
Abstract
Purpose: To summarize the evidence regarding screening, diagnosis, and treatment of early-infantile Krabbe disease in consideration of its addition to the core panel for newborn screening as has been done in New York state.
Methods: Systematic review of articles indexed in MEDLINE and Embase published between January 1988 and July 2009. Thirteen articles describing studies related to screening, diagnosis, or treatment were included in this review.
Results: Case series studies suggest that allogeneic hematopoietic stem-cell transplantation soon after the development of signs or symptoms of early-infantile Krabbe disease decreases early-childhood mortality and may improve neurodevelopment. However, limited data suggest there may be loss of motor function among some children who undergo transplantation. No long-term follow-up data are available from these case series. Of the ∼550,000 newborns reported to have been screened in New York, 25 tested positive. None of these were clinically recognized to have Krabbe disease prior these results. Four were considered to be high risk for early-onset Krabbe disease. Two were subsequently diagnosed and underwent stem-cell transplantation, of whom one died from complications. No data are available regarding the impact on families of a positive newborn screen.
Conclusions: Although early treatment with hematopoietic stem-cell transplant seems to alter early-childhood mortality and some of the morbidity associated with early-infantile Krabbe disease, significant gaps in knowledge exist regarding the accuracy of screening, the strategy for establishing diagnosis, the affect of a positive screen on families, the benefits and harms of treatment, and long-term prognosis.
Similar content being viewed by others

Main
Krabbe disease is an autosomal recessive lysosomal storage disorder (LSD) associated with mutations in the galactosylceramidase (GALC) gene.1 The inability to degrade certain galactolipids, found almost exclusively in the myelin sheath, leads to progressive damage in the white matter of the peripheral and central nervous systems.2 Krabbe disease also is referred to as globoid cell leukodystrophy because of its classic histologic finding.2
Krabbe disease has variable ages of onset and progression. Based on these differences, Krabbe disease is categorized into four subtypes: early infantile, late infantile, juvenile, and adult.2,3 Children with early-infantile Krabbe disease present with extreme irritability, spasticity, and developmental delay before 6 months of age and typically die before 2 years of age. Survey data collected from 334 families by the Hunter's Hope Foundation underscore the severity of early-infantile Krabbe disease.4 Commonly reported problems included extreme irritability, stiffness, and poor feeding, and a median survival of 17 months for those who developed symptoms before 6 months of life. Late-infantile Krabbe disease has an onset between 6 months and 3 years with death usually 2–3 years later. Children with juvenile Krabbe disease become symptomatic between 3 and 8 years and may have a longer survival time than those with either forms of infantile Krabbe disease.5 Adult-onset Krabbe disease is even more variable, and affected individuals may have a normal life span. In this study, we focus on early-infantile Krabbe disease, the subtype for which identification in the newborn period is thought to be most likely to improve outcomes and, therefore, could be most appropriate for newborn screening.
Diagnosis of early-infantile Krabbe disease is challenging because of the numerous known mutations in the GALC gene. Furthermore, GALC enzyme activity alone is not predictive of disease course.1,2,6 Only homozygosity for one particular deletion predicts early-infantile Krabbe disease.1 This 30-kb deletion may account for more than one third of mutant alleles.7
Across all subtypes, the average reported birth incidence before systematic case finding through population screening has been reported to be ∼1 per 100,000, with higher rates in specific populations (e.g., the Druze of northern Israel and the Muslim Arabs of villages outside Jerusalem).8 Prior epidemiologic work suggests that >85% of those with Krabbe disease have the early-infantile form.1,7 Allogeneic hematopoietic stem-cell transplant is the only available treatment; no enzymatic replacement for Krabbe disease is available.
Recent technical advances in newborn screening assays for Krabbe disease applied to dried blood spots yield the possibility of population-based screening. In 2006, the state of New York began screening all its newborns for Krabbe disease. At least two other states, Illinois and Missouri, are developing plans to begin newborn screening for Krabbe disease.
The Secretary of Health and Human Services Advisory Committee on Heritable Disorders in Newborns and Children (“Advisory Committee”) is charged with evaluating the best available scientific evidence to make recommendations about which conditions to include in states' newborn screening panels. To assist the Advisory Committee, we conducted a systematic evidence review9 regarding the potential benefits and harms of newborn screening for Krabbe disease.10 This report is a subset of the evidence review presented to the Advisory Committee, focusing specifically on three key issues considered by the Advisory Committee in its decision-making process regarding Krabbe disease.
-
1
Screening: What methods are available for screening using dried blood spots? What is the analytic validity of these methods? What is the clinical validity of these methods? How many potential cases are identified through population-based newborn screening?
-
2
Diagnosis: How is early-infantile Krabbe disease diagnosed among newborns and infants with a positive newborn screen? Can early-infantile Krabbe disease be diagnosed before the development of symptoms? Can early-infantile Krabbe disease be distinguished from other forms?
-
3
Treatment: Does earlier treatment of early-infantile Krabbe disease lead to improved clinical outcomes or quality of life?
METHODS
Data sources
We searched MEDLINE for studies published between January 1988 and July 2009 for all articles with the National Library of Medicine Medical subject heading term “leukodystrophy, globoid cell.” We combined this with keyword searches for “Krabbe disease” and “Krabbe's disease.” To capture articles that were not yet indexed, we also used the same search strategy within the OVID In-Process and Other Non-Indexed Citations Database. The search was limited to English language publications of human studies. The search strategy yielded a total of 330 articles. To assure completeness of our search strategy, we reviewed the bibliography of reviews and the included articles and searched MEDLINE by author of the studies that were included. We also repeated the search in Embase. No additional articles were identified.
Two investigators (A.R.K. and A.A.K) independently reviewed the abstracts of the articles to select those for inclusion in the review. We excluded reviews, editorials, and articles that did not address the key questions, and case series of four or fewer subjects. Disagreements were resolved through discussion, with an emphasis on inclusion of any potentially useful data. After this process, 29 articles were reviewed in detail for the full report to the Advisory Committee. Among these articles, none described an experimental intervention. We identified 1 cohort study, 4 case-control studies, 15 case series, and 9 cross-sectional studies. Of these 29 articles, 13 addressed the key questions for this review.
Data abstraction
Two reviewers (A.R.K. and A.A.K) abstracted all articles. Elements of the data abstraction included study location; study design; study inclusion and exclusion criteria; intervention, if any; sample size, age, and gender distribution; and outcome measures. A 20% subset of articles was abstracted by both reviewers to check for interobserver agreement. There were no significant differences in data abstraction between the reviewers.
RESULTS
Screening for early-infantile Krabbe disease
One study from 2004 evaluated the use of several protein and metabolic markers in dried blood spots to identify LSDs.11 Although markers could be used to identify some LSDs (e.g., Fabry disease and Tay-Sachs disease), Krabbe disease could not be detected. Two other reports from 2004 described the successful measurement of GALC enzyme activity in dried blood spots using tandem mass spectrometry (MS/MS).12,13 One of these demonstrated a multiplex system that also included screening for other LSDs.13 Unlike using MS/MS to screen for metabolic disorders by direct measurement of a marker (e.g., screening for medium chain acyl CoA dehydrogenase deficiency), the use of MS/MS to screen for LSDs (e.g., Krabbe disease) involves indirect measurement of the amount of enzyme by measuring a marker that is first generated in an independent enzymatic reaction. A report from 2008 documented a modified approach that may be somewhat easier for high-throughput use.14 New York state uses a further modified strategy to enhance throughput. A report from 200915 described the development of the laboratory approach used in the New York program, which also involves use of a GALC enzyme assay followed by MS/MS. Cutoff values for screening were based on the percentage daily mean activity (%DMA) of all samples processed on that day, which adjusts in part for variability in the test and testing conditions. Cutoffs were set to ensure high sensitivity for Krabbe disease.
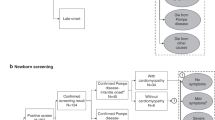
A report from 2009 summarized the New York State screening experience through June 2008.16 In Figure 1, we present a flow diagram describing the process from screening through diagnostic confirmation. This section focuses on screening through laboratory reporting of a positive or negative screen. Any dried blood spot with GALC enzyme activity ≤20%DMA is retested in duplicate. If the average of three samples from the same dried blood spot is ≤8%DMA, the sample undergoes full sequencing DNA testing but regardless of the result is considered to be a positive screen. If the average of three samples from the same dried blood spot is >8%DMA but ≤12%DMA, the sample undergoes full sequencing DNA testing. If there is at least one mutation in the GALC gene, the result is considered to be a positive screen. If no mutations are identified, the result is considered to be a negative screen. If the average of three samples from the same dried blood spot is >12%DMA, the result is considered to be a negative screen. By the end of June 2008, ∼550,000 newborns had been screened,16 of whom 25 had a positive screen reported by the laboratory (e.g., ∼1 per 22,000 screened newborns had a positive screen that was reported by the laboratory). No published data are available regarding the number of samples that required repeat testing or DNA testing or the pathway that was taken to a positive screen.

Diagnosis among those with a positive screen
Establishing the diagnosis of Krabbe disease in newborns is challenging because other than one specific mutation, there is poor genotype-phenotype correlation. Furthermore, GALC enzyme activity poorly predicts disease course. The New York State screening program has developed a risk-based follow-up system14 to identify infants and children for referral for allogeneic hematopoietic stem-cell transplant. Those with a positive screening result are stratified into three risk categories (high, medium, and low) for the development of infantile Krabbe disease based on GALC enzyme activity, which then is used to plan follow-up. For example, in the first year of life, those in the high-risk category are encouraged to have at least monthly neurologic examinations, quarterly neurodiagnostic studies (including magnetic resonance imaging [MRI], cerebrospinal fluid testing, brainstem auditory-evoked response, visual-evoked response, and nerve conduction studies), and annual neuropsychologic testing. In the first year of life, those in the moderate risk category are planned for at least quarterly neurologic examinations and annual neurodiagnostic studies and neuropsychologic testing. In the first year of life, those in the low-risk group are expected to have semiannual neurologic testing and annual neurodiagnostic studies and neuropsychologic testing. A point system is used to determine which children should be referred for allogeneic hematopoietic stem-cell transplant. The point system includes results of quantitative testing (e.g., protein in cerebrospinal fluid, nerve conduction, brain-stem auditory-evoked response, visual-evoked response, and homozygosity of the 30-kb GALC deletion) and findings from the neurologic examination. The system is weighted toward results of quantitative testing, with only 2 of 13 points assigned to neurologic examination findings. In contrast, homozygosity of the 30-kb GALC deletion is assigned 4 points. Referral for transplantation is considered for individuals with 4 or more points.
Of the 25 children with a positive screen, two children, both in the high-risk category, were diagnosed with Krabbe disease and referred for transplantation.16 The other two in the high-risk category had not developed symptoms by 8 and 16 months of age. No published data are available regarding the point distribution for these individuals. No information is available regarding the impact of a positive screen on family functioning or psychological status.
In the future, new imaging tests may help with earlier identification of infants who would benefit from transplantation. For example, diffusion tensor imaging of myelination patterns in the motor tracts may play a role in both early diagnosis and monitoring after diagnosis.17
Accuracy of screening
Because of diagnostic challenges, the limited time of follow-up for those who screened positive, and the lack of surveillance data among those who screened negative, it is not possible to calculate the screening test characteristics (i.e., sensitivity, specificity, positive, and negative predictive values). However, among the 25 children who screened positive, two were diagnosed and treated for infantile Krabbe disease, implying a positive predictive value of 8%. The positive predictive value may increase if other children who tested positive are diagnosed.
Benefit of early treatment
Of the two children referred for hematopoietic stem-cell transplantation from the New York State screening program, one died from a complication of transplantation.16 The other child was reported not to have signs or symptoms of Krabbe disease after transplant but was reported to be developmentally delayed. The age of that child was not included in the publication, and no further published information is available about the child's health status. No data are available to determine the degree to which the developmental delay was related to the Krabbe disease or the treatment the child received.
We identified five studies evaluating allogeneic hematopoietic stem-cell transplantation for Krabbe disease.3,18–21 One study specifically addressed both survival and functioning among children with infantile Krabbe disease.3 In this report, 11 children who were diagnosed with Krabbe disease because of having an affected older sibling and who underwent allogeneic hematopoietic stem-cell transplantation in early infancy were compared with 14 children who were diagnosed after developing symptoms and who were treated with transplantation after 4 months of age. No information was published about subject's GALC enzyme activity level before transplant or genotype (e.g., presence of the 30-kb deletion). All 11 children who received early treatment survived for 36 months, the length of follow-up reported in this study. In contrast, only 6 of the 14 children who were symptomatic survived for a median follow-up period of 41 months. Four of these deaths were attributed to progression of the Krabbe disease.
Neurodevelopmental outcomes were available for 10 of the 11 of those who received early treatment and 8 of the 14 children treated after becoming symptomatic.3 Summarizing the differences is difficult because of the small numbers; variable periods of follow-up, ranging from months to years; and changes in functioning over time. Those in the early-treatment group developed cognitive skills at a normal rate, two were below average for adaptive behavior skills, one was below average for receptive language, and two were below average for expressive language. In general, motor function outcomes were more variable, and symptoms could be progressively worse. For example, two children had severe fine motor function delay, four had mild or worse gross motor function delay, and two subjects lost gross motor skills by 3 years of age. Before the stem-cell transplantation, 7 of the 11 who received early treatment had some abnormal patterns seen by brain MRI; however, all 11 had normal brain myelination patterns documented by MRI after transplant. In contrast, none of the children who were treated after becoming symptomatic had improvement in neurodevelopmental outcome.
The other four studies we found did not report new neurodevelopmental outcome data. One study18 used the previously described data3 to develop a staging system to predict the progression of Krabbe disease. Another study19 reported the successful engraftment of three individuals after hematopoietic stem-cell transplantation of three individuals with Krabbe disease. No information was provided about the type of Krabbe disease; however, all received transplant after 6 years of age, making it unlikely that they had early-infantile Krabbe disease. The remaining two reports20,21 focused on improvement in myelination after transplant. One of these20 evaluated myelination through serial brain MRIs of three individuals with early-infantile Krabbe disease transplanted within the first month of life and four individuals with early-infantile Krabbe disease transplanted later in the first year of life. Six of these subjects had 1- and 2-year follow-ups. Overall, there was suggestion by the authors that transplantation led to “amelioration of the dysmyelinating process,” with better improvement in those in the early-treatment group. The remaining study21 demonstrated improvement in peripheral nerve conduction among 12 individuals with Krabbe disease (nine early infantile, two juvenile, and one late onset) after transplantation. The average follow-up among these subjects was 18 months.
DISCUSSION
Early-infantile Krabbe disease is a devastating condition, and without treatment, the prognosis is certain death in early childhood. Early treatment with allogeneic hematopoietic stem-cell transplantation, a difficult and risky procedure, was found to provide net benefits in decreasing early-childhood mortality, and some of the morbidity associated with early-infantile Krabbe disease in young children, at least in the short term. No long-term data are available regarding the benefit of treatment, and concern has been raised about the development of progressive neurologic impairment.3,22 Although screening is technologically possible, establishing the diagnosis of early-infantile Krabbe disease among those with a positive screen is challenging. In 2009, the Advisory Committee chose not to recommend adding Krabbe disease to the core panel of newborn screening tests offered by states because of uncertainties related to population-based screening, the diagnosis of early-infantile Krabbe disease, the impact of a positive screen on families, and treatment outcomes.
Diagnosing affected infants identified by newborn screen from a general population is particularly challenging. Children who have screened positive require repeated and sometimes invasive testing to confirm or rule out a diagnosis. No peer-reviewed published information is available regarding the completeness of this follow-up or the impact of follow-up on families. Currently, only experts in the care of Krabbe disease have been involved with the follow-up care after a positive screen. However, most academic medical centers have little experience in either the diagnosis or the treatment of Krabbe disease. No data are available regarding the ability of those without experience in caring for children with Krabbe disease to effectively administer the follow-up protocol. This increases the risk of either delayed treatment or inappropriate transplantation.
New York has demonstrated that it is feasible to screen for Krabbe disease, and other states are now preparing to add Krabbe disease to their newborn screening panel. States choosing to screen for Krabbe disease will need to assure the availability of resources, including clinical expertise, to provide follow-up care for both diagnosis and treatment. No data are available to guide newborn screening programs in how to provide counseling to families regarding the many uncertainties regarding diagnosis, treatment, and prognosis.
Developing strategies for the early detection and treatment of very rare conditions is challenging. We have identified several critical gaps in knowledge regarding newborn screening for Krabbe disease. The two most compelling gaps are (1) the inability to determine shortly after a positive screen which children would benefit from urgent transplantation and (2) the lack of long-term follow-up data for those children who have received transplants, especially neurodevelopmental outcomes.
As more data become available from New York and the other states that begin screening for Krabbe disease, the process of screening for and diagnosis of early-infantile Krabbe disease may be refined (e.g., new thresholds for a risk stratification, inclusion of new imaging methods for diagnosis, and evidence-based guidelines for follow-up based on risk). Therefore, we advocate that any screening for Krabbe disease be conducted within the framework of a research protocol. Enrollment of those with a positive newborn screen into registries and prospective clinical trials will be essential to addressing critical gaps such as the impact of a positive screen on families, including harms related to the uncertainty of diagnosis and the neurologic outcomes for those who receive early treatment. Until fundamental issues regarding screening, diagnosis, and treatment are resolved, the Advisory Committee would be unlikely to reconsider whether to recommend that all states should screen for Krabbe disease.
References
Wenger DA, Rafi MA, Luzi P, Datto J, Costantino-Ceccarini E . Krabbe disease: genetic aspects and progress toward therapy. Mol Genet Metab 2000; 70: 1–9.
Suzuki K . Globoid cell leukodystrophy (Krabbe's disease): update. J Child Neurol 2003; 18: 595–603.
Escolar ML, Poe MD, Provenzale JM, et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe's disease. N Engl J Med 2005; 352: 2069–2081.
Duffner PK, Jalal K, Carter RL . The Hunter's Hope Krabbe family database. Pediatr Neurol 2009; 40: 13–18.
Loonen MC, Van Diggelen OP, Janse HC, Kleijer WJ, Arts WF . Late-onset globoid cell leucodystrophy (Krabbe's disease). Clinical and genetic delineation of two forms and their relation to the early-infantile form. Neuropediatrics 1985; 16: 137–142.
Wenger DA, Suzuki K, Suzuki Y, Suzuki K, Galactosylceramide lipidosis: globoid cell leukodystrophy (Krabbe disease). In: Scriver CR, Sly WS (eds) The metabolic and molecular basis of inherited disease, 8th ed. New York, McGraw-Hill, 2001; 3669–3694.
Wenger DA Krabbe disease online NIH gene review. Available at: http://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=gene&part=krabbe. Accessed March 15, 2009.
Korn-Lubetzki I, Nevo Y . Infantile Krabbe disease. Arch Neurol 2003; 60: 1643–1644.
Perrin JM, Knapp AA, Browning MF, et al. An evidence development process for newborn screening. Genet Med 2010; 12: 131–134.
Advisory Committee on Heritable Disorders in Newborns and Children: External Evidence Review Workgroup. Krabbe evidence review final draft. Available at: http://www.hrsa.gov/heritabledisorderscommittee/reports/KrabbeEvidenceReviewFinalDraft.pdf. Accessed December 1, 2009.
Meikle PJ, Ranieri E, Simonsen H, et al. Newborn screening for lysosomal storage disorders: clinical evaluation of a two-tier strategy. Pediatrics 2004; 114: 909–916.
Li Y, Brockmann K, Turecek F, Scott CR, Gelb MH . Tandem mass spectrometry for the direct assay of enzymes in dried blood spots: application to newborn screening for Krabbe disease. Clin Chem 2004; 50: 638–640.
Li Y, Scott CR, Chamoles NA, et al. Direct multiplex assay of lysosomal enzymes in dried blood spots for newborn screening. Clin Chem 2004; 50: 1785–1796.
Zhang XK, Elbin CS, Chuang WL, et al. Multiplex enzyme assay screening of dried blood spots for lysosomal storage disorders by using tandem mass spectrometry. Clin Chem 2008; 54: 1725–1728.
Orsini JJ, Morrissey MA, Slavin LN, et al. Implementation of newborn screening for Krabbe disease: population study and cutoff determination. Clin Biochem 2009; 42: 877–884.
Duffner PK, Caggana M, Orsini JJ, et al. Newborn screening for Krabbe disease: the New York state model. Pediatr Neurol 2009; 40: 245–252.
Escolar ML, Poe MD, Smith JK, et al. Diffusion tensor imaging detects abnormalities in the corticospinal tracts of neonates with infantile Krabbe disease. AJNR Am J Neuroradiol 2009; 30: 1017–1021.
Escolar ML, Poe MD, Martin HR, Kurtzberg J . A staging system for infantile Krabbe disease to predict outcome after unrelated umbilical cord blood transplantation. Pediatrics 2006; 118: e879–e889.
Gaipa G, Dassi M, Perseghin P, et al. Allogeneic bone marrow stem cell transplantation following CD34+ immunomagnetic enrichment in patients with inherited metabolic storage diseases. Bone Marrow Transplant 2003; 31: 857–860.
McGraw P, Liang L, Escolar M, Mukundan S, Kurtzberg J, Provenzale JM . Krabbe disease treated with hematopoietic stem cell transplantation: serial assessment of anisotropy measurements–initial experience. Radiology 2005; 236: 221–230.
Siddiqi ZA, Sanders DB, Massey JM . Peripheral neuropathy in Krabbe disease: effect of hematopoietic stem cell transplantation. Neurology 2006; 67: 268–272.
Duffner PK, Caviness VS, Erbe RW, et al. The long-term outcomes of presymptomatic infants transplanted for Krabbe disease: report of the workshop held on July 11 and 12, 2008, Holiday Valley, New York. Genet Med 2009; 11: 450–454.
Acknowledgements
This work was supported by subcontracts to Columbia University, Duke University, and MassGeneral Hospital for Children under prime contract (HHSP23320045014XI) to Altarum Institute, from HHS, HRSA, and Maternal and Child Health Bureau (MCHB).
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure: None of the authors are involved in any research or clinical activity related to Krabbe disease. Other investigators at Alex Kemper's institution (Duke University) are involved with screening and treatment of Krabbe disease. All authors declare no other conflicts of interest.
Rights and permissions
About this article
Cite this article
Kemper, A., Knapp, A., Green, N. et al. Weighing the evidence for newborn screening for early-infantile Krabbe disease. Genet Med 12, 539–543 (2010). https://doi.org/10.1097/GIM.0b013e3181e85721
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1097/GIM.0b013e3181e85721
Keywords
This article is cited by
-
Health utilities and parental quality of life effects for three rare conditions tested in newborns
Journal of Patient-Reported Outcomes (2019)
-
Consensus guidelines for newborn screening, diagnosis and treatment of infantile Krabbe disease
Orphanet Journal of Rare Diseases (2018)
-
New in Newborn Screening
Current Genetic Medicine Reports (2017)
-
Ethical Considerations When Including Lysosomal Storage Disorders in Newborn Screening Programs
Current Genetic Medicine Reports (2015)
-
Lysosomal storage disorders: Present and future
Indian Pediatrics (2015)
