. K. & Bartolomei, M. S. Genomic imprinting: recognition and marking of imprinted loci. Curr. Opin. Genet. Dev. 22, 72–78 (2012).
Tomizawa, S. & Sasaki, H. Genomic imprinting and its relevance to congenital disease, infertility, molar pregnancy and induced pluripotent stem cell. J. Hum. Genet. 57, 84–91 (2012).
Beckwith, JB. Extreme cytomegaly of the adrenal fetal cortex, omphalocele, hyperplasia of kidneys and pancreas, and Leydig-cell hyperplasia: Another syndrome? Abstract, Western Society for Pediatric Research, Los Angeles, CA (1963).
Wiedemann, H. Complexe malformatif familial avec hernie ombilicale et macroglossia, un syndrome nouveau. J. Genet. Hum. 13, 223–232 (1964).
Thorburn, M. J., Wright, E. S., Miller, C. G. & Smith-Read, E. H. Exomphalos-macroglossia-gigantism syndrome in Jamaican infants. Am J Dis Child 119, 316–321 (1970).
Rump, P., Zeegers, M. P. & van Essen, A. J. Tumor risk in Beckwith-Wiedemann syndrome: a review and meta-analysis. Am. J. Med. Genet. A. 136, 95–104 (2005).
Lapunzina, P. Risk of tumorigenesis in overgrowth syndromes: a comprehensive review. Am. J. Med. Genet. C. Semin. Med. Genet. 137C, 53–71 (2005).
Elliott, M., Bayly, R., Cole, T., Temple, I. K. & Maher, E. R. Clinical features and natural history of Beckwith-Wiedemann syndrome: presentation of 74 new cases. Clin. Genet. 46, 168–174 (1994).
DeBaun, M. R. & Tucker, M. A. Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry. J. Pediatr. 132, 398–400 (1998).
Weksberg, R., Nishikawa, J., Caluseriu, O., Fei, Y. L., Shuman, C., Wei, C. et al. Tumor development in the Beckwith-Wiedemann syndrome is associated with a variety of constitutional molecular 11p15 alterations including imprinting defects of KCNQ1OT1. Hum. Mol. Genet. 10, 2989–3000 (2001).
Weksberg, R., Shuman, C. & Beckwith, J. B. Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 18, 8–14 (2010).
Choufani, S., Shuman, C. & Weksberg, R. Beckwith-Wiedemann syndrome. Am. J. Med. Genet. C. Semin. Med. Genet. 154C, 343–354 (2010).
Sasaki, K., Soejima, H., Higashimoto, K., Yatsuki, H., Ohashi, H., Yakabe, S. et al. Japanese and North American/European patients with Beckwith-Wiedemann syndrome have different frequencies of some epigenetic and genetic alterations. Eur. J. Hum. Genet. 15, 1205–1210 (2007).
Hao, Y., Crenshaw, T., Moulton, T., Newcomb, E. & Tycko, B. Tumour-suppressor activity of H19 RNA. Nature 365, 764–767 (1993).
Onyango, P. & Feinberg, A. P. A nucleolar protein, H19 opposite tumor suppressor (HOTS), is a tumor growth inhibitor encoded by a human imprinted H19 antisense transcript. Proc. Natl Acad. Sci. USA 108, 16759–16764 (2011).
Cai, X. & Cullen, B. R. The imprinted H19 noncoding RNA is a primary microRNA precursor. RNA 13, 313–316 (2007).
Li, E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat. Rev. Genet. 3, 662–673 (2002).
Sasaki, H. & Matsui, Y. Epigenetic events in mammalian germ-cell development: reprogramming and beyond. Nat. Rev. Genet. 9, 129–140 (2008).
Bell, A. C. & Felsenfeld, G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature 405, 482–485 (2000).
Hark, A. T., Schoenherr, C. J., Katz, D. J., Ingram, R. S., Levorse, J. M. & Tilghman, S. M. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405, 486–489 (2000).
Nativio, R., Wendt, K. S., Ito, Y., Huddleston, J. E., Uribe-Lewis, S., Woodfine, K. et al. Cohesin is required for higher-order chromatin conformation at the imprinted IGF2-H19 locus. PLoS Genet. 5, e1000739 (2009).
Nativio, R., Sparago, A., Ito, Y., Weksberg, R., Riccio, A. & Murrell, A. Disruption of genomic neighbourhood at the imprinted IGF2-H19 locus in Beckwith-Wiedemann syndrome and Silver-Russell syndrome. Hum. Mol. Genet. 20, 1363–1374 (2011).
Demars, J., Shmela, M. E., Rossignol, S., Okabe, J., Netchine, I., Azzi, S. et al. Analysis of the IGF2/H19 imprinting control region uncovers new genetic defects, including mutations of OCT-binding sequences, in patients with 11p15 fetal growth disorders. Hum. Mol. Genet. 19, 803–814 (2010).
Hori, N., Nakano, H., Takeuchi, T., Kato, H., Hamaguchi, S., Oshimura, M. et al. A dyad oct-binding sequence functions as a maintenance sequence for the unmethylated state within the H19/Igf2-imprinted control region. J. Biol. Chem. 277, 27960–27967 (2002).
Fedoriw, A. M., Stein, P., Svoboda, P., Schultz, R. M. & Bartolomei, M. S. Transgenic RNAi reveals essential function for CTCF in H19 gene imprinting. Science 303, 238–240 (2004).
Sparago, A., Russo, S., Cerrato, F., Ferraiuolo, S., Castorina, P., Selicorni, A. et al. Mechanisms causing imprinting defects in familial Beckwith-Wiedemann syndrome with Wilms’ tumour. Hum. Mol. Genet. 16, 254–264 (2007).
Cerrato, F., Sparago, A., Verde, G., De Crescenzo, A., Citro, V., Cubellis, M. V. et al. Different mechanisms cause imprinting defects at the IGF2/H19 locus in Beckwith-Wiedemann syndrome and Wilms’ tumour. Hum. Mol. Genet. 17, 1427–1435 (2008).
Higashimoto, K., Nakabayashi, K., Yatsuki, H., Yoshinaga, H., Jozaki, K., Okada, J. et al. Aberrant methylation of H19-DMR acquired after implantation was dissimilar in soma versus placenta of patients with Beckwith-Wiedemann syndrome. Am. J. Med. Genet. A. 158A, 1670–1675 (2012).
Gicquel, C., Rossignol, S., Cabrol, S., Houang, M., Steunou, V., Barbu, V. et al. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat Genet. 37, 1003–1007 (2005).
Eggermann, T., Begemann, M., Binder, G. & Spengler, S. Silver-Russell syndrome: genetic basis and molecular genetic testing. Orphanet. J. Rare Dis. 5, 19 (2010).
Bliek, J., Terhal, P., van den Bogaard, M. J., Maas, S., Hamel, B., Salieb-Beugelaar, G. et al. Hypomethylation of the H19 gene causes not only Silver-Russell syndrome (SRS) but also isolated asymmetry or an SRS-like phenotype. Am. J. Hum. Genet. 78, 604–614 (2006).
Mancini-Dinardo, D., Steele, S. J., Levorse, J. M., Ingram, R. S. & Tilghman, S. M. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev. 20, 1268–1282 (2006).
Pandey, R. R., Mondal, T., Mohammad, F., Enroth, S., Redrup, L., Komorowski, J. et al. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell. 32, 232–246 (2008).
Terranova, R., Yokobayashi, S., Stadler, M. B., Otte, A. P., van Lohuizen, M., Orkin, S. H. et al. Polycomb group proteins Ezh2 and Rnf2 direct genomic contraction and imprinted repression in early mouse embryos. Dev. Cell. 15, 668–679 (2008).
Mohammad, F., Mondal, T., Guseva, N., Pandey, G. K. & Kanduri, C. Kcnq1ot1 noncoding RNA mediates transcriptional gene silencing by interacting with Dnmt1. Development 137, 2493–2499 (2010).
Fitzpatrick, G. V., Pugacheva, E. M., Shin, J. Y., Abdullaev, Z., Yang, Y., Khatod, K. et al. Allele-specific binding of CTCF to the multipartite imprinting control region KvDMR1. Mol. Cell. Biol. 27, 2636–2647 (2007).
Shin, J. Y., Fitzpatrick, G. V. & Higgins, M. J. Two distinct mechanisms of silencing by the KvDMR1 imprinting control region. EMBO J. 27, 168–178 (2008).
Murakami, K., Oshimura, M. & Kugoh, H. Suggestive evidence for chromosomal localization of non-coding RNA from imprinted LIT1. J. Hum. Genet. 52, 926–933 (2007).
Soejima, H., Nakagawachi, T., Zhao, W., Higashimoto, K., Urano, T., Matsukura, S. et al. Silencing of imprinted CDKN1C gene expression is associated with loss of CpG and histone H3 lysine 9 methylation at DMR-LIT1 in esophageal cancer. Oncogene 23, 4380–4388 (2004).
Niemitz, E. L., DeBaun, M. R., Fallon, J., Murakami, K., Kugoh, H., Oshimura, M. et al. Microdeletion of LIT1 in familial Beckwith-Wiedemann syndrome. Am. J. Hum. Genet. 75, 844–849 (2004).
Algar, E., Dagar, V., Sebaj, M. & Pachter, N. An 11p15 imprinting centre region 2 deletion in a family with Beckwith Wiedemann syndrome provides insights into imprinting control at CDKN1C. PLoS One 6, e29034 (2011).
Du, M., Beatty, L. G., Zhou, W., Lew, J., Schoenherr, C., Weksberg, R. et al. Insulator and silencer sequences in the imprinted region of human chromosome 11p15.5. Hum. Mol. Genet. 12, 1927–1939 (2003).
Higashimoto, K., Urano, T., Sugiura, K., Yatsuki, H., Joh, K., Zhao, W. et al. Loss of CpG methylation is strongly correlated with loss of histone H3 lysine 9 methylation at DMR-LIT1 in patients with Beckwith-Wiedemann syndrome. Am. J. Hum. Genet. 73, 948–956 (2003).
Diaz-Meyer, N., Day, C. D., Khatod, K., Maher, E. R., Cooper, W., Reik, W. et al. Silencing of CDKN1C (p57KIP2) is associated with hypomethylation at KvDMR1 in Beckwith-Wiedemann syndrome. J. Med. Genet. 40, 797–801 (2003).
Zollino, M., Orteschi, D., Marangi, G., De Crescenzo, A., Pecile, V., Riccio, A. et al. A case of Beckwith-Wiedemann syndrome caused by a cryptic 11p15 deletion encompassing the centromeric imprinted domain of the BWS locus. J. Med. Genet. 47, 429–432 (2010).
Weksberg, R., Shuman, C., Caluseriu, O., Smith, A. C., Fei, Y. L., Nishikawa, J. et al. Discordant KCNQ1OT1 imprinting in sets of monozygotic twins discordant for Beckwith-Wiedemann syndrome. Hum. Mol. Genet. 11, 1317–1325 (2002).
Rossignol, S., Steunou, V., Chalas, C., Kerjean, A., Rigolet, M., Viegas-Pequignot, E. et al. The epigenetic imprinting defect of patients with Beckwith-Wiedemann syndrome born after assisted reproductive technology is not restricted to the 11p15 region. J. Med. Genet. 43, 902–907 (2006).
Azzi, S., Rossignol, S., Steunou, V., Sas, T., Thibaud, N., Danton, F. et al. Multilocus methylation analysis in a large cohort of 11p15-related foetal growth disorders (Russell Silver and Beckwith Wiedemann syndromes) reveals simultaneous loss of methylation at paternal and maternal imprinted loci. Hum. Mol. Genet. 18, 4724–4733 (2009).
Bliek, J., Verde, G., Callaway, J., Maas, S. M., De Crescenzo, A., Sparago, A. et al. Hypomethylation at multiple maternally methylated imprinted regions including PLAGL1 and GNAS loci in Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 17, 611–619 (2009).
Romanelli, V., Meneses, H. N., Fernández, L., Martínez-Glez, V., Gracia-Bouthelier, R., F Fraga, M. et al. Beckwith-Wiedemann syndrome and uniparental disomy 11p: fine mapping of the recombination breakpoints and evaluation of several techniques. Eur. J. Hum. Genet. 19, 416–421 (2011).
Bryke, C., Garber, A. & Israel, J. Evolution of a complex phenotype in a unique patient with a paternal uniparental disomy for every chromosome cell line and a normal biparental inheritance cell line. Abstract, The annual meeting of The American Society of Human Genetics, Toronto, Canada (2004).
Giurgea, I., Sanlaville, D., Fournet, J. C., Sempoux, C., Bellanné-Chantelot, C., Touati, G. et al. Congenital hyperinsulinism and mosaic abnormalities of the ploidy. J. Med. Genet. 43, 248–254 (2006).
Hoban, P. R., Heighway, J., White, G. R., Baker, B., Gardner, J., Birch, J. M. et al. Genome-wide loss of maternal alleles in a nephrogenic rest and Wilms’ tumour from a BWS patient. Hum. Genet. 95, 651–656 (1995).
Reed, R. C., Beischel, L., Schoof, J., Johnson, J., Raff, M. L. & Kapur, R. P. Androgenetic/biparental mosaicism in an infant with hepatic mesenchymal hamartoma and placental mesenchymal dysplasia. Pediatr. Dev. Pathol. 11, 377–383 (2008).
Romanelli, V., Nevado, J., Fraga, M., Trujillo, A. M., Mori, M., Fernández, L. et al. Constitutional mosaic genome-wide uniparental disomy due to diploidisation: an unusual cancer-predisposing mechanism. J. Med. Genet. 48, 212–216 (2011).
Wilson, M., Peters, G., Bennetts, B., McGillivray, G., Wu, Z. H., Poon, C. et al. The clinical phenotype of mosaicism for genome-wide paternal uniparental disomy: two new reports. Am. J. Med. Genet. A. 146A, 137–148 (2008).
Yamazawa, K., Nakabayashi, K., Matsuoka, K., Masubara, K., Hata, K., Horikawa, R. et al. Androgenetic/biparental mosaicism in a girl with Beckwith-Wiedemann syndrome-like and upd(14)pat-like phenotypes. J. Hum. Genet. 56, 91–93 (2011).
Inbar-Feigenberg, M., Choufani, S., Cytrynbaum, C., Chen, Y. A., Steele, L., Shuman, C. et al. Mosaicism for genome-wide paternal uniparental disomy with features of multiple imprinting disorders: diagnostic and management issues. Am. J. Med. Genet. A. 161A, 13–20 (2013).
Yamazawa, K., Nakabayashi, K., Kagami, M., Sato, T., Saitoh, S., Horikawa, R. et al. Parthenogenetic chimaerism/mosaicism with a Silver-Russell syndrome-like phenotype. J. Med. Genet. 47, 782–785 (2010).
Lee, M. H., Reynisdóttir, I. & Massagué, J. Cloning of p57KIP2, a cyclin-dependent kinase inhibitor with unique domain structure and tissue distribution. Genes. Dev. 9, 639–649 (1995).
Matsuoka, S., Edwards, M. C., Bai, C., Parker, S., Zhang, P., Baldini, A. et al. p57KIP2, a structurally distinct member of the p21CIP1 Cdk inhibitor family, is a candidate tumor suppressor gene. Genes Dev. 9, 650–662 (1995).
Borriello, A., Caldarelli, I., Bencivenga, D., Criscuolo, M., Cucciolla, V., Tramontano, A. et al. p57(Kip2) and cancer: time for a critical appraisal. Mol. Cancer Res. 9, 1269–1284 (2011).
Yokoo, T., Toyoshima, H., Miura, M., Wang, Y., Iida, K. T., Suzuki, H. et al. p57Kip2 regulates actin dynamics by binding and translocating LIM-kinase 1 to the nucleus. J. Biol. Chem. 278, 52919–52923 (2003).
Vlachos, P. & Joseph, B. The Cdk inhibitor p57(Kip2) controls LIM-kinase 1 activity and regulates actin cytoskeleton dynamics. Oncogene 28, 4175–4188 (2009).
Watanabe, H., Pan, Z. Q., Schreiber-Agus, N., DePinho, R. A., Hurwitz, J. & Xiong, Y. Suppression of cell transformation by the cyclin-dependent kinase inhibitor p57KIP2 requires binding to proliferating cell nuclear antigen. Proc. Natl Acad. Sci. USA 95, 1392–1397 (1998).
Hatada, I., Ohashi, H., Fukushima, Y., Kaneko, Y., Inoue, M., Komoto, Y. et al. An imprinted gene p57KIP2 is mutated in Beckwith-Wiedemann syndrome. Nat. Genet. 14, 171–173 (1996).
Romanelli, V., Belinchón, A., Benito-Sanz, S., Martínez-Glez, V., Gracia-Bouthelier, R., Heath, K. E. et al. CDKN1C (p57(Kip2)) analysis in Beckwith-Wiedemann syndrome (BWS) patients: Genotype-phenotype correlations, novel mutations, and polymorphisms. Am. J. Med. Genet. A. 152A, 1390–1397 (2010).
Yatsuki, H., Higashimoto, K., Jozaki, K., Koide, K., Okada, J., Watanabe, Y. et al. Novel mutations of CDKN1C in Japanese patients with Beckwith-Wiedemann syndrome. Genes Genom 35, 141–147 (2013).
Arboleda, V. A., Lee, H., Parnaik, R., Fleming, A., Banerjee, A., Ferraz-de-Souza, B. et al. Mutations in the PCNA-binding domain of CDKN1C cause IMAGe syndrome. Nat. Genet. 44, 788–792 (2012).
Waziri, M., Patil, S. R., Hanson, J. W. & Bartley, J. A. Abnormality of chromosome 11 in patients with features of Beckwith-Wiedemann syndrome. J. Pediatr. 102, 873–876 (1983).
Eggermann, T., Spengler, S., Bachmann, N., Baudis, M., Mau-Holzmann, U. A., Singer, S. et al. Chromosome 11p15 duplication in Silver-Russell syndrome due to a maternally inherited translocation t(11;15). Am. J. Med. Genet. A. 152A, 1484–1487 (2010).
Cardarelli, L., Sparago, A., De Crescenzo, A., Nalesso, E., Zavan, B., Cubellis, M. V. et al. Silver-Russell syndrome and Beckwith-Wiedemann syndrome phenotypes associated with 11p duplication in a single family. Pediatr. Dev. Pathol. 13, 326–330 (2010).
Hoovers, J. M., Kalikin, L. M., Johnson, L. A., Alders, M., Redeker, B., Law, D. J. et al. Multiple genetic loci within 11p15 defined by Beckwith-Wiedemann syndrome rearrangement breakpoints and subchromosomal transferable fragments. Proc. Natl Acad. Sci. USA 92, 12456–12460 (1995).
Lee, M. P., Hu, R. J., Johnson, L. A. & Feinberg, A. P. Human KVLQT1 gene shows tissue-specific imprinting and encompasses Beckwith-Wiedemann syndrome chromosomal rearrangements. Nat. Genet. 15, 181–185 (1997).
Smilinich, N. J., Day, C. D., Fitzpatrick, G. V., Caldwell, G. M., Lossie, A. C., Cooper, P. R. et al. A maternally methylated CpG island in KvLQT1 is associated with an antisense paternal transcript and loss of imprinting in Beckwith-Wiedemann syndrome. Proc. Natl Acad. Sci. USA 96, 8064–8069 (1999).
Kaltenbach, S., Capri, Y., Rossignol, S., Denjoy, I., Soudée, S., Aboura, A. et al. Beckwith-Wiedemann syndrome and long QT syndrome due to familial-balanced translocation t(11;17)(p15.5;q21.3) involving the KCNQ1 gene. Clin. Genet. (e-pub ahead of print 15 October 2012; doi:10.1111/cge.12038).
Smith, A. C., Suzuki, M., Thompson, R., Choufani, S., Higgins, M. J., Chiu, I. W. et al. Maternal gametic transmission of translocations or inversions of human chromosome 11p15.5 results in regional DNA hypermethylation and downregulation of CDKN1C expression. Genomics 99, 25–35 (2012).
Lee, M. P., DeBaun, M., Randhawa, G., Reichard, B. A., Elledge, S. J. & Feinberg, A. P. Low frequency of p57KIP2 mutation in Beckwith-Wiedemann syndrome. Am. J. Hum. Genet. 61, 304–309 (1997).
Gaston, V., Le Bouc, Y., Soupre, V., Burglen, L., Donadieu, J., Oro, H. et al. Analysis of the methylation status of the KCNQ1OT and H19 genes in leukocyte DNA for the diagnosis and prognosis of Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 9, 409–418 (2001).
Mummert, S. K., Lobanenkov, V. A. & Feinberg, A. P. Association of chromosome arm 16q loss with loss of imprinting of insulin-like growth factor-II in Wilms tumor. Genes. Chromosomes. Cancer 43, 155–161 (2005).
Satoh, Y., Nakadate, H., Nakagawachi, T., Higashimoto, K., Joh, K., Masaki, Z. et al. Genetic and epigenetic alterations on the short arm of chromosome 11 are involved in a majority of sporadic Wilms’ tumours. Br. J. Cancer 95, 541–547 (2006).
Honda, S., Arai, Y., Haruta, M., Sasaki, F., Ohira, M., Yamaoka, H. et al. Loss of imprinting of IGF2 correlates with hypermethylation of the H19 differentially methylated region in hepatoblastoma. Br J Cancer 99, 1891–1899 (2008).
Cox, G. F., Bürger, J., Lip, V., Mau, U. A., Sperling, K., Wu, B. L. et al. Intracytoplasmic sperm injection may increase the risk of imprinting defects. Am. J. Hum. Genet. 71, 162–164 (2002).
DeBaun, M. R., Niemitz, E. L. & Feinberg, A. P. Association of in vitro fertilization with Beckwith-Wiedemann syndrome and epigenetic alterations of LIT1 and H19. Am. J. Hum. Genet. 72, 156–160 (2003).
Gicquel, C., Gaston, V., Mandelbaum, J., Siffroi, J. P., Flahault, A. & Le Bouc, Y. In vitro fertilization may increase the risk of Beckwith-Wiedemann syndrome related to the abnormal imprinting of the KCN1OT gene. Am. J. Hum. Genet. 72, 1338–1341 (2003).
Manipalviratn, S., DeCherney, A. & Segars, J. Imprinting disorders and assisted reproductive technology. Fertil. Steril. 91, 305–315 (2009).
Doherty, A. S., Mann, M. R., Tremblay, K. D., Bartolomei, M. S. & Schultz, R. M. Differential effects of culture on imprinted H19 expression in the preimplantation mouse embryo. Biol. Reprod. 62, 1526–1535 (2000).
Fauque, P., Jouannet, P., Lesaffre, C., Ripoche, M. A., Dandolo, L., Vaiman, D. et al. Assisted reproductive technology affects developmental kinetics, H19 Imprinting control region methylation and H19 gene expression in individual mouse embryos. BMC Dev. Biol. 7, 116 (2007).
Zaitseva, I., Zaitsev, S., Alenina, N., Bader, M. & Krivokharchenko, A. Dynamics of DNA-demethylation in early mouse and rat embryos developed in vivo and in vitro. Mol. Reprod. Dev. 74, 1255–1261 (2007).
Rivera, R. M., Stein, P., Weaver, J. R., Mager, J., Schultz, R. M. & Bartolomei, M. S. Manipulations of mouse embryos prior to implantation result in aberrant expression of imprinted genes on day 9.5 of development. Hum. Mol. Genet. 17, 1–14 (2008).
Young, L. E., Fernandes, K., McEvoy, T. G., Butterwith, S. C., Gutierrez, C. G., Carolan, C. et al. Epigenetic change in IGF2R is associated with fetal overgrowth after sheep embryo culture. Nat. Genet. 27, 153–154 (2001).
Sato, A., Otsu, E., Negishi, H., Utsunomiya, T. & Arima, T. Aberrant DNA methylation of imprinted loci in superovulated oocytes. Hum. Reprod. 22, 26–35 (2007).
Hiura, H., Okae, H., Miyauchi, N., Sato, F., Sato, A., Van De Pette, M. et al. Characterization of DNA methylation errors in patients with imprinting disorders conceived by assisted reproduction technologies. Hum. Reprod. 27, 2541–2548 (2012).
Kobayashi, H., Sato, A., Otsu, E., Hiura, H., Tomatsu, C., Utsunomiya, T. et al. Aberrant DNA methylation of imprinted loci in sperm from oligospermic patients. Hum. Mol. Genet. 16, 2542–2551 (2007).
Poplinski, A., Tüttelmann, F., Kanber, D., Horsthemke, B. & Gromoll, J. Idiopathic male infertility is strongly associated with aberrant methylation of MEST and IGF2/H19 ICR1. Int. J. Androl. 33, 642–649 (2010).
Chang, A. S., Moley, K. H., Wangler, M., Feinberg, A. P. & Debaun, M. R. Association between Beckwith-Wiedemann syndrome and assisted reproductive technology: a case series of 19 patients. Fertil. Steril. 83, 349–354 (2005).
Lim, D., Bowdin, S. C., Tee, L., Kirby, G. A., Blair, E., Fryer, A. et al. Clinical and molecular genetic features of Beckwith-Wiedemann syndrome associated with assisted reproductive technologies. Hum. Reprod. 24, 741–747 (2009).
Mackay, D. J., Boonen, S. E., Clayton-Smith, J., Goodship, J., Hahnemann, J. M., Kant, S. G. et al. A maternal hypomethylation syndrome presenting as transient neonatal diabetes mellitus. Hum. Genet. 120, 262–269 (2006).
Boonen, S. E., Pörksen, S., Mackay, D. J., Oestergaard, E., Olsen, B., Brondum-Nielsen, K. et al. Clinical characterisation of the multiple maternal hypomethylation syndrome in siblings. Eur. J. Hum. Genet. 16, 453–461 (2008).
Turner, C. L., Mackay, D. M., Callaway, J. L., Docherty, L. E., Poole, R. L., Bullman, H. et al. Methylation analysis of 79 patients with growth restriction reveals novel patterns of methylation change at imprinted loci. Eur. J. Hum. Genet. 18, 648–655 (2010).
Demars, J. & Gicquel, C. Epigenetic and genetic disturbance of the imprinted 11p15 region in Beckwith-Wiedemann and Silver-Russell syndromes. Clin. Genet. 81, 350–361 (2012).
Mackay, D. J., Callaway, J. L., Marks, S. M., White, H. E., Acerini, C. L., Boonen, S. E. et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat. Genet. 40, 949–951 (2008).
Boonen, S. E., Hahnemann, J. M., Mackay, D., Tommerup, N., Brøndum-Nielsen, K., Tümer, Z. et al. No evidence for pathogenic variants or maternal effect of ZFP57 as the cause of Beckwith-Wiedemann Syndrome. Eur. J. Hum. Genet. 20, 119–121 (2012).
Quenneville, S., Verde, G., Corsinotti, A., Kapopoulou, A., Jakobsson, J., Offner, S. et al. In embryonic stem cells, ZFP57/KAP1 recognize a methylated hexanucleotide to affect chromatin and DNA methylation of imprinting control regions. Mol. Cell. 44, 361–372 (2011).
Zuo, X., Sheng, J., Lau, H. T., McDonald, C. M., Andrade, M., Cullen, D. E. et al. Zinc finger protein ZFP57 requires its co-factor to recruit DNA methyltransferases and maintains DNA methylation imprint in embryonic stem cells via its transcriptional repression domain. J. Biol. Chem. 287, 2107–2118 (2012).
Messerschmidt, D. M., de Vries, W., Ito, M., Solter, D., Ferguson-Smith, A. & Knowles, B. B. Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science 335, 1499–1502 (2012).
Drenth, J. P. & van der Meer, J. W. The inflammasome—a linebacker of innate defense. N. Engl. J. Med. 355, 730–732 (2006).
Meyer, E., Lim, D., Pasha, S., Tee, L. J., Rahman, F., Yates, J. R. et al. Germline mutation in NLRP2 (NALP2) in a familial imprinting disorder (Beckwith-Wiedemann Syndrome). PLoS Genet. 5, e1000423 (2009).
Murdoch, S., Djuric, U., Mazhar, B., Seoud, M., Khan, R., Kuick, R. et al. Mutations in NALP7 cause recurrent hydatidiform moles and reproductive wastage in humans. Nat. Genet. 38, 300–302 (2006).
Parry, D. A., Logan, C. V., Hayward, B. E., Shires, M., Landolsi, H., Diggle, C. et al. Mutations causing familial biparental hydatidiform mole implicate c6orf221 as a possible regulator of genomic imprinting in the human oocyte. Am. J. Hum. Genet. 89, 451–458 (2011).
Zhao, Z., Tavoosidana, G., Sjölinder, M., Göndör, A., Mariano, P., Wang, S. et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat. Genet. 38, 1341–1347 (2006).
Acknowledgements
This study was supported, in part, by a Grant for Research on Intractable Diseases from the Ministry of Health, Labor, and Welfare; a Grant for Child Health and Development from the National Center for Child Health and Development; a Grant-in-Aid for Challenging Exploratory Research; and, a Grant-in-Aid for Young Scientists (B) from the Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Soejima, H., Higashimoto, K. Epigenetic and genetic alterations of the imprinting disorder Beckwith–Wiedemann syndrome and related disorders. J Hum Genet 58, 402–409 (2013). https://doi.org/10.1038/jhg.2013.51
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2013.51
Keywords
This article is cited by
-
Widespread allele-specific topological domains in the human genome are not confined to imprinted gene clusters
Genome Biology (2023)
-
Genome-wide DNA methylation patterns reveal clinically relevant predictive and prognostic subtypes in human osteosarcoma
Communications Biology (2022)
-
Megabase-scale methylation phasing using nanopore long reads and NanoMethPhase
Genome Biology (2021)
-
A Beckwith-Wiedemann syndrome case with de novo 24 Mb duplication of chromosome 11p15.5p14.3
Molecular Cytogenetics (2021)
-
Large parental differences in chromatin organization in pancreatic beta cell line explaining diabetes susceptibility effects
Nature Communications (2021)
