
Abstract
Purpose:
This report describes a fast online tool to accelerate and improve clinical interpretation of single nucleotide polymorphism array results for diagnostic purposes, when consanguinity or inbreeding is identified.
Methods:
We developed a web-based program that permits entry of regions of homozygosity and, using OMIM, UCSC, and NCBI databases, retrieves genes within these regions as well as their associated autosomal recessive disorders. Relevant OMIM Clinical Synopses can be searched, using key clinical terms permitting further filtering for candidate genes and disorders.
Results:
The tool aids the clinician by arriving at a short list of relevant candidate disorders, guiding the continued diagnostic work-up. Its efficacy is illustrated by presenting seven patients who were diagnosed using this tool.
Conclusion:
The online single nucleotide polymorphism array evaluation tool rapidly and systematically identifies relevant genes and associated conditions mapping to identified regions of homozygosity. The built-in OMIM clinical feature search allows the user to further filter to reach a short list of candidate conditions relevant for the diagnosis, making it possible to strategize more focused diagnostic testing. The tabulated results can be downloaded and saved to the desktop in an Excel format. Its efficacy is illustrated by providing a few clinical examples.
Genet Med 2013:15(5):354–360
Similar content being viewed by others


Introduction
The molecular basis of Mendelian traits continues to be clarified as new technologies improve the classic strategies of gene mapping and mutation detection, producing insights with just a few patients with a rare disorder.1,2 This increase in knowledge is a promising development, fueling the expectation that patients and their families will benefit from unraveling the molecular basis of these disorders. However, this improved knowledge is frequently not met by an increased capability of the clinician to recognize, identify, and diagnose patients with these many rare disorders.3 This conundrum, in our opinion, is largely due to the fact that the genetics evaluation is still predominantly phenotype-oriented. Therefore, the clinician’s experience and engagement are important variables, contributing to the lack of recognition of signs and symptoms (patterns) if the phenotype is rare or atypical. Clearly, innovative and more effective approaches are needed to achieve a timely diagnosis, to optimize management, to identify action for surveillance and anticipatory guidance, to communicate recurrence risk, and to counsel regarding reproductive options.
Most medical genetics units serve families with obvious consanguinity and from communities with geographic isolation expected to result in inbreeding. Consanguinity is an ancient cultural practice that is common in many regions of the world, especially in the Middle East and parts of Asia.4 Individuals born into such families typically have long runs (or regions) of homozygosity (ROHs), with the combined length a function of the degree of relatedness of the parents, expressed as ROHtotal = F × sizehg (sizehg = size of the haploid human genome, or ~3,200 Mb). The coefficient of inbreeding, F, is defined as the fraction of the individual’s genome expected to be homozygous by descent. Because F is 1/16 in the offspring of first-cousin parents, the calculated ROHtotal is 200 Mb. It is well known that such individuals are at an increased risk for autosomal recessive disorders, with the mutated locus being typically homozygous and expected to reside in a ROH. This risk (α) can actually be estimated using α = F/(F + (1 − F)q), where q is the frequency of the mutant allele.5,6 In an inbred or consanguineous family, q will be small as compared with F, and α therefore close to 1, which implies that the disease-causing mechanism is highly likely due to homozygosity by descent.5,6 The single nucleotide polymorphism (SNP) array, a genomic microarray platform, can identify these various ROHs. Through analysis of these ROHs, we can evaluate for autosomal recessive disorders associated with genes that map to these regions. This would therefore constitute a meaningful approach to identify candidate genes and associated disorders. In Saudi Arabia, where consanguinity is common, the usefulness of an SNP array analysis early in the diagnostic evaluation of a phenotype with genetic heterogeneity has been demonstrated, thus making the diagnosis in a more targeted manner and with less cost.7 However, it can take a skilled genetics professional several hours to query genetic databases to evaluate ROHs that total 200 Mb for candidate genes and associated disorders.
On the basis of our clinical experience and realizing that the time needed to manually interrogate all ROHs thoroughly using current databases is prohibitive, we developed a computer algorithm to systematically search through relevant genetic databases, including the Online Mendelian Inheritance in Man (OMIM) database, the University of California at Santa Cruz Genome Browser (UCSC), and the National Center for Biotechnology Information (NCBI) database, to rapidly identify the genes mapping to the ROHs (as given in the original SNP array report), to enumerate associated autosomal recessive clinical disorders and their clinical features, and to match the clinical features of the patient being evaluated against these phenotypes. We further demonstrate the clinical utility in seven recent patients, accrued in just a few months. Another case has been reported elsewhere.8
Materials and Methods
Our online SNP array evaluation tool, based on the Common Gateway Interface, uses Practical Extraction and Report Language (Perl) to handle hypertext transfer protocol (HTTP) requests and responses. The graphic user interface is implemented using HyperText Markup Language (HTML), cascading style sheets, and JavaScript and delivered to client servers using an Apache 2 HTTP server. The approach chosen in our tool is quite different from the conventional way of using various individual online genetics browsers, such as the Database of Genomic Variants and the UCSC Genome Browser, where users manually scrutinize candidate genes for a single ROH at a time; in contrast, our tool can systematically search candidate genes on multiple (theoretically unlimited) ROHs, using several genetic databases. Currently, login privileges are granted by e-mail registration at http://www.ccs.miami.edu/ROH.
To conduct a search ( Figure 1 ), after clinical evaluation and receipt of a SNP array report, preferably as an electronic file to facilitate “cut” and “paste” of the nucleotide addresses, the user enters the coordinates of the various ROHs (in bases, kb, or Mb) and selects the Human Genome Assembly (hg) version stated in the report. The tool then automatically converts the coordinates to hg19 if an older hg version was used in the SNP array report. The user picks one depth of the search: (i) all genes, (ii) OMIM-annotated genes, (iii) OMIM-annotated genes associated with disorders (Morbid Map genes), or (iv) Morbid Map genes associated with autosomal dominant traits or Morbid Map genes associated with autosomal recessive traits. For the last three options, the user can provide the patient’s key clinical features (phenotype) to refine the search, using Boolean operators “AND,” “OR,” and “NOT” to formulate an efficient search string from the “OMIM Clinical Synopsis.” Because some OMIM entries have no Clinical Synopsis (and hence also no documented mode of inheritance), a search through annotation text for clinical features in OMIM genes is an available, although less reliable option. Separately, a special option permits entry of specific genes of interest, using either the official gene symbol or gene identification number. This is an option for users who have “favorite gene” lists, for example, for conditions with locus heterogeneity (e.g., retinitis pigmentosa and Bardet–Biedl syndrome).

Input of relevant data into the search page of the single nucleotide polymorphism (SNP) array evaluation tool. In this example, three regions of homozygosity (ROHs) identified by SNP array analysis are placed into the text box, one ROH per line, after which the user selects the location unit (base, kb, and/or Mb) and the version of the Human Genome Assembly as stated in the SNP array analysis report. The user then selects the query type, here ROH (microdeletion/microduplication option not discussed here). The user then selects the query depth, typically for autosomal recessive disorders in the setting of consanguinity. The user may filter further by performing a clinical features search using an OMIM Clinical Synopsis search string (using search terms, often using wildcards, combined with Boolean operators).
The report of the search ( Figure 2 ), returned in HyperText Markup Language, is downloadable in an Excel spreadsheet format with tabs corresponding to the result sections. The result page also provides the calculated coefficients of inbreeding (F) and consanguinity (f) using the formulae F = ROHtotal/sizehg (sizehg = 3,138 Mb in hg19) and f = 2F. Also provided are the genes identified (given a certain search depth), their associated phenotypes, and hypertext links to the OMIM entries with the NCBI and UCSC annotations. In our experience, using relevant clinical features, the user usually arrives at a short list of candidate genes and disorders for review and ranking. The user can then strategize the continued diagnostic approach, now focused on a small selection of likely relevant genes and disorders.

single nucleotide polymorphism array evaluation tool report of search. The report of the search, returned in hypertext markup language and downloadable in a tabulated Excel spreadsheet format, provides coefficients of inbreeding (F) and consanguinity (f), the genes identified (given a certain search depth), their associated phenotypes and hypertext links to the OMIM genes and their disorders. University of California at Santa Cruz and National Center for Biotechnology Information annotations.
Cases solved through the use of the SNP array evaluation tool were not collected systematically, as the SNP array evaluation tool went through various stages of development, making cases hard to compare even if accrued in one institution. One case was recruited from another institution as particularly illustrative. Sanger sequencing of relevant genes was performed in commercial or academic, US-based, Clinical Laboratory Improvement Amendments (CLIA)-certified laboratories unless stated otherwise.
Results
Principles and procedures are illustrated on the basis of seven recent patients and their families ( Table 1 ). The patient group, ranging from newborns to 12-year-olds, presented with common issues for clinical geneticists: abnormal newborn screening results, hypotonia, developmental delay, failure to thrive, neurologic regression, or obesity. A few patients had other features that suggested a specific condition (polydactyly and hypogonadism consistent with Bardet–Biedl syndrome) or category of metabolic disorder (hyperammonemia suggesting a urea cycle defect; coarse facies pointing to a storage disorder). For the two cases of Bardet–Biedl syndrome, the tool correctly identified the one candidate gene that lay within the ROH out of 18, obviating a tedious, expensive search by serially sequencing all candidate genes. In all cases, the diagnostic odyssey ended and families were counseled regarding the diagnosis, the recurrence risk, and the availability of prenatal diagnosis for future pregnancies. In one case (patient 6), the newly assigned diagnosis led to change in management, followed by improved metabolic control and linear growth.
Patient 1
A male newborn with prenatal onset of ascites was the fourth child of first cousin parents. The three siblings were healthy. He was hypotonic, and examination results were otherwise normal. Elevation of very long chain fatty acids and elevated erythrocyte plasmalogen led to the diagnosis of Zellweger syndrome. PEX genes were considered. SNP array revealed 191 Mb of ROHs > 8 Mb (a total of 191 Mb of homozygosity when considering only ROHs > 8 Mb in length, if including shorter ROHs as requested from the laboratory, totaling 363 Mb of ROHs > 1 Mb), with PEX1 and PEX6 mapping within the ROHs. Sequencing of PEX1 revealed no mutations, and sequencing of PEX6 was not available commercially. Having reached an impasse, more biochemical studies were performed; enzymatic activity from fibroblast culture revealed normal catalase activity and intracellular location, suggesting a single peroxisomal enzyme defect instead of a form of Zellweger syndrome. The genomic SNP array evaluation tool, with the clinical feature search (hypoton* AND ascites) revealed two further genes (GBE1 and HSD17B4), but only the latter had peroxisomal location. Novel homozygous mutations in HSD17B4 were identified by the Laboratory Genetic Metabolic Diseases, Academic Medical Center of the University of Amsterdam, The Netherlands: c.296insA (p.N99KfsX12), predicted to result in a truncated protein. Final diagnosis was D-bifunctional protein deficiency (OMIM no. 261515). The patient died at the age of 18 months.
Patient 2
A male newborn was referred because an abnormal newborn screen revealed elevated C5OH acylcarnitine species (0.82 μmol/l initially and 0.94 μmol/l on a repeat sample 10 days later; normal cutoff <0.80 μmol/l). He was the second child of first-cousin parents. Elevation of C5OH in plasma was confirmed, and urine organic acid studies revealed elevations predominantly of 3-methylglutaconic acid. Due to locus heterogeneity of 3-methylglutaconic acidurias, a SNP array was performed revealing 261 Mb of ROHs > 8 Mb (374 Mb of ROHs > 1 Mb). The genomic SNP array evaluation tool, with the clinical feature search using two wildcards (*glutacon*), revealed two genes: AUH (3-methylglutaconic aciduria type 1, OMIM no. 250950) and OPA3 (3-methylglutaconic aciduria type 3, Costeff syndrome). Costeff syndrome was deemed unlikely because it is mostly seen in individuals of Iraqi–Jewish descent. Novel homozygous mutations in AUH were identified: c.373C→T (p.R125W), with the p.Arg125 highly conserved from fruitfly to humans, and predicted to be damaging by Polyphen2(ref.9) and SIFT.10 He was started on L-carnitine and mild protein restriction and is doing well at the age of 15 months.
Patient 3
A 3-month-old boy was evaluated for developmental delay, hypogonadism, and polydactyly. Pertinent family history included first-cousin parents, and a brother and sister manifesting similar signs and symptoms, in addition to obesity, both without diagnosis at the time. SNP array revealed 207 Mb of ROHs > 8 Mb (316 Mb of ROHs > 1 Mb). The genomic SNP array evaluation tool, with the clinical feature search (polydact* AND (delay OR retard*)), identified TTC8 as the only candidate gene. Sequencing revealed homozygosity for a known pathogenic mutation in TTC8: c.624+1G→A, predicted to abolish the universal donor splice site of exon 7, securing the diagnosis of Bardet–Biedl syndrome (OMIM no. 209900).
Patient 4
A 30-month-old girl was evaluated for a history of regression of milestones, progressive weakness, hypotonia, hyperreflexia, and loss of speech starting at the age of 1 year. Brain magnetic resonance imaging and ophthalmological examination were normal at 26 months. The parents denied consanguinity but were from the same community. Initially, a full genetic, metabolic, and endocrine evaluation was normal, including a karyotype, methylation studies for Angelman, MECP2 testing, creatine kinase level, and lysosomal enzyme testing for GM1 gangliosidosis, metachromatic leukodystrophy, and Tay–Sachs and Krabbe diseases. SNP array revealed 179 Mb of ROHs > 8 Mb (311 Mb of ROHs > 1 Mb). The genomic SNP array evaluation tool, with the clinical features search (hypoton* AND regress*), identified eight candidate disorders, six of which had already been ruled out by specific studies. Infantile neuroaxonal dystrophy (OMIM no. 256600) was considered the likely diagnosis in the two remaining candidate disorders, and sequencing of PLA2G6 revealed homozygosity for c.2098C→T, predicted to lead to a premature stop codon at p.700.
Patient 5
A 7-year-old boy, whose parents were second cousins, was seen for developmental delay. He had mildly coarse facial features, as compared with his younger brother. Urinary glucosaminoglycans showed normal levels. SNP array revealed 38 Mb of ROHs > 8 Mb (134 Mb of ROHs > 1 Mb). Searching for recessive disorders with the clinical features search ((delay OR retard*) AND coarse) within the ROHs identified Sanfilippo syndrome B as a candidate disorder. Lysosomal studies revealed markedly reduced α-N-acetylglucosaminidase activity. Novel homozygous mutations c.1811C→T, p.P604L in NAGLU were identified. The p.P604 is highly conserved from zebrafish to human. Final diagnosis was Sanfilippo syndrome B (OMIM no. 252920).
Patient 6
A 9-year-old girl underwent hospital evaluation for failure to thrive, hepatomegaly, osteopenia, and episodic hyperammonemia. She had been diagnosed in the past with autoimmune hepatitis based on liver biopsies and had been unsuccessfully treated with corticosteroids and immune modulators. Parents were first cousins and first cousins once removed; a younger sibling was healthy. A urea cycle disorder with relatively mild features was suspected. SNP array revealed 299 Mb of ROHs > 8 Mb (435 Mb of ROHs > 1 Mb). Of five of the relevant recessive urea cycle and other relevant disorders, only ASL (argininosuccinic aciduria) and PCCA (propionic aciduria) mapped to the ROHs, but these diagnostic possibilities had been ruled out by biochemical studies. Searching for other relevant recessive disorders, using the clinical features search ((hyperammon* OR ammon*) AND hepatomegaly AND thrive), revealed lysinuric protein intolerance (OMIM no. 222700) as a candidate diagnosis, which was subsequently confirmed by studies of plasma and urinary amino acids. She was placed on a protein-restricted diet and started on citrulline supplementation; she had significantly improved (catch-up growth, no further hyperammonemic episodes) until she was lost to follow-up when the family moved out of the state. Mutation studies could not be performed.
Patient 7
A 12-year-old boy was evaluated for developmental delay. Parents were first cousins once removed. He had obesity, hypogonadism, and postaxial polydactyly, consistent with Bardet–Biedl syndrome. SNP array revealed 145 Mb of ROHs > 8 Mb (287 Mb of ROHs > 1 Mb). Searching for relevant genes of the clinical features search (polydact* AND (delay OR retard*)) revealed BBS1 to be the only gene of Bardet–Biedl syndrome within the ROHs. Sequencing revealed homozygosity for a known pathogenic mutation: c.1169T→G, p.M390R. Final diagnosis was Bardet–Biedl syndrome (OMIM no. 209900).
Discussion
As with any bioinformatics approach, reliable results depend on high-quality laboratory reports of the individual patient and the completeness and validity of the underlying databases, including OMIM, especially the OMIM Clinical Synopsis database, UCSC and NCBI ( Figure 3 ). Clearly, if there is a high degree of consanguinity, as seen in offspring of incestuous relationships, the ROHtotal may take up 25% of the genome, reducing the success rate of the tool. On the other hand, in cases where parents are only remotely related, the ROHtotal will be relatively low, and the probability of a disorder being caused by mechanisms other than “identity by descent” will be increased. To date, our impression is that the SNP array evaluation tool functions optimally when ROHtotal is between 50 and 400 Mb. Obviously, nonspecific phenotypes as a learning disability or a seizure disorder will necessarily produce a large number of results, although the combination of two nonspecific findings by the Boolean “AND” will likely produce a tractable short list.

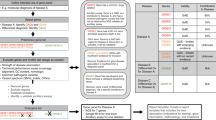
Algorithm used by single nucleotide polymorphism (SNP) array evaluation tool to identify candidate genes and disorders searching within regions of homozygosity (ROHs). Genetic evaluation identifies patient at risk for autosomal recessive disorders by pedigree analysis. SNP array analysis identifies genomic coordinates flanking various ROHs. The tool filters at desired depth (here for autosomal recessive disorders). The user can further filter by matching the clinical features of these disorders with key clinical features of the patient. In this way, a short list of candidate gene(s) and disorder(s) is created for review, ranking, and further evaluation. Reaching a diagnosis can be strategized using relevant tests (Sanger sequencing, biochemical testing, radiography, and pathological examination of biopsy specimens). This process is completed once a diagnosis is reached, moving to treatment and counseling. If the strategy does not result in an actionable list or diagnosis, the assumptions have to be reconsidered, including the possibility of an as yet unmapped disorder.
Our experience suggests room for improvement in the Clinical Synopses and common vocabulary of OMIM. Sometimes OMIM Clinical Synopses for even well-known disorders are not available, resulting in such disorders inadvertently not being included among the results, as the inheritance pattern (documented in the OMIM Clinical Synopsis) is then also absent. In our opinion, a clinical synopsis should always be available and regularly updated in a timely manner. As to precision and recall (e.g., “lack of vision” vs. “blindness”, or “developmental delay” vs. “mental retardation”), OMIM clearly suffers from lack of standardized, hierarchically structured terminology and could benefit from collaborating with existing endeavors, such as the Unified Medical Language System, Systematized Nomenclature of Medicine Clinical Terms, or Human Phenotype Ontology. Human Phenotype Ontology may be especially useful as it provides standardized vocabulary of phenotypic abnormalities encountered in human disease, initially developed using information from OMIM.11
A clinical geneticist’s expert judgment and experience will likely improve results by detecting the patient’s key symptoms and signs and by deciding on the most informative search terms. Some laboratories report only relatively long ROHs (longer than 8 or 10 Mb), although short ROHs may also carry valuable information. Although homozygous pathogenic mutations were all on ROHs > 10 Mb in our selected cases, such occurrence in ROHs < 10 Mb has been documented.12 Because consanguinity is a cultural practice, the presence of long and short ROHs identified in a patient is reflective of multigenerational consanguinity, presumably as many ROHs have shortened due to recombination. Actually, in such populations, the background level of homozygosity is increased by ~5% over and above that predicted by simple models of consanguinity.12 In our experience, the laboratories performing SNP array testing make these short ROHs available electronically, if requested. Because interrogating a large number of ROHs is not a problem for our tool, a genetics professional can analyze multiple ROHs each as low as 1 Mb in length. Although we emphasize the benefit of SNP analysis in patients with recognized consanguinity or inbreeding, as many as 93% of homozygous mutations in the offspring of outbred families affected by rare diseases reflect identity by descent, so even short ROHs in outbred matings could be informative.13 Finally, having used the approach as outlined above without arriving at a diagnosis against a background of consanguinity, such negative finding adds to the suspicion that the disorder may not have been documented before or, more likely, that the causative locus has not yet been mapped. In such a case, the causative locus may be identified using other, currently more expensive technologies such as the whole-exome sequencing.
In summary, we have demonstrated that during the genetics evaluation of an individual affected by a rare disorder in the setting of consanguinity, a SNP array analysis should be considered, unless the diagnosis is obvious. It is our opinion that our SNP array evaluation tool can greatly facilitate the diagnostic process, as it allows the clinician to rapidly and systematically filter both genomic and phenotypic information for candidate genes and disorders.
Disclosure
The authors declare no conflict of interest.
References
Collins FS, Morgan M, Patrinos A . The Human Genome Project: lessons from large-scale biology. Science 2003;300:286–290.
Coppola G, Geschwind DH . Technology Insight: querying the genome with microarrays–progress and hope for neurological disease. Nat Clin Pract Neurol 2006;2:147–158.
Deeble VJ, Roberts E, Jackson A, Lench N, Karbani G, Woods CG . The continuing failure to recognise Alström syndrome and further evidence of genetic homogeneity. J Med Genet 2000;37:219.
Jaber L, Halpern GJ, Shohat M . The impact of consanguinity worldwide. Community Genet 1998;1:12–17.
Lander ES, Botstein D . Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science 1987;236:1567–1570.
Huqun, Fukuyama S, Morino H, et al. A quantitatively-modeled homozygosity mapping algorithm, qHomozygosityMapping, utilizing whole genome single nucleotide polymorphism genotyping data. BMC Bioinformat 2010;11(suppl 7):S5.
Alkuraya FS . Homozygosity mapping: one more tool in the clinical geneticist’s toolbox. Genet Med 2010;12:236–239.
Winters KA, Jiang Z, Xu W, et al. Re-assigned diagnosis of D4ST1-deficient Ehlers-Danlos syndrome (adducted thumb-clubfoot syndrome) after initial diagnosis of Marden-Walker syndrome. Am J Med Genet A 2012; doi:10.1002/ajmg.a.35613.
Adzhusei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Meth 2010;7:248–249.
Kumar P, Henikoff S, Ng PC . Predicting the effects of coding nonsynonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4:1073–1081.
Robinson PN, Köhler S, Bauer S, Seelow D, Horn D, Mundlos S . The Human Phenotype Ontology: a tool for annotating and analyzing human hereditary disease. Am J Hum Genet 2008;83:610–615.
Woods CG, Cox J, Springell K, et al. Quantification of homozygosity in consanguineous individuals with autosomal recessive disease. Am J Hum Genet 2006;78:889–896.
Hildebrandt F, Heeringa SF, Rüschendorf F, et al. A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS Genet 2009;5:e1000353.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Wierenga, K., Jiang, Z., Yang, A. et al. A clinical evaluation tool for SNP arrays, especially for autosomal recessive conditions in offspring of consanguineous parents. Genet Med 15, 354–360 (2013). https://doi.org/10.1038/gim.2012.136
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2012.136
Keywords
This article is cited by
-
Azoospermia and reciprocal translocations: should whole-exome sequencing be recommended?
Basic and Clinical Andrology (2021)
-
Analytical validation and chromosomal distribution of regions of homozygosity by oligonucleotide array comparative genomic hybridization from normal prenatal and postnatal case series
Molecular Cytogenetics (2019)
-
Isolated proteinuria due to CUBN homozygous mutation – challenging the investigative paradigm
BMC Nephrology (2019)
-
Novel phenotype of 5p13.3-q11.2 duplication resulting from supernumerary marker chromosome 5: implications for management and genetic counseling
Molecular Cytogenetics (2018)
-
Increased homozygosity in the first Hispanic patient with plantar lipomatosis, unusual facies, and developmental delay (Pierpont syndrome): a case report
Journal of Medical Case Reports (2016)
