
Abstract
Bacterial interactions with plants and animals have been examined for many years; differently, only with the new millennium the study of bacterial–fungal interactions blossomed, becoming a new field of microbiology with relevance to microbial ecology, human health and biotechnology. Bacteria and fungi interact at different levels and bacterial endosymbionts, which dwell inside fungal cells, provide the most intimate example. Bacterial endosymbionts mostly occur in fungi of the phylum Mucoromycota and include Betaproteobacteria (Burkhoderia-related) and Mollicutes (Mycoplasma-related). Based on phylogenomics and estimations of divergence time, we hypothesized two different scenarios for the origin of these interactions (early vs late bacterial invasion). Sequencing of the genomes of fungal endobacteria revealed a significant reduction in genome size, particularly in endosymbionts of Glomeromycotina, as expected by their uncultivability and host dependency. Similar to endobacteria of insects, the endobacteria of fungi show a range of behaviours from mutualism to antagonism. Emerging results suggest that some benefits given by the endobacteria to their plant-associated fungal host may propagate to the interacting plant, giving rise to a three-level inter-domain interaction.
Similar content being viewed by others

Introduction
In their introduction to a special issue of Science entitled ‘Manipulating the microbiota’, Ash and Mueller (2016) wrote ‘No man is an island,’ quoting the poet John Donne. In recent years, we have learned that animals and plants host thousands of microbes, many beneficial, some essential and only a few deleterious. Next-generation sequencing approaches have enabled in-depth investigations of the microbial communities associated with animals and plants, asking ‘Who is there?’, and ‘What is it doing?’. Most surveys of animal- and plant-associated microbes so far have focused on bacteria and have demonstrated that they mainly participate in immune regulation and barrier defense (Haney and Ausubel, 2015). Conversely, fungi have been mostly neglected, even though recent studies demonstrated the unique biological and ecological role of fungal communities (Shakya et al., 2013; Coleman-Derr et al., 2016).
Since the discovery of antibiotics, fungi and bacteria have been assumed to interact antagonistically, but new ideas have emerged in microbial studies: as elements of the same microbiota, fungi and bacteria may interact non-antagonistically (Olsson et al., 2017). Indeed, emerging work has discovered an increasing number of cooperative bacterial–fungal associations, giving rise to a new field of microbiology (Frey-Klett et al., 2011). These inter-domain interactions occur in different ways: bacteria may loosely associate with the hyphal surface or may show some partner specificity, indicating potential metabolic complementation (Schroeckh et al., 2009; Olsson et al., 2017). The most intimate interaction takes place when bacteria live inside the fungal cells as endobacteria. Irrespective of their genetic and functional diversity, fungus-associated bacterial communities constitute a novel type of microbiota, the fungal microbiota (Desirò et al., 2014).
This mini-review summarizes our current knowledge on the endobacteria of fungi, with particular attention to Glomeromycotina (arbuscular mycorrhizal fungi (AMF)), and uses these AMF as a paradigm to better understand the diversity, origins and functions of fungal endobacteria.
The places they call home: distribution and diversity of fungal endobacteria
In 1970, Barbara Mosse reported the presence of bacteria-like organisms in the cytoplasm of Glomeromycotina spores (Mosse, 1970). In the words of Neil Armstrong, who landed in the moon the year before, we could say: ‘One small step for a woman, one giant leap for science’. Indeed, Mosse’s finding offered the first glimpse of this intimate, inter-domain interaction. In later decades, before the invention of PCR, many pioneering studies provided additional morphological evidence of the presence of these bacteria-like organisms in Endogone flammicorona (Bonfante and Scannerini, 1976) and several AMF species (references in Scannerini and Bonfante, 1991), demonstrating that endobacteria can be found widely in fungi. Only with the new millennium, the study of bacterial–fungal interactions blossomed, enjoying a boost from the suitable-for-all availability of molecular biological techniques.
Numerous recent studies have identified endobacteria in several fungal lineages (Olsson et al., 2017). Ascomycota and Basidiomycota were shown to harbour Gammaproteobacteria (Arendt et al., 2016) and Alphaproteobacteria (Sharma et al., 2008; Glaeser et al., 2016), respectively. However, most of the fungal endobacteria hitherto described were identified in members of the phylum Mucoromycota (Spatafora et al., 2016). The endobacteria of Mucoromycota mainly involve Betaproteobacteria (Burkhoderia-related) and Mollicutes (Mycoplasma-related). One exception is the cyanobacterium Nostoc puntiforme that resides within Geosiphon pyriformis (Schüßler and Kluge, 2001).
One of the better-known example involving a betaproteobacterium is the rice pathogenic fungus Rhizopus microsporus (Mucoromycotina), the causal agent of rice seedling blight, whose pathogenicity is due to the presence of the endosymbiont Burkholderia rhizoxinica (Partida-Martinez and Hertweck, 2005). Indeed, B. rhizoxinica produces the rhizoxin used as a virulence factor by the fungus (Partida-Martinez and Hertweck, 2007). Furthermore, this facultative endobacterium affects the vegetative reproduction of the fungal host, controlling the formation of sporangia and spores (Partida-Martinez et al., 2007). Much recent attention has been devoted to the interaction between a Burkholderia-related endobacterium (BRE) and several strains of Mortierella elongata (Mortierellomycotina) (Sato et al., 2010; Ohshima et al., 2016; Uehling et al., 2017). This obligate endosymbiont was named Mycoavidus cysteinexigens, as it requires cysteine to grow without the fungal host (Ohshima et al., 2016). Interestingly, further new studies (Li et al., 2017; Uehling et al., 2017) demonstrated that the presence of this endobacterium can strongly affect the metabolism of M. elongata: the wild-type strain with endobacteria showed a lower growth rate, compared with the strain that was cured (that is, devoid of endobacteria), suggesting that the fungus experiences a metabolic cost for accommodating Mycoavidus.
Also fungi of Glomeromycotina interact with Betaproteobacteria. An obligate rod-shaped BRE named Candidatus Glomeribacter gigasporarum (CaGg; Bianciotto et al., 2003) was detected in several species of the family Gigasporaceae (Mondo et al., 2012; Desirò et al., 2014). Its phylogenetically closest relative turned out to be Mycoavidus (Ohshima et al., 2016; Uehling et al., 2017). Similar to Mycoavidus (Sato et al., 2010), diverse genetic variants of CaGg were identified, thus casting doubt on the existence of a unique CaGg species (Desirò et al., 2014). Notwithstanding the different CaGg phylotypes detected, each fungal host strain harbours a genetically uniform CaGg population (Mondo et al., 2012; Desirò et al., 2014). Curiously this genetic uniformity was observed in the Betaproteobacteria populations found in Rhizopus (Lackner et al., 2011) and Mortierella (Sato et al., 2010).
A second type of endosymbiont resides within the spores and hyphae of Glomeromycotina, and this coccoid endobacterium represents a taxon of Mollicutes/Mycoplasma-related endobacteria (MRE) (Naumann et al., 2010; Desirò et al., 2013, 2014; Toomer et al., 2015). This novel bacterial taxon, whose biology is still little known, has been recently accommodated in the novel genus Candidatus Moeniiplasma and named Candidatus Moeniiplasma glomeromycotorum (CaMg) (Naito et al., 2017). Similar to CaGg, CaMg does not appear to be able to grow outside of its fungal host, a feature that places it among the obligate endosymbionts. CaMg occurs widely across Glomeromycotina and, unlike BRE, multiple CaMg populations can inhabit a single fungal strain: up to three highly dissimilar 16S rDNA CaMg phylotypes were identified in a single spore (Naumann et al., 2010; Desirò et al., 2014; Toomer et al., 2015). Notwithstanding the remarkable level of diversity, all the CaMg 16S rDNA sequences retrieved from AMF cluster within a monophyletic clade, sister to Mycoplasmatales and Entomoplasmatales. Regardless of the internal phylogenetic structure, the more accurate multigene phylogenetic reconstructions placed this motley lineage of Mollicutes within the Mycoplasmataceae, close to the Mycoplasma species (Naito et al., 2015; Torres-Cortés, et al., 2015). The impressive diversity that characterizes these enigmatic microbes seems to be determined by several factors, such as ultrarapid mutation rate, vertical transmission, activity of mobile genetic elements, active recombination machinery and apparent retention of the ability to conduct horizontal gene transfer (Naito et al., 2015; Toomer et al., 2015; Naito and Pawlowska, 2016a, 2016b).
Glomeromycotina are not the only fungi that harbour MRE, as CaMg-related endobacteria were identified in fruiting bodies of several strains of Endogone (Mucoromycotina) (Desirò et al., 2015a) and recent results reported their presence in Sphaerocreas pubescens (Mucoromycotina) (Takashima et al., 2015; Desirò et al., 2015b). Similar to the AMF-associated MRE, multiple and highly dissimilar populations can co-exist in the same Mucoromycotina fungal strain. Despite their striking diversity (up to 21.8% sequence divergence with AMF-associated MRE phylotypes), the 16S rDNA phylogeny placed Endogone-associated MRE with those living in Glomeromycotina. Interestingly, MRE from Endogone cluster within a new phylogenetic group, clearly distinguishable from the AMF-associated MRE groups (Desirò et al., 2015a). The finding of a novel group of MRE in another fungal lineage, Mucoromycotina, suggests that these endosymbionts are likely more widespread than expected. It would therefore be extremely interesting to know whether the third Mucoromycota subphylum, Mortierellomycotina, host MRE.
Conjectures about the origin of fungal endosymbiosis
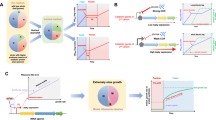
The presence of phylogenetically related endosymbionts in different fungal hosts offers insights about the origin and evolution of these inter-domain interactions. The CaGg symbiosis appears to have existed for at least 400 million years (Mondo et al., 2012), beginning before the diversification of the family Gigasporaceae (VanKuren et al., 2013), and suggesting a scenario where a BRE was already associated with the common ancestor of Gigasporaceae. Similarly, the Mycoavidus–Mortierella association seems to have originated in the mid-Paleozoic, most likely >350 million years ago (MYA) (Uehling et al., 2017). Further phylogenomic analyses and divergence time estimations placed the separation of the CaGg and Mycoavidus lineage from the free-living Burkholderia some 350 MYA, while their fungal hosts (Glomeromycotina and Mortierellomycotina, respectively) diverged at 358–508 MYA (Uehling et al., 2017). On the one hand, CaGg and Mycoavidus seem to have diverged from a common ancestor; on the other hand, B. rhizoxinica seems to have had an independent origin, sharing with the free-living Burkholderia its most recent common ancestor. Therefore, it could be hypothesized that a free-living Burkholderia-related bacterium (BRB) diversified into two ancestral lineages, which evolved as (i) mammalian and plant pathogens, saprotrophic species, plant-associated microbes (see references in Estrada-de Los Santos et al., 2013) and facultative endosymbionts of fungi (B. rhizoxinica) or (ii) obligate endobacteria that mostly lost their capacity to grow outside of their fungal hosts (CaGg and Mycoavidus). Accordingly, it is fascinating to hypothesize that the common ancestor of CaGg and Mycoavidus was already dwelling within the mycelium of the ancestral fungal lineage that later produced Glomeromycotina and Mortierellomycotina, thereby implying an early bacterial invasion of the fungal host (Figure 1). However, the rather limited distribution of these endosymbionts could undermine the early bacterial invasion hypothesis. Indeed, we would expect the broad presence of BRE in most of the fungal species that originated following the invasion. The presence of Mycoavidus exclusively within M. elongata may be attributed to limited investigation of Mortierellomycotina; however, Gigasporaceae seem to represent the only ‘fungal environment’ where CaGg can be retrieved. The absence of CaGg in most Glomeromycotina lineages might be the result of secondary losses of the bacterial partner. Alternatively, it might suggest a different scenario entailing a late bacterial invasion, which may have occurred when the evolutionary lines leading to Glomeromycotina and Mortierellomycotina had already separated (Figure 1).

Schematic representation of the two hypothetical scenarios of bacterial invasion (early vs late) at the basis of the origin of the association between BRE (in red) and MRE (in blue) and members of Mucoromycota. BRE: The early bacterial invasion (left side) assumes the existence of an ancestral free-living BRB that invaded the common ancestor of Glomeromycotina and Mortierellomycotina. During the separation of fungal hosts, BRE have been diversified into Candidatus Glomeribacter gigasporarum and Mycoavidus cysteinexigens and maintained only in Gigasporaceae (a family of Diversisporales) and Mortierella elongata, respectively. On the contrary, the late bacterial invasion (right side) entails a subsequent invasion by an ancestral free-living BRB, which may have occurred when the evolutionary lines leading to Glomeromycotina and Mortierellomycotina had already separated but before the diversification of Gigasporaceae. Differently, Burkholderia rhizoxinica has had an independent origin, sharing with the free-living Burkholderia its most recent common ancestor. Irrespective of the bacterial invasion scenario, CaGg is absent in some Gigasporaceae strains and most of Glomeromycotina lineages, and that might be the result of secondary losses of the endobacterial partner. MRE: The early bacterial invasion (left side) assumes the existence of an ancestral free-living/animal-associated MRB that invaded the ancestor of Mucoromycota and began its evolutionary path toward obligate mutualism. During the diversification of Mucoromycota, MRE have been maintained in Glomeromycotina and Mucoromycotina, whereas it is still unknown whether Mortierellomycotina maintained these coccoid endosymbionts. By contrast, the late bacterial invasion (right side) entails a subsequent invasion by an ancestral free-living/animal-associated MRB, which may have occurred when the evolutionary lines leading to the three Mucoromycota subphyla had already separated. However, regardless of the bacterial invasion scenario, MRE are absent in several fungal lineages or strains and that might be the result of secondary losses of the endobacterial partner. Legend: Burkholderia rhizoxinica (Br); Candidatus Glomeribacter gigasporarum (CaGg); Mycoavidus cysteinexigens (Mc); bacterial invasion event (arrow); presence of bacteria/endobacteria (thick line); absence of endobacteria in at least one fungal strain or lineage (species, genus, family or order) (thin line); unknown/there are no data available about the presence/absence of endobacteria (double thin line with a question mark).
The mechanisms of invasion also remain unknown. AMF hyphae can be damaged by other fungi (Lace et al., 2015) or by grazing soil fauna (Hedlund et al., 1991; Gange, 2000). This could have allowed an ancestral free-living BRB to invade the fungus through breaks in its wall. At the same time, we cannot exclude a more direct role of the bacterium in breaking the fungal wall and colonizing the fungus: indeed, there are several examples of Betaproteobacteria with chitinolitic capacities (Shimosaka et al., 2001; Shu-Chang et al., 2004). Further, a crucial role in the invasion process of Rhizopus was attributed to the B. rhizoxinica type II secretion system (T2SS), which releases chitinolytic enzymes (Moebius et al., 2014). Similarly, CaGg possesses T2SS and T3SS, which are differentially expressed through the fungal life cycle (Ghignone et al., 2012). However, bacterial chitinases were not detected in the currently available CaGg genome, suggesting that nowadays CaGg would not be able to invade a fungus using such a mechanism.
We can make similar conjectures about the origin and evolution of MRE. The widespread distribution of these endobacteria in Glomeromycotina suggested that the MRE invasion occurred before Glomeromycotina radiation, >400 MYA (Naumann et al., 2010). The finding that Endogone harbours MRE pushed Desirò et al. (2015a) to hypothesize that this inter-domain interaction was even older and originated before the split between Glomeromycotina and Mucoromycotina (Figure 1). This scenario assumes the existence of an ancestral MRE in the common ancestor of these two fungal lineages, thereby implying an early bacterial invasion of the fungal host. However, this scenario can be undermined by divergence time estimations. The divergence of Mycoplasmataceae was estimated at 410 MYA (Maniloff, 2002), whereas Mucoromycotina are believed to have split from the ancestral Glomeromycotina–Mortierellomycotina lineage some 500–600 MYA (Chang et al., 2015; Uehling et al., 2017), before the appearance of MRE. These results support the alternative scenario of a late bacterial invasion, which may have occurred when the evolutionary lines leading to Glomeromycotina, Mortierellomycotina and Mucoromycotina were already separated (Figure 1).
Naito et al. (2015) suggested that the origin of MRE resulted from a host-switching event from animals to fungi by an ancestral MRE. Other lineages of Mollicutes, such as Entomoplasma, Mesoplasma and Spiroplasma can be associated with animals, such as arthropods (Tully et al., 1993), which, in turn, are known to graze on fungal hyphae (Hedlund et al., 1991; Gange, 2000). Thus, in support of the host-switching hypothesis (Naito et al., 2015), it is intriguing to suppose that ancestral soil invertebrates, already associated with the MRE ancestor, may have acted as vectors, allowing the invasion of fungi. This scenario provides an interesting parallel with Phytoplasma, another lineage of uncultivable Mollicutes, which are insect-transmitted pathogenic agents of numerous plant species (Weintraub and Beanland, 2006). However, assuming the existence of an ancestral free-living Mollicutes/Mycoplasma-related bacterium (MRB), it also needs to take into consideration the possibility that the colonization of fungal hosts may have occurred directly from the soil, when the mycelium was damaged by other fungi or soil invertebrates.
Regardless of the nature of the symbiosis and how and when bacteria settled within fungi, these inter-domain associations may be included among the oldest interactions on Earth. Furthermore, they all involve plant-associated fungi, therefore raising questions about the influence of endobacteria on the history and evolution of plant–fungal symbiosis. It is intriguing to hypothesize that endobacteria may have had a role, as their fungal hosts (Field et al., 2015), during one of the major turning points in the evolution of the planet, the conquest of land by plants.
Keeping the home fires burning: analysis of endobacterial genomes to understand endobacterial functions
One common feature of the endobacteria so far identified in AMF is their vertical transmission, a modality used by these endosymbionts, which obligately depend on their host, to move from one fungal generation to the next. By contrast, the relationship is facultative for the fungus; for example, Gigaspora margarita can proliferate in the absence of CaGg (Lumini et al., 2007) and several other AMF strains devoid of CaMg can be propagated under laboratory conditions (Naumann et al., 2010). Vertical transmission and obligate dependence on the host may imply that endobacteria complement their metabolism using metabolites from their partner. The genome sequencing of both CaGg (Ghignone et al., 2012) and CaMg (Naito et al., 2015; Torres-Cortés et al., 2015) has largely confirmed the fungal–host dependency hypothesis. The size of the CaGg genome (1.7–1.9 Mb) and the more strongly reduced genomes of CaMg (0.7–1.3 Mb) are consistent with their uncultivable status. Indeed, they have a smaller genome when compared with B. rhizoxinica (3.7 Mb) (Lackner et al., 2011) and M. cysteinexigens (~2.6 Mb) (Fujimura et al., 2014; Uehling et al., 2017) that can grow independently, outside of their fungal hosts. The possibility to maintain B. rhizoxinica in pure culture allowed Moebius et al. (2014) to provide experimental evidences on the mechanisms underlying the colonization process of Rhizopus.
AMF endobacteria do not possess metabolic pathways producing essential amino acids. For example, CaGg lacks the ability to biosynthesize arginine, isoleucine, leucine, methionine, phenylalanine, tryptophan, histidine and valine, while CaMg has even more strongly reduced capacities. At the same time, CaGg contains the full operon for vitamine B12 synthesis and is equipped with many amino-acid permeases and transporters (Ghignone et al., 2012). By contrast, only a few putative nutrient transporters were annotated in CaMg genomes, such as a putative arginine–ornithine antiporter (Naito et al., 2015; Torres-Cortés et al., 2015).
Interestingly, when a hierarchical clustering analysis of KEGG metabolic pathways was applied to CaGg and 28 other bacterial genomes, the genomic features grouped CaGg together with insect endosymbionts, including Baumannia cicadellinicola and Wolbachia spp. (Ghignone et al., 2012). The same analysis applied to CaMg placed it with obligate endosymbionts of insects with reduced metabolic capacities (Torres-Cortés et al., 2015), including Ca. Carsonella ruddii and Ca. Sulcia muelleri, which possess some of the smallest characterized bacterial genomes (McCutcheon and Moran, 2012). These findings indicate that the genomes of phylogenetically distinct lineages of endobacteria have been likewise shaped by the selection pressure generated within diverse eukaryotic hosts, providing evidence of convergent evolutionary adaptation to an intracellular lifestyle (Ghignone et al., 2012). Curiously, B. cicadellinicola and Ca. Sulcia muelleri may co-reside in the same host, Homalodisca coagulata, where they reveal functional complementation (Cottret et al., 2010). Similarly, CaGg and CaMg have been found to coexist within the same fungal strain (Desirò et al., 2014). Thus, further work is required to address whether functional complementation also occurs between the two endosymbionts when they dwell together inside the same fungal niche. Moreover, the identification of mobile genetic elements in CaGg (Ghignone et al., 2012) and CaMg (Naito and Pawlowska, 2016b) offered new insights into the possibility of gene exchange between the two endobacterial lineages.
Although the genome analysis of AMF endobacteria has nicely revealed the mechanistic basis of their dependency on the host, the reasons why many fungal strains have maintained their bacterial guests for hundreds of million years have remained unknown. Using a stable endosymbiont-free strain of Gigaspora margarita (B−), multiple morphological and ‘omics’ approaches have been applied to directly compare the B− line with the wild-type strain hosting the endobacterium (B+). In spite of its success in colonizing the plant host, the B− line is impaired in the mycelial growth, has a different spore wall structure and sometimes produces fewer spores than the B+ line (Lumini et al., 2007). Consistent with these features, transcriptome analysis showed that CaGg has a stronger effect on the presymbiotic than the symbiotic phase of the fungal host (Salvioli et al., 2016). The coupling of transcriptomics and proteomics with physiological and cell biological approaches demonstrated that CaGg raises the bioenergetic capacity of the fungus, increasing its ATP production and respiration, and eliciting mechanisms to detoxify reactive oxygen species (Salvioli et al., 2016; Vannini et al., 2016). In this scenario, many proteins specifically involved in endogenous reactive oxygen species detoxification were not only found to be upregulated in the B+ line, which indeed produced more H2O2, but also had higher antioxidant capacities (Vannini et al., 2016). The fungal mitochondrion and its main metabolic pathways (ATP synthesis, respiration, reactive oxygen species metabolism) appear therefore to be particularly sensitive to the presence of the bacteria.
Much experimental evidence has offered insights into the complex molecular events that directed the evolution of endosymbionts into contemporary organelles (Dyall et al., 2004). In line with these observations and acknowledging that the distinction between endosymbiont and organelle is not always clear-cut, all the data generated so far might suggest a scenario where CaGg seems to act as a ‘mitochondrion-like organelle’. In addition, the antioxidant activities elicited in the fungus by CaGg were also observed in the plant when colonized by the B+ strain (Vannini et al., 2016). Thus, even if the B− line is able to maintain its mycorrhizal capacities, it might be hypothesized that the ecological functionality of the AM symbiosis is positively affected by the presence of CaGg (Figure 2). This hypothesis provides a further element to the idea that, similar to their animal counterparts, plants are never alone!

Schematic comparison of the colonization success of Gigaspora margarita with (B+) and without (B−) its endosymbiont (CaGg). When compared with the B+ strain, the growth of the germinating mycelium from a B− spore is slower and, when the host plant root is relatively distant (~10 cm from the spore), it stops after reaching a few centimetres (5 cm) (Lumini et al., 2007). Further, the B− strain often produces a lower number of spores than the B+ strain (Salvioli et al., 2016). Thus, in the words of Charles Darwin ‘survival of the fittest’, these differences make the B+ strain the fittest one. AMF are obligate biotrophs, that is, they need a plant host to complete their life cycle. In natural conditions, the capacity to grow faster and for a longer distance/time may provide the B+ strain a greater chance of success in finding and reaching a host plant root and then reproducing. On the contrary, the B− strain may have more difficulties in contacting plant host roots and, accordingly, completing its life cycle (grey spores). As a consequence, over the generations (from left to right), a decrease of the B− lines and a predominance of B+ lines in the soil might occur.
Home sweet home: conclusions
In the past few years, the scientific community has made giant strides in the study of bacterial–fungal interactions. Today, this new field of microbiology has assumed substantial relevance not only to microbial ecology but also to human health and biotechnology (Frey-Klett et al., 2011; Netzker et al., 2015). In this changing context, interest in the endobacteria of fungi, which have long been considered an oddity, has blossomed. Indeed, the combination of different ‘omics’ approaches is revealing an unexpectedly widespread distribution and is allowing us to gradually understand the biological functions and evolution of these microbes.
Here, we have described examples of endobacterial–fungal interactions that involve fungi in Mucoromycota. These fungi possess a coenocytic mycelium (that is, they lack or have few transverse septa), a feature that endobacteria might prefer. Indeed, the absence of physical barriers could facilitate bacterial movement along the hyphal network (Desirò et al., 2014; 2015a). Thus, we wonder whether Mucoromycota, with their coenocytic mycelium, represent the more suitable niche where endobacteria can thrive or whether endobacteria could also inhabit other groups of fungi, such as Dikarya. From the bacterial side, and irrespective of the host range, the diversity of fungal endobacteria discovered so far is mostly limited to two bacterial lineages, Burkholderia and Mycoplasma. Are they the only ones? Or do fungal endobacteria cover a broader taxonomic range, as for endobacteria–insect associations (Moran, 2001; Wernegreen, 2002).
The rather limited diversity and distribution of fungal endobacteria cannot yet be compared with the one of insect endosymbionts; however, their biological roles offer interesting parallels. Insect endosymbioses show a continuity of behaviours from true symbionts, through weak pathogens, to sex manipulators (Moran et al., 2008; McLean et al., 2016). Fungal endosymbioses behave likewise: B. rhizoxinica seems to be a true endosymbiont, with a positive effect on Rhizopus, as well as CaGg, which has a positive effect on G. margarita. By contrast, based on predictions of evolutionary theory, Toomer et al. (2015) suggested that CaMg may behave as antagonist of AMF. Last, even if it cannot be strictly defined as a sex manipulator, B. rhizoxinica affects the vegetative reproduction of its fungal partner (Partida-Martinez et al., 2007).
Similar to some obligate insect endosymbionts, such as Buchnera, endobacteria of fungi sequenced so far (particularly the AMF endobacteria) have revealed relevant genome reductions that entail host dependency and, as a consequence, a range of difficulties in growing these bacteria in pure culture. As for insects, the interaction is facultative for the fungus and, moreover, has only been maintained in some strains/lineages, suggesting that endobacteria are not essential for the evolutionary success of the hosts. Again this is true for endobacteria–insect interactions. For example, Wolbachia is one of the most widespread endobacteria, being present in around 40% of arthropod species. However, within a given species, usually Wolbachia infects most or only a few individuals (Zug and Hammerstein, 2012). Interestingly, when Wolbachia is introduced into Aedes aegypti it has the potential to suppress the ability of the mosquito to transmit pathogens such as dengue (Hoffmann et al., 2011).
Compared with these mature fields, studies of endobacterial–fungal interactions are in their infancy. However, it is worthwhile to note that insects and fungi share common metabolic pathways, such as chitin biosynthesis, suggesting that the shift from a free-living lifestyle to obligate mutualism inside eukaryotic cells overcame similar structural barriers. Thus, we can ask whether the endobacteria of insects, which made an evolutionary transition from a free-living lifestyle to obligate mutualism (Hosokawa et al., 2016), faced challenges similar to the ones we hypothesized for fungal endobacteria.
Focussing on endobacterial–fungal interactions, we can draw a portrait of an ancient scenario where soil is the main character, operating as a microbial reservoir where myriad organisms thrive together. Thus, it may be hypothesized that soil, with its living components, has acted as a facilitator in transferring free-living bacteria inside fungi. This has offered important opportunities for horizontal gene transfer in both directions, contributing to shaping the fungal genomes. The taking home message is that fungal endobacteria are probably active tenants, rather than silent occupants, of their fungal homes.
References
Arendt KR, Hockett KL, Araldi-Brondolo SJ, Baltrus DA, Arnold AE . (2016). Isolation of endohyphal bacteria from foliar Ascomycota and in vitro establishment of their symbiotic associations. Appl Environmental Microbiol 10: 2943–2949.
Ash C, Mueller K . (2016). Manipulating the microbiota. Science 352: 530–531.
Bianciotto V, Lumini E, Bonfante P, Vandamme P . (2003). 'Candidatus Glomeribacter gigasporarum' gen. nov., sp. nov., an endosymbiont of arbuscular mycorrhizal fungi. Int Syst Evol Microbiol 53: 121–124.
Bonfante P, Scannerini S . (1977). Cytological observations on the mycorrhiza Endogone flammicorona-Pinus strobus. Allionia 22: 23–34.
Bonfante P, Scannerini S . (1976). The ultrastructure of the zygospore in Endogone flammicorona Trappe and Gerdemann. Mycopathologia 59: 117–123.
Chang Y, Wang S, Sekimoto S, Aerts AL, Choi C, Clum A et al. (2015). Phylogenomic analyses indicate that early fungi evolved digesting cell walls of algal ancestors of land plants. Genome Biol Evol 7: 1590–1601.
Coleman-Derr D, Desgarennes D, Fonseca-Garcia C, Gross S, Clingenpeel S, Woyke T et al. (2016). Plant compartment and biogeography affect microbiome composition in cultivated and native Agave species. New Phytol 209: 798–811.
Cottret L, Milreu PV, Acuña V, Marchetti-Spaccamela A, Stougie L, Charles H et al. (2010). Graph-based analysis of the metabolic exchanges between two co-resident intracellular symbionts, Baumannia cicadellinicola and Sulcia muelleri, with their insect host, Homalodisca coagulata. PLoS Comput Biol 6: e1000904.
Desirò A, Faccio A, Kaech A, Bidartondo MI, Bonfante P . (2015a). Endogone, one of the oldest plant-associated fungi, host unique Mollicutes-related endobacteria. New Phytol 205: 1464–1472.
Desirò A, Bonfante P, Bonito G . (2015b), The fungal microbiome: a peek at plant-associated fungi and their endobacteria. 8th International Conference on Mycorrhiza; Flagstaff, AZ, USA.
Desirò A, Salvioli A, Ngonkeu EL, Mondo SJ, Epis S, Faccio A et al. (2014). Detection of a novel intracellular microbiome hosted in arbuscular mycorrhizal fungi. ISME J 8: 257–270.
Desirò A, Naumann M, Epis S, Novero M, Bandi C, Genre A et al. (2013). Mollicutes-related endobacteria thrive inside liverwort-associated arbuscular mycorrhizal fungi. Environ Microbiol 15: 822–836.
Dyall SD, Brown MT, Johnson PJ . (2004). Ancient invasions: from endosymbionts to organelles. Science 304: 253–257.
Estrada-de Los Santos P, Vinuesa P, Martínez-Aguilar L, Hirsch AM, Caballero-Mellado J . (2013). Phylogenetic analysis of Burkholderia species by multilocus sequence analysis. Curr Microbiol 67: 51–60.
Field KJ, Rimington WR, Bidartondo MI, Allinson KE, Beerling DJ, Cameron DD et al. (2015). First evidence of mutualism between ancient plant lineages (Haplomitriopsida liverworts) and Mucoromycotina fungi and its response to simulated Palaeozoic changes in atmospheric CO2. New Phytol 205: 743–756.
Frey-Klett P, Burlinson P, Deveau A, Barret M, Tarkka M, Sarniguet A . (2011). Bacterial-fungal interactions: hyphens between agricultural, clinical, environmental, and food microbiologists. Microbiol Mol Biol Rev 75: 583–609.
Fujimura R, Nishimura A, Ohshima S, Sato Y, Nishizawa T, Oshima K et al. (2014). Draft genome sequence of the betaproteobacterial endosymbiont associated with the fungus Mortierella elongata FMR23-6. Genome Announc 2: e01272–14.
Gange A . (2000). Arbuscular mycorrhizal fungi, Collembola and plant growth. Trends Ecol Evol 15: 369–372.
Ghignone S, Salvioli A, Anca I, Lumini E, Ortu G, Petiti L et al. (2012). The genome of the obligate endobacterium of an AM fungus reveals an interphylum network of nutritional interactions. ISME J 6: 136–145.
Glaeser S, Imani J, Alabid I, Guo H, Kumar N, Kämpfer P et al. (2016). Non-pathogenic Rhizobium radiobacter F4 deploys plant beneficial activity independent of its host Piriformospora indica. ISME J 10: 871–884.
Haney CH, Ausubel FM . (2015). Plant microbiome blueprints. Science 349: 788–789.
Hedlund K, Boddy L, Preston CM . (1991). Mycelial responses of the soil fungus, Mortierella isabellina, to grazing by Onychiurus armatus (Collembola). Soil Biol Biochem 23: 361–366.
Hoffmann AA, Montgomery BL, Popovici J, Iturbe-Ormaetxe I, Johnson PH, Muzzi F et al. (2011). Successful establishment of Wolbachia in Aedes populations to suppress dengue transmission. Nature 476: 454–457.
Hosokawa T, Ishii Y, Nikoh N, Fujie M, Satoh N, Fukatsu T . (2016). Obligate bacterial mutualists evolving from environmental bacteria in natural insect populations. Nat Microbiol 11: 15011.
Lace B, Genre A, Woo S, Faccio A, Lorito M, Bonfante P . (2015). Gate crashing arbuscular mycorrhizas: in vivo imaging shows the extensive colonization of both symbionts by Trichoderma atroviride. Environ Microbiol Rep 7: 64–77.
Lackner G, Moebius N, Partida-Martinez L, Hertweck C . (2011). Complete genome sequence of Burkholderia rhizoxinica, an endosymbiont of Rhizopus microsporus. J Bacteriol 193: 783–784.
Li Z, Yao Q, Dearth SP, Entler MR, Castro Gonzales HF, Uehling JK et al. (2017). Integrated proteomics and metabolomics suggests symbiotic metabolism and multimodal regulation in a fungal-endobacterial system. Environ Microbiol; epub ahead of print 21 November 2016; doi:10.1111/1462-2920.13605.
Lumini E, Bianciotto V, Jargeat P, Novero M, Salvioli A, Faccio A et al. (2007). Presymbiotic growth and sporal morphology are affected in the arbuscular mycorrhizal fungus Gigaspora margarita cured of its endobacteria. Cell Microbiol 9: 1716–1729.
Maniloff J . (2002) Phylogeny and evolution. In: Razin S, Herrmann R (eds). Molecular Biology and Pathogenicity of Mycoplasmas. Springer: Heidelberg, Germany, pp 31–44.
McCutcheon JP, Moran NA . (2012). Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol 10: 13–26.
McLean AH, Parker BJ, Hrček J, Henry LM, Godfray HC . (2016). Insect symbionts in food webs. Phil Trans R Soc B 371: 20150325.
Moebius N, Üzüm Z, Dijksterhuis J, Lackner G, Hertweck C . (2014). Active invasion of bacteria into living fungal cells. Elife 3: e03007.
Mondo SJ, Toomer KH, Morton JB, Lekberg Y, Pawlowska TE . (2012). Evolutionary stability in a 400-million-year-old heritable facultative mutualism. Evolution 66: 2564–2576.
Moran NA, McCutcheon JP, Nakabachi A . (2008). Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet 42: 165–190.
Moran NA . (2001). Bacterial menageries inside insects. Proc Natl Acad Sci USA 98: 1338–1340.
Mosse BA . (1970). Honey-coloured, sessile Endogone spores: II. Changes in fine structure during spore development. Arch Microbiol 74: 129–145.
Naito M, Pawlowska TE . (2016a). Defying Muller’s ratchet: heritable endobacteria escape extinction through recombination and genome plasticity. mBio 7: e02057-15.
Naito M, Pawlowska TE . (2016b). The role of mobile genetic elements in evolutionary longevity of heritable endobacteria. Mob Genet Elements 6: e1136375.
Naito M, Morton JB, Pawlowska TE . (2015). Minimal genomes of mycoplasma-related endobacteria are plastic and contain host-derived genes for sustained life within Glomeromycota. Proc Natl Acad Sci USA 112: 7791–7796.
Naito M, Desirò A, Gonzales JB, Tao G, Morton JB, Bonfante P et al. (2017). Candidatus Moeniiplasma glomeromycotorum’, an endobacterium of arbuscular mycorrhizal fungi. Int J Syst Evol Microbiol; epub ahead of print 10 January 2017; doi:10.1099/ijsem.0.001785.
Naumann M, Schüβler A, Bonfante P . (2010). The obligate endobacteria of arbuscular mycorrhizal fungi are ancient heritable components related to the Mollicutes. ISME J 4: 862–871.
Netzker T, Fischer J, Weber J, Mattern DJ, König CC, Valiante V et al. (2015). Microbial communication leading to the activation of silent fungal secondary metabolite gene clusters. Front Microbiol 6: 299.
Ohshima S, Sato Y, Fujimura R, Takashima Y, Hamada M, Nishizawa T et al. (2016). Mycoavidus cysteinexigens gen. nov., sp. nov., an endohyphal bacterium isolated from a soil isolate of the fungus Mortierella elongata. Int J Syst Evol Microbiol 66: 2052–2057.
Olsson S, Bonfante P, Pawlowska TE . (2017) Ecology and evolution of fungal-bacterial interactions. In: Dighton J, Oudemans P (eds). The Fungal Community: Its Organization and Role in the Ecosystem, 4th edn. CRC Press Taylor & Francis: Boca Raton, FL, USA, pp 563–583.
Partida-Martinez LP, Hertweck C . (2007). A gene cluster encoding rhizoxin biosynthesis in Burkholderia rhizoxinica, the bacterial endosymbiont of the fungus Rhizopus microsporus. Chembiochem 8: 41–45.
Partida-Martinez LP, Monajembashi S, Greulich KO, Hertweck C . (2007). Endosymbiont-dependent host reproduction maintains bacterial-fungal mutualism. Curr Biol 17: 773–777.
Partida-Martinez LP, Hertweck C . (2005). Pathogenic fungus harbours endosymbiotic bacteria for toxin production. Nature 437: 884–888.
Salvioli A, Ghignone S, Novero M, Navazio L, Venice F, Bagnaresi P et al. (2016). Symbiosis with an endobacterium increases the fitness of a mycorrhizal fungus, raising its bioenergetic potential. ISME J 10: 130–144.
Sato Y, Narisawa K, Tsuruta K, Umezu M, Nishizawa T, Tanaka K et al. (2010). Detection of betaproteobacteria inside the mycelium of the fungus Mortierella elongata. Microbes Environ 25: 321–324.
Scannerini S, Bonfante P . (1991) Bacteria and bacteria-like objects in endomycorrhizal fungi. In: Margulis L Fester R (eds). Symbiosis as a Source of Evolutionary Innovation: Speciation and Morphogenesis. MIT Press: Cambridge, MA, USA, pp 273–287.
Schroeckh V, Scherlach K, Nutzmann HW, Shelest E, Schmidt-Heck W, Schuemann J et al. (2009). Intimate bacterial–fungal interaction triggers biosynthesis of archetypal polyketides in Aspergillus nidulans. Proc Natl Acad Sci USA 106: 14558–14563.
Schüßler A, Kluge M . (2001) Geosiphon pyriforme, an endocytosymbiosis between fungus and cyanobacteria, and its meaning as a model system for arbuscular mycorrhizal research. In: Hock B (ed). The Mycota. Vol. IX. Fungal Associations. Springer-Verlag: Berlin, Germany, pp 151–161.
Shakya M, Gottel N, Castro H, Yang ZK, Gunter L, Labbé J et al. (2013). A multifactor analysis of fungal and bacterial community structure in the root microbiome of mature Populus deltoides trees. PLoS One 8: e76382.
Sharma M, Schmid M, Rothballer M, Hause G, Zuccaro A, Imani J et al. (2008). Detection and identification of bacteria intimately associated with fungi of the order Sebacinales. Cell Microbiol 10: 2235–2246.
Shimosaka M, Fukumori Y, Narita T, Zhang XY, Kodaira R, Nogawa M et al. (2001). The bacterium Burkholderia gladioli strain CHB101 produces two different kinds of chitinases belonging to families 18 and 19 of the glycosyl hydrolases. J Biosci Bioeng 91: 103–105.
Shu-Chang C, Wang JT, Vandamme P, Hwang JH, Chang PS, Chen WM . (2004). Chitinimonas taiwanensis gen. nov., sp. nov., a novel chitinolytic bacterium isolated from a freshwater pond for shrimp culture. Syst Appl Microbiol 27: 43–49.
Spatafora JW, Chang Y, Benny GL, Lazarus K, Smith ME, Berbee ML et al. (2016). A phylum-level phylogenetic classification of zygomycete fungi based on genome-scale data. Mycologia 108: 1028–1046.
Takashima Y, Yamamoto K, Seto K, Degawa Y, Narisawa K . (2015), Detection of Mollicutes-related endobacteria from putative saprotrophic Endogone spp. and Sphaerocreas pubescens 8th International Conference on Mycorrhiza: Flagstaff, AZ, USA.
Toomer KH, Chen X, Naito M, Mondo SJ, den Bakker HC, VanKuren NW et al. (2015). Molecular evolution patterns reveal life history features of mycoplasma-related endobacteria associated with arbuscular mycorrhizal fungi. Mol Ecol 24: 3485–3500.
Torres-Cortés G, Ghignone S, Bonfante P, Schüβler A . (2015). Mosaic genome of endobacteria in arbuscular mycorrhizal fungi: transkingdom gene transfer in an ancient mycoplasma-fungus association. Proc Natl Acad Sci USA 112: 7785–7790.
Tully JG, Bové JM, Laigret F, Whitcomb RF . (1993). Revised taxonomy of the class Mollicutes: proposed elevation of a monophyletic cluster of arthropod-associated Mollicutes to ordinal rank (Entomoplasmatales ord. nov.), with provision for familial rank to separate species with nonhelical morphology (Entomoplasmataceae fam. nov.) from helical species (Spiroplasmataceae), and emended descriptions of the order Mycoplasmatales, family Mycoplasmataceae. Int J Syst Evol Microbiol 43: 378–385.
Uehling J, Gryganskyi A, Hameed K, Tschaplinski T, Misztal PK, Wu S et al. (2017). Genome sequencing reveals origins of a unique bacterial endosymbiosis in the earliest lineages of terrestrial fungi. Environ Microbiol; doi:10.1111/1462-2920.13669.
VanKuren NW, den Bakker HC, Morton JB, Pawlowska TE . (2013). Effective population size, rRNA gene diversity, and evolutionary longevity of asexual Glomeromycota. Evolution 67: 207–224.
Vannini C, Carpentieri A, Salvioli A, Novero M, Marsoni M, Testa L et al. (2016). An interdomain network: the endobacterium of a mycorrhizal fungus promotes antioxidative responses in both fungal and plant hosts. New Phytol 211: 265–275.
Weintraub PG, Beanland L . (2006). Insect vectors of phytoplasmas. Annu Rev Entomol 51: 91–111.
Wernegreen JJ . (2002). Genome evolution in bacterial endosymbionts of insects. Nat Rev Genet 3: 850–861.
Zug R, Hammerstein P . (2012). Still a host of hosts for Wolbachia: analysis of recent data suggests that 40% of terrestrial arthropod species are infected. PLoS One 7: e38544.
Acknowledgements
We thank Alessandra Salvioli, Mara Novero and Stefano Ghignone as well as all the colleagues who, during the years, have contributed to the development of the project Endobacteria in AMF, and whose names have been quoted in the references. We also thank Jennifer M Mach for the critical reading and three anonymous referees for their helpful suggestions. Research illustrated in this review was funded by the Ateneo Project (ex 60%) and by the CSP-Ateneo Mycoplant project to PB.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Bonfante, P., Desirò, A. Who lives in a fungus? The diversity, origins and functions of fungal endobacteria living in Mucoromycota. ISME J 11, 1727–1735 (2017). https://doi.org/10.1038/ismej.2017.21
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2017.21
This article is cited by
-
Interplay of biotic and abiotic factors shapes tree seedling growth and root-associated microbial communities
Communications Biology (2024)
-
Klebsiella michiganensis: a nitrogen-fixing endohyphal bacterium from Ustilago maydis
AMB Express (2023)
-
A symbiotic footprint in the plant root microbiome
Environmental Microbiome (2023)
-
Characterization of microbial communities during Grifola frondosa (maitake) wood log cultivation
Journal of Wood Science (2023)
-
Detection and isolation of a new member of Burkholderiaceae-related endofungal bacteria from Saksenaea boninensis sp. nov., a new thermotolerant fungus in Mucorales
IMA Fungus (2023)
