
Abstract
Purpose:
Copy-number variants have been associated with a variety of diseases, especially cancer, autism, schizophrenia, and developmental delay. The majority of clinically relevant events occur de novo, necessitating the interpretation of novel events. In this light, we present the Scripps Genome ADVISER CNV annotation pipeline and Web server, which aims to fill the gap between copy number variant detection and interpretation by performing in-depth annotations and functional predictions for copy number variants.
Methods:
The Scripps Genome ADVISER CNV suite includes a Web server interface to a high-performance computing environment for calculations of annotations and a table-based user interface that allows for the execution of numerous annotation-based variant filtration strategies and statistics.
Results:
The annotation results include details regarding location, impact on the coding portion of genes, allele frequency information (including allele frequencies from the Scripps Wellderly cohort), and overlap information with other reference data sets (including ClinVar, DGV, DECIPHER). A summary variant classification is produced (ADVISER score) based on the American College of Medical Genetics and Genomics scoring guidelines. We demonstrate >90% sensitivity/specificity for detection of pathogenic events.
Conclusion:
Scripps Genome ADVISER CNV is designed to allow users with no prior bioinformatics expertise to manipulate large volumes of copy-number variant data. Scripps Genome ADVISER CNV is available at http://genomics.scripps.edu/ADVISER/.
Genet Med 17 9, 714–718.
Similar content being viewed by others

Introduction
Copy-number variants (CNVs) are defined as segments of the genome larger than 1 kb (ref. 1) that are either deleted or duplicated. CNVs can be benign polymorphic variations or can significantly affect phenotypic variability, gene expression, disease, and cancer.2 De novo CNV events are enriched in aggregate, especially in autism,3 schizophrenia,4 and developmental delay.5 Although extremely large (>1 Mb) de novo pathogenic CNVs can be readily recognized due to their rarity in nondiseased individuals, large CNVs (100 kb–1 Mb) are relatively frequent in both diseased and nondiseased individuals.5 For example, ~10% of nondiseased individuals carry a CNV of at least 400 kb, as compared with ~25% of individuals with intellectual disability and/or developmental delay.5 Because these CNVs occur de novo, the physical boundaries of CNVs causing similar diseases can vary greatly. Moreover, because of the relative rarity of each individual event, the critical region of many known pathogenic CNVs is not well defined.5 Although individual events are rare, large (>100 kb) de novo CNV events are not rare in aggregate (1.2 × 10−2 events per genome per transmission).6 Thus, the variability in observed CNV boundaries, the rarity of individual pathogenic CNVs, the relatively frequent occurrence of de novo CNV events, and the presence of large CNVs in healthy individuals warrant the use of computational tools to quickly annotate and prioritize potential pathogenic versus benign CNVs.
At the genome scale, CNVs can be detected using a variety of methods, including microarray-based methods,7 high-throughput sequencing-based read-depth approaches,8 and paired-end mapping approaches.9 With the rapid evolution of different CNV detection methodologies and increased resolution of CNV detection, it will be increasingly possible to detect small single-gene or even single-exon CNVs down to single–base pair resolution. These smaller CNVs are present in abundance in healthy individuals but also have a demonstrated role in mediating disease.10,11 The de novo mutation rate for these smaller CNVs is not well established but is expected to be higher than that for large CNVs.6 As the resolution for CNV detection continues to improve, tools for the computational prioritization of pathogenic CNVs will be necessary because the ultimate functional implication of these small CNVs will need to be evaluated in the context of the affected genes and their relationship to disease, as well as the nature of the CNV overlap with the gene (e.g., intronic, in-frame, truncating).
Thus, we introduce the Scripps Genome Annotation and Distributed Variant Interpretation Server (SG-ADVISER) CNV, which takes as input CNV calls and automatically produces both known and predicted information about each CNV and its relationship with disease. SG-ADVISER combines information about known pathogenic CNVs, known gene–disease relationships and the predicted functional impact of CNVs on these genes, and the allele frequency of CNVs observed in various reference populations in order to ultimately score CNVs for their potential pathogenicity.
Materials and Methods
SG-ADVISER CNV is a multicomponent system including a Web server that accepts and returns genomic data and annotations, a high-performance computing system that utilizes both precomputation databases and parallel computation to produce variant annotation rapidly, and a local client graphical user interface (UI) that allows the filtration of variants based on annotations and comparisons of multiple genomes using custom as well as predefined variant filtration strategies. The overall goal is to provide near-comprehensive CNV annotation without the burden of complex software or intense client-side computing capabilities.
SG-ADVISER CNV accepts variant files in CNVnator,8 VCF, Complete Genomics, PennCNV,12 or plain tab-delimited file formats. The computational infrastructure underlying SG-ADVISER CNV allows rapid turnover of CNV annotations. We evaluated the performance of SG-ADVISER by annotating CNV calls for 10 Wellderly genomes, sequenced by Complete Genomics. With an average of 3,782 CNVs per genome, the average time of completion was 6 ± 3 minutes per genome. For more information on input formats and error reporting, see https://genomics.scripps.edu/ADVISER/Input_Desc_CNV.jsp.
Data are encrypted during transfer to the Web server via a Secure Sockets Layer (SSL 3.0) to a secure computational cluster maintained by the Scripps Research Institute. Thus, SG-ADVISER CNV is compliant with the dbGaP Security Best Practices for controlled access data. In addition, variant files uploaded to the Web server, as well as the resulting annotation file, are destroyed 30 days after the variant file is uploaded.
Data output format
Annotations are output in a tab-separated file, in which the first six columns contain information about the physical location and type of variant being annotated and the rest of the columns are annotations produced by the SG-ADVISER CNV pipeline. The notes column of the output is used to store quality, dosage, and/or other metrics (e.g., P value of detection, normalized read depth) produced by the calling algorithm and can be utilized in downstream analyses for variant filtration. The annotation pipeline itself assumes that input CNVs and their boundaries are accurately determined, although many of the results are of robust to ambiguous breakpoint determination. Variants are presented as a single line per variant, yet complete annotations are produced for each individual transcript and overlap influenced by a variant; thus the format of each annotation column depends on whether the annotation is relative to the gene or transcript it impacts or relative to the physical location of the variant. Any column containing annotations produced relative to a gene or transcripts is further subdivided by triple slashes (“///”). Across annotation columns, “///”-separated values correspond to one another, i.e., annotations in the same position relative to “///”-separated values within a column influence the same transcript. Annotations not directly relevant to a particular transcript, for example, transcription factor binding sites or the conservation of the position, are also “///”-separated, but that separation corresponds to a related column. For example, transcription factor binding sites influenced by a variant are “///”-separated, and the calculation of the impact of the variant on binding of the “///” factor is presented in a separate “///”-separated column. When an annotation is not applicable to a variant or transcript, a null value is represented by a “-” character, often in the format of the column. For example, a column in which entries are formatted as “Value1~Value2”, if null, will receive a value of “-~-.” This is required because of partially complete outputs. For a more thorough description of the annotation types and output format, see http://genomics.scripps.edu/ADVISER/Result_Desc_CNV.jsp.
Results
Annotation categories
SG-ADVISER CNV is an automated computational system for producing known and predicted variant annotation information about CNVs. SG-ADVISER CNV produces four major classes of variant annotation, including
-
1. Residence within known or inferred genomic elements, as well as the predicted impact on those elements (e.g., exons, promoters, conserved elements, protein domains).
-
2. Reciprocal overlaps with reference CNVs and their allele frequency information from the 1000 Genomes Project,13 the Database of Genomic Variants (http://projects.tcag.ca/variation/), and the Scripps Translational Science Institute Wellderly cohort.
-
3. Prior knowledge from the Human Gene Mutation Database,14 OMIM,15 DECIPHER,16 Mitelman Database,17 ClinVar,18 and known cancer CNV genes19 is provided.
-
4. A synthesis of the above produces an American College of Medical Genetics and Genomics–like ADVISER variant classification schema for known and predicted disease association.
For a more thorough description of the annotation types and output format, see https://genomics.scripps.edu/ADVISER/Result_Desc_CNV.jsp.
Wellderly CNVs
Although a more thorough description of CNVs identified in the Wellderly population is presented elsewhere, we present a brief evaluation of the utility of these annotations. The Wellderly CNVs (identified using the standard Complete Genomics CNV calls) were compared with CNVs identified in the 1000 Genomes population.20 Eight percent (602 of 7,657) achieved >50% reciprocal overlap with a CNV identified in the 1000 Genomes population. Although a low degree of overlap is expected due to the allele frequency spectrum of CNVs in general (the majority of CNVs are rare), this number is potentially decreased further by differences in CNV detection sensitivity across sequencing platforms. Regardless, this demonstrates that Wellderly CNVs are an independent source of useful information above and beyond those catalogued in the 1000 Genomes Project. Moreover, certain CNVs classified as pathogenic in ClinVar overlap frequently with CNVs in the Wellderly cohort; for example, nsv529460 and nsv530281 have >80% reciprocal overlap with CNVs observed relatively frequently (>10% allele frequency) in the Wellderly data set, suggesting these data are useful for differentiating between benign and pathogenic CNVs in regions of interest.
ADVISER classification schema
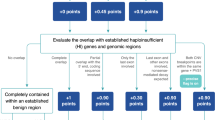
A variant classification is produced (ADVISER score) based on the American College of Medical Genetics and Genomics scoring guidelines with categories 1–5. In brief, category 1 variants are rare and completely encompass CNVs previously reported to be pathogenic, whereas category 2 variants are rare and predicted to be pathogenic based on a high degree of overlap with reported pathogenic CNVs or deleterious effect on genes known to be associated with disease. For genes not fully encompassed by a CNV, the effect on individual genes is predicted by determining whether the CNV produces an in-frame or out-of-frame deletion/duplication, what fraction of the conserved portion of the protein sequence is removed or amplified by the CNV, and whether alternative start codons may rescue N-terminal truncations. Reciprocal overlap with previously observed common CNVs in the 1000 Genomes and Wellderly cohorts are used to exclude CNVs that are unlikely to be pathogenic because of elevated allele frequency in the population at large. Table 1 displays the various thresholds required to achieve each ADVISER score tier.
The performance of ADVISER classification schema was evaluated using data from International Standards for Cytogenomic Arrays (ISCA). The ISCA data used to test the SG-ADVISER score performance were not included in the prior knowledge used to generate the scores, although there is a high degree of overlap between ISCA and ClinVar. We annotated 5,104 pathogenic and 5,822 benign CNVs and evaluated performance with and without the use of prior knowledge (e.g., known pathogenic CNVs). Known pathogenic CNVs are derived from ClinVar,18 Decipher,16 and other sources, including manual curation.5 To determine performance with prior knowledge excluded, we modified the classifier to rely solely on predictions (thus, class 1 scores are not possible). Predictions are based on the overlap and predicted impact of CNVs on specific genes known to be associated with disease, as described in Table 1 (ref. 14). Performance is evaluated as cumulative for each threshold ADVISER class or lower (more pathogenic). Thus, true-positive classifications occur when an International Collaboration for Clinical Genomics pathogenic variant achieves a threshold ADVISER class or lower, and true-negative classifications occur when ISCA benign CNVs do not achieve the threshold ADVISER class or lower (as delineated in Table 2 ).
As can be seen in Table 2 , ADVISER CNV achieved excellent 94% balanced accuracy for ADVISER class 1 and maintained this overall accuracy while improving sensitivity at lower threshold scores. This sensitivity–specificity profile is ideal for stepping through use cases with different sensitivity versus specificity requirements. For example, clinical applications require maximized specificity to reduce false-positive results, whereas research applications may require maximized sensitivity to prioritize all potential pathogenic CNVs (without the undue introduction of false positives). When all prior knowledge regarding known pathogenic CNVs was removed, ADVISER CNV was still able to achieve 90% balanced accuracy, demonstrating that SG-ADVISER CNV can accurately identify novel pathogenic CNVs and discriminate them from benign CNVs.
Comparison with other methods
CNVs are increasingly recognized as crucial contributors to genome diversity and disease risk; however, to the best of our knowledge, only a couple of tools exist to annotate CNVs.21,22,23 Although these tools compute overlaps of CNVs with known pathogenic CNVs, neither tool produces an ultimate pathogenicity classification. Thus, a direct comparison of predictive performance is not possible. SG-ADVISER CNV is unique in that it annotates CNVs using several different methods, databases, and algorithms, and it combines this information into a score that can discriminate benign from pathogenic CNVs.
User interface
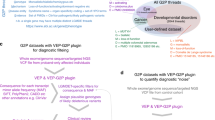
To facilitate the interpretation of SG-ADVISER CNV output, we added new functionality to the existing SG-ADVISER UI for small variants ( Figure 1 ). The UI is a visualization tool that allows scientists with little or no programming experience to easily and quickly view, manipulate, sort, and filter the SG-ADVISER CNV output file. For example, it is possible to sort or filter CNVs based on whether they are known disease-causative variants, they impact protein coding genes, or they have been previously observed in specific databases. Moreover, important downstream filtration functions—such as filtration on quality information, familial inheritance, or dosage—can be executed in the UI. These functionalities are necessary to differentiate between, for example, a pathogenic CNV that can be expected to result in carrier status and those that would be expected to result in an overt expressed phenotype. At any point during the process the tool can provide summary statistics and output the filtered results to a new file for further manipulation or presentation in other software tools. The tool is written in Java and uses multithreaded architecture and paginated view to make data analysis of large files (≥3 GB) possible on a desktop computer.

User interface: the SG-ADVISER user interface is pictured with useful functionalities highlighted. These functionalities include (1) sorting; (2) custom filters; (3) saving to file; (4) statistics; (5) help menu; (6) simple filter; (7) advanced filter; (8) stepping backward and forward through executed filters; and (9) adding and saving comments.
Discussion
Numerous studies have documented CNVs in a genome-wide fashion, and their impact on disease and evolution is clearly established. Remarkable improvements have been made to CNV detection and calling algorithms from sequence-based applications,8,9 yet tools to subsequently assign functional and potential clinical implications of each individual event remain limited and relegated to specialty laboratories. The overall goal of the SG-ADVISER CNV suite of tools is to put computational power and bioinformatics expertise into the hands of individuals with little to no computational proficiency but with the biological and/or clinical expertise to interpret CNVs when appropriately filtered.
There is one major caveat that should be highlighted when evaluating SG-ADVISER CNV output. SG-ADVISER CNV assumes that the breakpoints provided to the system are accurately determined, so that functional impacts can be precisely calculated. However, even with some sequence-based assays, single–base pair resolution of breakpoints is not always possible. Therefore, we have tiered our CNV scoring scheme so that it is robust to errors in breakpoint determination. The high-specificity to high-sensitivity accuracy profile of the ADVISER score ensures this; a functional CNV that is incorrectly classified due to breakpoint determination errors will be caught in the higher-sensitivity tiers of the SG-ADVISER score. Many of these predictions are actually unchanged, even when the breakpoint is not determined at single–base pair resolution, for example, CNVs encompassing whole exons would be accurately classified regardless of where within the adjacent introns the breakpoints lie. However, splicing predictions would be more dramatically influenced by errors in breakpoint determination. Nonetheless, SG-ADVISER CNV annotations anticipate the day when CNV detection at single–base pair resolution is routine practice.
Finally, SG-ADVISER will continue to be updated and expanded to provide access to new annotations/predictions as new versions become available, and improvements to the interactive UI and the Web interface will be made. Questions and requests for specific annotations can be made on the Biostar forum (http://www.biostars.org/).
Disclosure
A.T. is a cofounder and equity owner of Cypher Genomics. The other authors declare no conflict of interest.
References
Feuk L, Carson AR, Scherer SW . Structural variation in the human genome. Nat Rev Genet 2006;7:85–97.
Zhang F, Gu W, Hurles ME, Lupski JR . Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet 2009;10:451–481.
Sebat J, Lakshmi B, Malhotra D, et al. Strong association of de novo copy number mutations with autism. Science 2007;316:445–449.
Xu B, Roos JL, Levy S, van Rensburg EJ, Gogos JA, Karayiorgou M . Strong association of de novo copy number mutations with sporadic schizophrenia. Nat Genet 2008;40:880–885.
Cooper GM, Coe BP, Girirajan S, et al. A copy number variation morbidity map of developmental delay. Nat Genet 2011;43:838–846.
Itsara A, Wu H, Smith JD, et al. De novo rates and selection of large copy number variation. Genome Res 2010;20:1469–1481.
Pinto D, Darvishi K, Shi X, et al. Comprehensive assessment of array-based platforms and calling algorithms for detection of copy number variants. Nat Biotechnol 2011;29:512–520.
Abyzov A, Urban AE, Snyder M, Gerstein M . CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 2011;21:974–984.
Handsaker RE, Korn JM, Nemesh J, McCarroll SA . Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat Genet 2011;43:269–276.
Krumm N, O’Roak BJ, Karakoc E, et al. Transmission disequilibrium of small CNVs in simplex autism. Am J Hum Genet 2013;93:595–606.
Ching MS, Shen Y, Tan WH, et al.; Children’s Hospital Boston Genotype Phenotype Study Group. Deletions of NRXN1 (neurexin-1) predispose to a wide spectrum of developmental disorders. Am J Med Genet B Neuropsychiatr Genet 2010;153B:937–947.
Wang K, Li M, Hadley D, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 2007;17:1665–1674.
Abecasis GR, Auton A, Brooks LD, et al.; 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature 2012;491:56–65.
Stenson PD, Ball E, Howells K, Phillips A, Mort M, Cooper DN . Human Gene Mutation Database: towards a comprehensive central mutation database. J Med Genet 2008;45:124–126.
Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA . Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res 2005;33(Database issue):D514–D517.
Swaminathan GJ, Bragin E, Chatzimichali EA, et al. DECIPHER: web-based, community resource for clinical interpretation of rare variants in developmental disorders. Hum Mol Genet 2012;21(R1):R37–R44.
Mitelman F, Johansson B, Mertens F . The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer 2007;7:233–245.
Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014;42(Database issue):D980–D985.
Forbes SA, Bindal N, Bamford S, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res 2011;39(Database issue):D945–D950.
Mills RE, Walter K, Stewart C, et al.; 1000 Genomes Project. Mapping copy number variation by population-scale genome sequencing. Nature 2011;470:59–65.
Gai X, Perin JC, Murphy K, et al. CNV Workshop: an integrated platform for high-throughput copy number variation discovery and clinical diagnostics. BMC Bioinformatics 2010;11:74.
Zhao M, Zhao Z . CNVannotator: a comprehensive annotation server for copy number variation in the human genome. PLoS One 2013;8:e80170.
CNVAnalysisToolkit 1.3 [computer program]. 2014. http://statgen.org/.
Acknowledgements
This work was supported by the National Human Genome Research Institute Genome Sequencing Informatics Tools Program via National Institutes of Health grant U01 HG006476 (to A.T.). Further information is available at http://iseqtools.org. Further support was provided by Scripps Genomic Medicine, a National Institutes of Health–National Center for Advancing Translational Sciences Clinical and Translational Science Award (5 UL1 RR025774) to the Scripps Translational Science Institute.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Erikson, G., Deshpande, N., Kesavan, B. et al. SG-ADVISER CNV: copy-number variant annotation and interpretation. Genet Med 17, 714–718 (2015). https://doi.org/10.1038/gim.2014.180
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2014.180
Keywords
This article is cited by
-
ParseCNV2: efficient sequencing tool for copy number variation genome-wide association studies
European Journal of Human Genetics (2023)
-
ClassifyCNV: a tool for clinical annotation of copy-number variants
Scientific Reports (2020)
-
Genome-wide common and rare variant analysis provides novel insights into clozapine-associated neutropenia
Molecular Psychiatry (2017)
-
CNV analysis in the Lithuanian population
BMC Genetics (2016)
-
Whole-genome mutational burden analysis of three pluripotency induction methods
Nature Communications (2016)
