
Abstract
The epidemiology of multiple sclerosis suggests that a complex interaction of genes and environment contribute to susceptibility. To enrich for families with large genetic effects and to potentially reduce genetic heterogeneity, we screened a sample of 18,794 probands and identified forty families with four or more affected individuals. Within these 40 families, HLA DRB1*15 was present in 70% of affected individuals; the transmission disequilibrium test showed a significant excess in transmission of DRB1*15 alleles to affected individuals (47 transmitted, 19 untransmitted, χ 2 = 11.9, p = 0.00057). A 10 cM genome scan was performed and analyzed for linkage under a parametric model with heterogeneity. No excess of significant sharing was observed (HLOD > 3.3) in the parametric multipoint analysis. No region exceeded that for marker GATA8A05 with an HLOD = 1.11. Follow-up genotyping with 17 microsatellites revealed a significant two-point parametric HLOD = 3.99 at marker D4S1597. Transmission disequilibrium tests for markers in this candidate region showed no transmission distortion. A scan for variants in a gene adjacent to D4S1597, PALLD, was negative for synonymous or nonsynonymous changes. A final multipoint scan incorporating all microsatellites in the region provided an HLOD = 1.30. The inability to find significant linkage in these highly penetrant families suggests that linkage is not the optimal tool for dissecting the inheritance of MS.
Similar content being viewed by others


Introduction
Epidemiological studies of families with multiple sclerosis have revealed a complex inheritance pattern. Recurrence risks to first, second and third degree relatives initially suggested an oligogenic inheritance pattern consistent with other complex diseases (Sadovnick and Baird 1988). However, recent half-sibling data (Ebers et al. 2004) would not fit with this inheritance model, since the half-sibling rate is greater than expected relative to the full-sibling rate. Adopted relatives of MS patients fail to demonstrate increased risk of MS compared to the general population (Ebers et al. 1995) and it is therefore unlikely that a transmissible agent is responsible for familial aggregation in MS. Siblings of MS patients have an increased risk of developing MS of approximately 20–40-fold compared to the population and approximately 3.5% for siblings overall (Sadovnick et al. 1988). Twin studies have shown that the MZ twin concordance rate is approximately tenfold greater than the risk to siblings (Willer et al. 2003), and in small numbers the DZ rate is double that for sibs. In addition, evidence for an asymmetric parent of origin effect (Ebers et al. 2004) and for birth timing (Willer et al. 2005) all suggest that susceptibility is determined by a complex interplay between genes and both external and maternal environment. These studies hint at the underlying complexity and difficulty involved in studying genetic and environmental factors in small samples when underlying interactions are difficult to observe.
Further illustration of the difficulty involved in determining the exact nature of MS inheritance is provided by the disappointing linkage results from affected relative pair studies (Ebers et al. 1996; Haines et al. 1996; Kuokkanen et al. 1996; Sawcer et al. 1996; Kuokkanen et al. 1997; Oturai et al. 1999; Broadley et al. 2001; Coraddu et al. 2001; Transatlantic Multiple Sclerosis Genetics Cooperative 2001; Akesson et al. 2002; Ban et al. 2002; Goedde et al. 2002; Dyment et al. 2004; Kenealy et al. 2004). The major histocompatibility complex locus (MHC) has been consistently implicated as playing a role in MS inheritance. The HLA-DRB1*15 allele is associated with MS (Winchester et al. 1975), but the mechanism of its effect on risk is unclear (Hillert 1994). Evidence for increased sharing in sibling pairs with MS has been observed in families that do not carry the HLA-DRB1*15 allele (Ligers et al. 2001), and the observation of effects in trans as well as protection and varying relative risks of susceptibility alleles suggests that the effect of the MHC on susceptibility is more complex than simply antigen presentation (Dyment et al. 2004).
Many other non-MHC loci have been implicated in previous genome scans but none of these linkage regions have yet been unambiguously replicated in independent samples. Several regions of the genome are suggestively linked in multiple studies, but large datasets will be required to confirm and refine linkage regions and identify specific variants associated with susceptibility (Cardon and Bell 2001). Meta-analysis has not resolved the complexity (Transatlantic Multiple Sclerosis Genetics Cooperative 2001). Furthermore, no areas of putative linkage have been successfully fine-mapped to identify a susceptibility gene. The lack of positive findings using sibling pair linkage analysis may be due to locus heterogeneity and/or the lack of power to detect a relatively minor effect size. However, even within the context of such genetic complexity, it is still possible that there are some families where susceptibility is determined by a single locus with major effects which may be detectable with parametric linkage analysis.
Here we present genome scan results in a large cohort of multigenerational, multicase (n ≥ 4) families derived from a population-based ascertainment scheme. These families are unique in terms of the number of affected individuals per pedigree. If rare high-penetrance mutations exist, the selection of families with a large number of affected individuals was hypothesized to maximize the potential for identifying such mutations. If a gene of major effect is acting to increase susceptibility, these families have the advantage of being amenable to traditional linkage and haplotype analysis to refine regions of interest and to identify risk-conferring variants. Although parametric analysis methods using large families are more susceptible to misspecification of inheritance, they can also be powerful tools for detection of linkage.
Methods
Families were ascertained by the Canadian Collaborative Project on the Genetic Susceptibility to Multiple Sclerosis (Sadovnick et al. 1998). At the time of family selection, over 18,000 families had been ascertained and DNA samples from >1,500 families had been obtained. Informed consent was obtained from all subjects and the experiments performed for this study comply with current guidelines and ethics as set out by the respective clinic sites participating in the Canadian Collaborative Project on the genetic susceptibility to multiple sclerosis.
For this study, a subset of multicase families was selected based upon the following criteria: (1) DNA was available from a minimum of four affected individuals in each pedigree (or DNA from parents, siblings, spouses and offspring were available for genotype reconstruction); (2) affected individuals were present in more than one generation. Eighty-six families met these minimum criteria and simulations were performed on each of these families to determine the relative power of each pedigree to independently detect linkage. The SLINK package (Terwilliger et al. 1993) was used to generate 1,000 linked replicates of markers with equally frequent alleles, assuming a dominant model with a penetrance of 60% in females and 30% in males and with 100%, 75% or 50% of families linked. The simulated datasets were then analyzed by MSIM and associated programs (Terwilliger et al. 1993). The 40 families with the greatest power to detect linkage were used for the genotyping stage of the project.
Genotyping of the 40 selected families was performed by the Marshfield NHLBI Mammalian Genotyping Service using Marshfield Screening Set 10; the details of the 392 microsatellite markers have been previously published (Weber and Broman 2001). HLA genotyping was performed using an allele-specific primer method (Olerup and Zetterquist 1991). Additional microsatellite markers for follow-up study were identified from the Marshfield maps. PCR for these markers was performed with Taq Gold and fluorescently-labeled primers and products were separated on ABI 373, 377 or 3,700 genotyping platforms. Scoring was performed using Genescan and Genotyper v3.7 software (ABI PRISM, Applied Biosystems, Foster City, CA, USA).
Pedigree relationship were verified by the software GRR (Abecasis et al. 2001). Mendelian errors were identified by the program PEDCHECK (O’Connell and Weeks 1998) and the genotype most likely containing the error (based upon other genotype information in the pedigree) was scored as a missing genotype (i.e., 0, 0). When the genotype error could not be determined without ambiguity, both individuals were scored as unknown genotype.
Marker maps were obtained from the Marshfield website (http://www.marshfieldclinic.org/) and compared with the human genome sequence to confirm the chromosomal location, order and approximate distance between microsatellite markers. When order discrepancies arose, the sequence map was taken to be true and distances were approximated from the physical distance.
Multipoint parametric analyses were performed using SIMWALK2 Version 2.83 (Sobel and Lange 1996; Mukhopadhyay et al. 1999), with file preparation using MEGA2 (Version 2.5R2; Mukhopadhyay et al. 1999). Parameters for multipoint parametric analysis were specified as follows: penetrance of 0.60 for females and 0.30 for males, dominant model, disease allele frequency of 0.003 and phenocopy rate of 0.001. We set the penetrance rate to be higher in females than males since MS is more prevalent in females in the general population and also in these multicase families. The disease was specified as a dominant model since there was no evidence of recessive transmission in these families and because these families were selected as having the most power when analyzed under a dominant model. The disease allele frequency was set to be relatively rare to model transmission of a single disease-causing allele through each family, and the phenocopy rate was set at the population prevalence of the disease. The disease allele frequency is difficult to estimate in these families, and 0.003 was chosen in order to approximate the disease frequency given the penetrances modeled. Two-point parametric analyses were performed with the same model parameters using the LINKAGE package (Lathrop et al. 1984).
Models incorporating heterogeneity (HLODS) defined by the proportion of families linked at a particular locus (α) were calculated using the heterogeneity option of parametric analysis in SIMWALK2 (Sobel and Lange 1996). Multipoint parametric analysis with heterogeneity was used to analyze the genome screen results with SIMWALK2. In a complex disease, the disease-related parameters in the sample being analyzed are difficult to estimate, and for this reason we incorporated heterogeneity into the analysis, to allow for subsets of families to be linked to the particular region. Multipoint nonparametric analysis was performed using the NPL-all option of SIMWALK2.
Mutation screening of the palladin gene (PALLD) was performed by designing primers to amplify all exons and 20 bases of flanking intron. Potential promoters were identified by screening 10,000 bases upstream of the first exon using Softberry and PromotorScan. Heterozygotes were identified by exon amplification, denaturing and annealing and analyzed by DHPLC (transgenomic wave) methods for temperature-modulated heteroduplex detection of heterozygotes. Any suspected polymorphisms were confirmed by sequence analysis. Seqscape software v1.0 (ABI PRISM) was used for sequence analysis.
Results
The families
We selected 40 multigenerational, multicase families for genotyping and genome-wide linkage analysis. The complete pedigrees, including founders, contained 1,020 individuals, and DNA was available from 550 individuals. Since all pedigrees extend 3–9 generations back from individuals with MS, many of the individuals in the top generations of the pedigrees were deceased. We attempted to obtain blood samples from all healthy individuals informative for linkage analysis that were over the age of 14. There were 220 individuals considered to be affected with MS by Poser’s criteria (Poser et al. 1983), and DNA was available from 180 (81.8%). Nine additional affected individuals had at least one genotyped offspring (mean = 3.3 offspring) and could be partially informative for linkage analysis. Any individuals with signs of MS who did not fulfill the criteria for definite or probable diagnosis (Poser et al. 1983) were considered to have unknown affection status (N = 15). Of the confirmed affected individuals, 137 were female and 83 were male, giving a gender ratio of 1.65:1, which is significantly less biased towards females than that observed in the entire CCPGSMS dataset of 19,938 cases of definite MS (N female = 14,081, N male = 5,857, ratio = 2.4:1, χ 2 = 8.12, p = 0.0043). The average age of onset was 29.8 (SE = 0.9) years of age. There was no correlation for age of onset in first, second, or third degree related pairs (n = 139, r = −0.06, p = 0.47).
At the stipulated parameters, the forty selected families had an average maximum LOD score of 25.50 when all families were linked to the same locus. When 50% of families were linked to a single locus, a LOD score of 7.74 was expected. A LOD score of 3.3 was attainable with 40% of families linked.
HLA-DRB1
HLA-DRB1*15 was present in 117/172 affected individuals (68.0%). A transmission disequilibrium test (TDT) was performed and DRB1*15 was transmitted 49 times versus 17 times untransmitted (χ 2=15.5; p = 8.2×10−5). Two-point parametric linkage analysis at the HLA locus revealed a LOD score of 2.55 at a recombination value (θ) of 0.20, but multipoint HLOD analysis was maximal at a LOD of only 0.23 with a predicted 4% of families linked.
Genome screen: parametric analysis
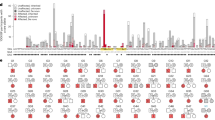
Multipoint parametric linkage analysis incorporating heterogeneity showed no significant linkage (HLOD > 3.3) for the initial genome screen. The threshold of 0.83 was used as an arbitrary cut-off. There were four regions that met this criterion (Table 1). The greatest evidence for linkage in the initial genome scan was for marker GATA8A05 at chromosome 4q32 (HLOD = 1.11) with an estimated 11% of families linked.
We divided the families into two smaller subsets; ten families with 50% or fewer of the cases carrying at least one copy of HLA-DRB1*15 and fifteen families where all genotyped cases had at least one copy of HLA-DRB1*15. Amongst the six regions with the highest HLOD in all families (Table 1), two regions had higher HLODs in the high HLA-DRB1*15 frequency families (4q32 and 5q32, Table 2) and two regions had higher HLODs in the low HLA-DRB1*15 frequency families (5p14 and 7p14). Two regions not highlighted by the analysis in all families had HLOD values >0.83 in the low HLA-DRB1*15 frequency families (6p12 and 8p23) and four regions had HLOD values >0.83 in the high HLA-DRB1*15 frequency subset of families (1p32, 3p24, 3q27, and 16p12). However, there were no striking results after analysis of these family subsets, with the highest HLOD of 1.51 observed at 5q32 which was highlighted as one of the top all family HLODs.
When each family was assessed individually, only three locations in three separate families had a multipoint parametric LOD score >2. These locations were on chromosome 1p32 (GATA26G09, family Mx16, lod = 2.07), chromosome 3p24 (GATA73D01, family Mx26, lod = 2.31), and chromosome 9q21 (GATA7D12, family Mx14, 2.12).
Genome screen: nonparametric analysis
Due to the size of these pedigrees and the long running time for analysis (∼1 week with several chromosomes running in parallel), it was impossible to perform simulations to assess the significance of the nonparametric statistics in the full pedigrees. We did perform simulations in reduced versions of the pedigrees, where eight families required trimming of unaffected individuals and two families required breaking into two smaller pedigrees each. Simulation of the original genome scan markers with the same number of alleles and frequencies as the true markers was performed and analyzed using MERLIN (Abecasis et al. 2002). In the reduced pedigrees, the top nonparametric LOD score was 1.57 for the HLA region. We observed 10 loci with NPL >1, which was also observed in two of the 20 simulated genome scans. The fifth-best result in the real data had a NPL of 1.2. None of the twenty simulated genome scans had five loci with NPL >1.2.
A complete nonparametric analysis of the full pedigrees was also performed using Simwalk2. The regions with the strongest evidence for linkage using a nonparametric method were at 6p22, 3q13, 1q23, 2p21 and 22q13 (Table 3).
Follow-up analysis
Seventeen additional microsatellites were genotyped in the region of chromosome 4q32, which was the most promising region based on the HLOD analysis. The additional markers resulted in high-density coverage of the susceptibility region with a 2.1 cM average spacing over 42 cM on chromosome 4q (from 143.3 cM to 181.9 cM). Among the additional markers, the evidence for linkage was maximal at D4S1597 with a single point HLOD = 3.99, θ = 0.15 and 95% of families linked. The follow-up multipoint HLOD was substantially lower (HLOD = 1.30).
Transmission disequilibrium at the candidate region
Analysis of transmission distortion using the QTDT package (Abecasis et al. 2000) revealed no evidence for significant association between MS affection status and autosomal microsatellite markers (n = 378) after adjusting for the number of alleles tested. There was also no evidence for significant association with microsatellite markers spanning the candidate region (n = 17).
Variant screening
There are four genes present in the candidate region at 4q; Annexin A10, an RNA helicase, a hypothetical helicase and palladin (PALLD). Of the four, palladin was thought to be an attractive candidate due to its expression in the axonal growth cone in rats (Hwang et al. 2001) and because D4S1597 is present within one of its introns. Screening for variants was performed in PALLD gene, based on exons present in mRNA AB023209, in 43 affected individuals, nine married-in unaffected parents and eight unrelated controls. All affected individuals from the ten families showing the most evidence for linkage at this locus were chosen for sequencing. The unaffected parents and unrelated controls were added as controls to help determine whether any variants seen in sequence analysis might be segregating with disease status in these pedigrees. There were no nonsynonymous or synonymous amino acid changes that segregated with the disease and were not present in controls. There were two adjacent nonsynonymous changes that resulted in methionine to isoleucine in exon 2 (M224I, rs7655494 and rs7671781) that segregated with disease in three of ten families, but were also present in 4/8 of the unrelated controls and 4/9 married-in parents.
Discussion
Despite overwhelming evidence from epidemiological studies that genes determine susceptibility to multiple sclerosis, progress in identifying them has been slow. This has been variably attributed to complexity and heterogeneity and the small effect size of individual genes. Studies have largely focused on affected sibling pairs with little success, and no non-MHC locus has been significantly replicated in an independent sample of families. Other studies have focused on linkage in 1–2 families with 5–8 affected relatives in the hopes of reducing genetic heterogeneity (Vitale et al. 2002; Haghighi et al. 2006). Again, nonsignificant evidence for linkage was observed and the results must be interpreted in the context of reduced power when studying so few families. Given the slow rate of progress, we have reasoned that many families with four or more affected members in different generations could be more homogeneous and would have larger gene effects and therefore aid in the identification of susceptibility genes.
The accumulation of this family material has required the entire resource of the Canadian Collaborative Project on the Genetic Susceptibility to Multiple Sclerosis (CPGSMS). There are 19 MS centers across Canada that contribute to this long-standing project. A large-scale ascertainment strategy is a necessity given the rarity of pedigrees with four or more affected members; this is in spite of families residing in a nation with a relatively high prevalence of MS (Beck et al. 2005). At the time of ascertainment, close to 20,000 families had been screened and only 86 families (0.4%) met the original criteria. Of these, only 15/86 had six or more relatives affected with MS, the largest of which (MS2) had 11 affected relatives.
The affected members of these families were remarkably similar to sporadic and sibling pair MS. The average age of onset was 29.8 years of age and similar to the 31 years of age of onset observed in sibling pairs (data taken from CCPGSMS database) and also sporadic cases (Paty and Ebers 1997). All forms of MS were present in the families. No single MS phenotype (e.g., relapsing–remitting, primary progressive, or secondarily progressive) segregated exclusively within an individual family. One area of difference was observed in the gender ratio of cases. In the families with four or more cases, the sex ratio was 1.6:1, different from the 2.4:1 ratio seen in the Canadian population.
Another difference between cases in these families compared to sibling-pair families was the frequency of the known susceptibility allele HLA DRB1*15. In these families, 119 of 169 cases genotyped for HLA DRB1 were positive for HLA-DRB1*15 (70.8%). Sibling pairs ascertained as part of the CCPGSMS showed a lesser frequency of HLA-DRB1*15 at 59% (χ 2 = 13.8; p = 0.002). As has been speculated for another large family segregating MS as an autosomal dominant trait, the size effect of DRB1*15 appears to be greater in families with many affected individuals (Dyment et al. 2002). The two-point LOD score of 2.25 at DRB1 was one of the highest LOD scores observed in this investigation.
Overall, the genome screen showed no evidence for significant linkage (HLOD > 3.3). This may reflect underlying heterogeneity or complexity of multiple sclerosis genetics. The presence of heterogeneity, phenocopies, a misspecified penetrance or disease allele frequency may all contribute to a decreased power to detect significant linkage. This is despite our efforts to homogenize our sample by selecting those families with a high prevalence of MS and a seemingly autosomal dominant mode of inheritance. The greatest evidence in the initial multipoint scan was at marker GATA8A05, with a modest HLOD = 1.11.
After follow-up with additional markers, the highest parametric score was obtained for marker D4S1597 on chromosome 4q, located within an intron of the palladin gene. A previous genome-wide association study in Icelandic MS patients found its second-most significant genome-wide p value for a microsatellite marker located intragenic to the PALLD gene (Jonasdottir et al. 2003). Palladin is reportedly involved in stabilization of the actin cytoskeleton and is highly expressed in the axonal growth cone (Boukhelifa et al. 2001; Hwang et al. 2001). Unfortunately, the two-point HLOD score of HLOD = 3.99 decreased substantially to a multipoint HLOD = 1.30. While this may have been due to map misspecification in the dense set of markers, it may also represent a more accurate reflection of haplotype sharing at this locus. Transmission disequilibrium with the microsatellites as well as a screen for variants showed no further evidence to implicate the PALLD gene; however, additional association analysis testing in large case-control cohorts would be informative as to the effect of this locus.
This cohort represents a more powerful and potentially informative collection of families than has been presented for genetic analysis of MS. The lack of significant findings in the original genome screen may be due to locus heterogeneity between or within the families, or to inadequate marker coverage in some regions of the genome. However it must now be considered that the inheritance paradigm has been incorrect.
Additional information
Marshfield marker set ten information: http://research.marshfieldclinic.org/genetics/sets/combo.html
Marshfield marker maps:
http://research.marshfieldclinic.org/genetics/Map_Markers/maps/IndexMapFrames.html
Goldenpath web server:
Softberry: http://www.softberry.com
References
Abecasis GR, Cardon LR, Cookson WO (2000) A general test of association for quantitative traits in nuclear families. Am J Hum Genet 66:279–292
Abecasis GR, Cherny SS, Cookson WO, Cardon LR (2001) GRR: graphical representation of relationship errors. Bioinformatics 17:742–743
Abecasis GR, Cherny SS, Cookson WO, Cardon LR (2002) Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30:97–101
Akesson E, Oturai A, Berg J, Fredrikson S, Andersen O, Harbo HF, Laaksonen M, Myhr KM, Nyland HI, Ryder LP, Sandberg-Wollheim M, Sorensen PS, Spurkland A, Svejgaard A, Holmans P, Compston A, Hillert J, Sawcer S (2002) A genome-wide screen for linkage in Nordic sib-pairs with multiple sclerosis. Genes Immun 3:279–285
Ban M, Stewart GJ, Bennetts BH, Heard R, Simmons R, Maranian M, Compston A, Sawcer SJ (2002) A genome screen for linkage in Australian sibling-pairs with multiple sclerosis. Genes Immun 3:464–469
Beck CA, Metz LM, Svenson LW, Patten SB (2005) Regional variation of multiple sclerosis prevalence in Canada. Mult Scler 5:516–519
Boukhelifa M, Parast MM, Valtschanoff JG, LaMantia AS, Meeker RB, Otey CA (2001) A role for the cytoskeleton-associated protein palladin in neurite outgrowth. Mol Biol Cell 12:2721–2729
Broadley S, Sawcer S, D’Alfonso S, Hensiek A, Coraddu F, Gray J, Roxburgh R, Clayton D, Buttinelli C, Quattrone A, Trojano M, Massacesi L, Compston A (2001) A genome screen for multiple sclerosis in Italian families. Genes Immun 2:205–210
Cardon LR, Bell JI (2001) Association study designs for complex diseases. Nat Rev Genet 2:91–99
Coraddu F, Sawcer S, D’Alfonso S, Lai M, Hensiek A, Solla E, Broadley S, Mancosu C, Pugliatti M, Marrosu MG, Compston A (2001) A genome screen for multiple sclerosis in Sardinian multiplex families. Eur J Hum Genet 9:621–626
Dyment DA, Cader MZ, Willer CW, Sadovnick AD, Ebers GC (2002) A multigenerational family with multiple sclerosis. Brain 125:1474–1482
Dyment DA, Sadovnick AD, Willer CJ, Armstrong H, Cader ZM, Wiltshire S, Kalman B, Risch N, Ebers GC (2004) An extended genome scan in 442 Canadian multiple sclerosis-affected sibships: a report from the Canadian Collaborative Study Group. Hum Mol Genet 13:1005–1015
Dyment DA, Herrera BM, Cader MZ, Willer CJ, Lincoln M, Sadovnick AD, Risch N, Ebers GC (2005) Complex interactions among MHC haplotypes in multiple sclerosis: susceptibility and resistance. Hum Mol Genet 14:2019–2026
Ebers GC, Sadovnick AD, Risch NJ (1995) A genetic basis for familial aggregation in multiple sclerosis. Canadian Collaborative Study Group. Nature 377:150–151
Ebers GC, Kukay K, Bulman DE et al. (1996) A full genome search in multiple sclerosis. Nat Genet 13:472–476
Ebers G, Sadovnick A, Dyment D, Yee I, Willer C, Risch N, Group ftCCS (2004) A parent of origin effect in multiple sclerosis: observations in half siblings. Lancet 363:847–850
Goedde R, Sawcer S, Boehringer S, Miterski B, Sindern E, Haupts M, Schimrigk S, Compston A, Epplen JT (2002) A genome screen for linkage disequilibrium in HLA-DRB1*15-positive Germans with multiple sclerosis based on 4666 microsatellite markers. Hum Genet 111:270–277
Haghighi S, Andersen O, Nilsson S, Rydberg L, Walhstrom J (2006) A linkage study in two families with multiple scelrosis and healthy members with oligoclonal CSF immunopathy. Mult Scler 12:723–730
Haines JL, Ter-Minassian M, Bazyk A et al. (1996) A complete genomic screen for multiple sclerosis underscores a role for the major histocompatability complex. The Multiple Sclerosis Genetics Group. Nat Genet 13:469–471
Hillert J (1994) Human leukocyte antigen studies in multiple sclerosis. Ann Neurol 36:S15–S17
Hwang SJ, Pagliardini S, Boukhelifa M, Parast MM, Otey CA, Rustioni A, Valtschanoff JG (2001) Palladin is expressed preferentially in excitatory terminals in the rat central nervous system. J Comp Neurol 436:211–224
Jonasdottir A, Thorlacius T, Fossdal R, Benediktsson K, Benedikz J, Jonsson HH, Sainz J, Einarsdottir H, Sigurdardottir S, Kristjansdottir G, Sawcer S, Compston A, Stefansson K, Gulcher J (2003) A whole genome association study in Icelandic multiple sclerosis patients with 4804 markers. J Neuroimmunol 143:88–92
Kenealy SJ, Babron MC, Bradford Y, Schnetz-Boutaud N, Haines JL, Rimmler JB, Schmidt S, Pericak-Vance MA, Barcellos LF, Lincoln RR, Oksenberg JR, Hauser SL, Clanet M, Brassat D, Edan G, Yaouanq J, Semana G, Cournu-Rebeix I, Lyon-Caen O, Fontaine B (2004) A second-generation genomic screen for multiple sclerosis. Am J Hum Genet 75:1070–1078
Kuokkanen S, Sundvall M, Terwilliger JD, Tienari PJ, Wikstrom J, Holmdahl R, Pettersson U, Peltonen L (1996) A putative vulnerability locus to multiple sclerosis maps to 5p14–p12 in a region syntenic to the murine locus Eae2. Nat Genet 13:477–480
Kuokkanen S, Gschwend M, Rioux JD, Daly MJ, Terwilliger JD, Tienari PJ, Wikstrom J, Palo J, Stein LD, Hudson TJ, Lander ES, Peltonen L (1997) Genomewide scan of multiple sclerosis in Finnish multiplex families. Am J Hum Genet 61:1379–1387
Lathrop GM, Lalouel J-M, Julier C, Ott J (1984) Strategies for multilocus analysis in humans. Proc Natl Acad Sci USA 81:3443–3446
Ligers A, Dyment DA, Willer CJ, Sadovnick AD, Ebers G, Risch N, Hillert J (2001) Evidence of linkage with hla-dr in drb1*15-negative families with multiple sclerosis. Am J Hum Genet 69:900–903
Mukhopadhyay N, Almasy L, Schroeder M, Mulvihill WP, Weeks DE (1999) Mega2, a data-handling program for facilitating genetic linkage and association analyses. Am J Hum Genet 65:A436
O’Connell JR, Weeks DE (1998) PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 63:259–266
Olerup O, Zetterquist H (1991) HLA-DRB1*01 subtyping by allele-specific PCR amplification: a sensitive, specific and rapid technique. Tissue Antigens 37:197–204
Oturai A, Larsen F, Ryder LP, Madsen HO, Hillert J, Fredrikson S, Sandberg-Wollheim M, Laaksonen M, Koch-Henriksen N, Sawcer S, Fugger L, Sorensen PS, Svejgaard A (1999) Linkage and association analysis of susceptibility regions on chromosomes 5 and 6 in 106 Scandinavian sibling pair families with multiple sclerosis. Ann Neurol 46:612–616
Paty DW, Ebers GC (1997) Multiple sclerosis. FA Davis Company, Philadelphia, PA
Poser CM, Paty DW, Scheinberg L, McDonald WI, Davis FA, Ebers GC, Johnson KP, Sibley WA, Silberberg DH, Tourtellotte WW (1983) New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol 13:227–231
Sadovnick AD, Baird PA (1988) The familial nature of multiple sclerosis: age-corrected empiric recurrence risks for children and siblings of patients. Neurology 38:990–991
Sadovnick AD, Baird PA, Ward RH (1988) Multiple sclerosis: updated risks for relatives. Am J Med Genet 29:533–541
Sawcer S, Jones HB, Feakes R, Gray J, Smaldon N, Chataway J, Robertson N, Clayton D, Goodfellow PN, Compston A (1996) A genome screen in multiple sclerosis reveals susceptibility loci on chromosome 6p21 and 17q22. Nat Genet 13:464–468
Sobel E, Lange K (1996) Descent graphs in pedigree analysis: applications to haplotyping, location scores, and marker-sharing statistics. Am J Hum Genet 58:1323–1337
Tabor HK, Risch NJ, Myers RM (2002) Candidate-gene approaches for studying complex genetic traits: practical considerations. Nat Rev Genet 3:391–397
Terwilliger JD, Ott J (1994) Handbook of human genetic linkage. Johns Hopkins University Press, Baltimore, MD
Terwilliger JD, Speer M, Ott J (1993) Chromosome-based method for rapid computer simulation in human genetic linkage analysis. Genet Epidemiol 10:217–224
Transatlantic Multiple Sclerosis Genetics Cooperative (2001) A meta-analysis of genomic screens in multiple sclerosis. Mult Scler 7:3–11
Vitale E, Cook S, Sun R, Specchia C, Subramanian K, Rocchi M, Nathanson D, Schwalb M, Devoto M, Rohowsky-Kochan C (2002) Linkage analysis conditional on HLA status in a large North American pedigree supports the presence of a multiple sclerosis susceptibility locus on chromosome 12p12. Hum Mol Genet 11:295–300
Weber JL, Broman KW (2001) Genotyping for human whole-genome scans: past, present, and future. Adv Genet 42:77–96
Willer CJ, Dyment DA, Risch NJ, Sadovnick AD, Ebers GC (2003) Twin concordance and sibling recurrence rates in multiple sclerosis. Proc Natl Acad Sci USA 100:12877–12882
Willer CJ, Dyment DA, Sadovnick AD, Rothwell PM, Murray TJ, Ebers GC (2005) Timing of birth and risk of multiple sclerosis: population based study. BMJ 330:120
Acknowledgments
The authors would like to thank the participants who kindly participated in this research. We would also like to acknowledge John Broxholme for assistance with analysis. Special thanks to Holly Armstrong and Beverly Scott for sample preparation and laboratory assistance. This work was funded by the Multiple Sclerosis Society of Canada Scientific Research Foundation. ADS is a Michael Smith Foundation for Health Research Distinguished Scholar.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Willer, C.J., Dyment, D.A., Cherny, S. et al. A genome-wide scan in forty large pedigrees with multiple sclerosis. J Hum Genet 52, 955–962 (2007). https://doi.org/10.1007/s10038-007-0194-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-007-0194-6
Keywords
This article is cited by
-
An Assessment of Genetic Counseling Services for Individuals with Multiple Sclerosis
Journal of Genetic Counseling (2015)
