
Abstract
Purpose: We previously reported a deletion of the Galactose-1-Phosphate Uridyl Transferase (GALT) gene. This deletion can cause apparent homozygosity for variants located on the opposite allele, potentially resulting in a discrepancy between the biochemical phenotype and the apparent genotype in an individual. The purpose of this study was to determine the deletion breakpoints, allowing the development of a rapid and reliable molecular test for the mutation.
Methods: A Polymerase Chain Reaction walking strategy was used to map the 5′ and 3′ breakpoints. The junction fragment was amplified and sequenced to precisely characterize the deletion breakpoints.
Results: The deletion has a bipartite structure involving two large segments of the GALT gene, while retaining a short internal segment of the gene. Molecular characterization allowed the development of a deletion specific Polymerase Chain Reaction-based assay. In 25 individuals who had a biochemical carrier galactosemia phenotype, but tested negative for 8 common GALT gene variants, 3 carried this deletion.
Conclusion: This deletion occurs at an appreciable frequency and should be considered when there is a discrepancy between the genotype and biochemical phenotype. Many of the individuals carrying the allele were of Ashkenazi Jewish ancestry suggesting that the deletion may be a common cause of galactosemia in that population.
Similar content being viewed by others

Main
Classic galactosemia refers to severe galactose-1-phosphate uridyltransferase (GALT) deficiency and manifests within the first week of life with poor feeding, jaundice, vomiting, liver dysfunction, increased bleeding tendency and septicemia, leading to death if left untreated.1 The disease can be easily managed by lactose restriction, although some long term complications may not be prevented. Diagnostic work-up for classic galactosemia is often initiated following abnormal newborn screening. Classic galactosemia can be confirmed by extremely low or absent GALT enzymatic activity in the hemolysates; while variant forms or carrier status may require confirmation by further biochemical analysis and/or molecular genotyping of the GALT gene. Our laboratory performs both biochemical phenotyping and molecular genotyping to confirm the diagnosis of galactosemia.
The biochemical analysis consists of assessment of GALT enzyme activity, measurement of galactose-1-phosphate (Gal-1-P; the GALT enzyme substrate) and GALT protein isozyme analysis in red cells. The first tier of molecular analysis consists of genotyping for 9 common GALT variants (Q188R, N314D, L218L, S135L, K285N, L195P, T138M, Y209C, and IVS2-2A>G). These mutations account for the vast majority of galactosemia alleles in various populations.2 The second tier of testing consists of GALT gene sequencing (the exons and the intron/exon junctions) that will detect less common coding and splice junction mutations. These combined biochemical and molecular approaches provide a comprehensive characterization of the patient's biochemical phenotype and genotype to guide nutritional management and genetic counseling.
Our combined approach also enables identification of variants that may otherwise go undetected or lead to erroneous diagnosis. For example, the discrepancy between biochemical phenotype and molecular genotype due to the presence of a deletion on one of the alleles results in apparent homozygosity for the variant on the opposite allele. Previously, a large deletion of about 5 kb in the GALT gene was identified because it resulted in discordance between biochemical phenotype and the apparent genotype.3,4 We have encountered this mutation numerous times during our testing for galactosemia. Here we describe the molecular characterization of this mutation, describe a simple Polymerase Chain Reaction (PCR)-based assay to score for the presence of the deletion, and provide a preliminary report on its frequency in our population of galactosemia patients.
MATERIALS AND METHODS
DNA samples
Patient samples were submitted for both biochemical and molecular diagnostic testing for galactosemia. All patient DNA samples were isolated from whole blood using a salting-out procedure5 or by using the automated Qiagen DNA extraction system.
PCR amplification and DNA sequencing
Segments of the GALT gene and its flanking sequences were amplified using 2.5 units of HotStar Taq (Qiagen) in the presence of 10 mM Tris-HCl (pH 8.3), 1.5 mM MgCl2, 50 mM KCl, 0.125 mM dNTPs, 0.2 mM each of forward and reverse primers. The primer sequences and locations relative to the GALT gene are available upon request. The PCR parameters used for all reactions were: 95°C for 15 minutes [(95°C for 1 minute, 59°C for 1 minute, 72°C for 1 minute) repeated 40 times], 72°C for 7 minutes. For amplification and sequencing of the junction fragment the following primers were used: -1164F 5′-AGTACCAGGGGAGGAATTAATTTGAATTTT-3′; +4795R 5′-GTGTGATTTCCCCACCCACAGG-3′.
A 5-μl aliquot of the PCR sample amplified with the primers -1164F and 4795R was treated with ExoSAP-IT (USB) at 37°C for 60 minutes followed by heat inactivation at 80°C for 15 minutes. The ExoSAP treated product was sequenced in forward and reverse directions using the −1164F and 4795R primers. The sequence data were analyzed using the SeqScape software package (ABI). To score for the presence of a nondeleted GALT allele, a third primer (+4116F primer-5′-CTTGTGT-CTTGGTTGTGGCTGGAGG-3′) was added to the PCR reaction.
Biochemical analysis
The GALT enzymatic assay in erythrocyte extracts used modifications of previously described methods for the conversion of 14C-galactose-1-phosphate to UDP-galactose.6–8 Isozymes of GALT were defined using isoelectric focusing.9
Southern analysis
Genomic DNA was digested with EcoRI, Bam HI, Hind III, followed by electrophoresis on a 0.8% agarose gel, transferred to a nylon membrane, and hybridized with radioactively labeled human full-length GALT cDNA probes.
RESULTS
The deletion can be detected by Southern analysis using a GALT cDNA probe, as shown in Figure 1. This deletion is predicted to be 5.5 kb in size, since it results in the reduction of an ∼10.5 kb EcoRI band to approximately 5 kb. Importantly, deletion of GALT on one allele would result in a heterozygous mutation on the opposite allele scoring as homozygous during molecular analysis, resulting in a potential discrepancy between the genotype and biochemical phenotype.3,4 However, an individual carrying a null galactosemia allele (e.g., Q188R) on one GALT allele and the deletion on the other GALT allele would score as a homozygote for the null mutation and would not cause a discrepancy between the apparent genotype and the biochemical phenotype.

Southern analysis of the GALT deletion. Genomic DNA from an individual who was homozygous for the deletion, and from a wild-type individual, were digested with EcoR1, BamHI or Hind III. After transfer, the membrane was probed with a GALT cDNA probe. The marker sizes are indicated on the left side of the panel.
Identification of deletion breakpoints by PCR walking
In order to develop a rapid and reliable molecular test for the deletion, we need to further characterize this deletion. Previously, we identified a patient by Southern blot analysis who was homozygous for the GALT deletion3 (DEL/DEL in Fig. 1). Using this patient's DNA as a template, a PCR walking strategy was employed to map both the 5′ and 3′ deletion breakpoints. Using genomic DNA sequence from a segment of chromosome 9 (NT_008413), we designed PCR primer pairs targeting DNA sequences flanking the 5′ end of the GALT gene. We started by targeting a region that is located 10.5 kb upstream of the GALT transcription start site. We designed primer pairs that generated 500 bp products that were spaced approximately every 2,000 bp apart, at 10.5 kb, 8.5 kb, 6.5 kb, 4.5kb, 2.5 kb, and 0.5 kb upstream of the transcription initiation site. In DNA from the GALT deletion homozygote, all of these primer pairs generated PCR products, except the primer pair targeting sequence 500 bp upstream of the GALT gene (−674F to −174R). This indicated that the 5′ breakpoint was located in a region between −2,174 bp and −174 bp of the GALT transcription start. By using additional primer pairs, we were then able to refine further the 5′ breakpoint of the deletion to a region between −2,174 and −1,001 bp upstream of the GALT gene. Using a similar approach, we mapped the 3′ breakpoint of the deletion to a segment located between 300 bp and 2,800 bp downstream of the transcription termination signal of the GALT gene.
Using a forward primer that anneals 1,164 bp upstream of the GALT transcription start site (−1164F) and a reverse primer located 865 bp downstream of the polyadenylation signal (+4795R) we were able to amplify an 500-bp DNA segment that spans the breakpoints of the GALT deletion. Sequencing of this amplification product revealed a complex deletion in which two segments of the GALT gene were absent; a 3,163-bp segment of DNA containing the 5′ end of the gene, including the gene promoter, and a 2,295-bp segment from the 3′ end of the GALT gene. An internal 117-bp segment of the GALT gene is retained that contains portions of exon 8 and intron 8 (Fig. 2). In addition, 12 bp of sequence of unknown origin was found immediately downstream of the exon8/intron 8 segment. The restriction digestion products predicted from the deleted GALT allele were entirely consistent with the DEL/DEL patient Southern results (Figs. 1 and 3).

Sequence of the junction fragment. Electropherogram of GALT DNA sequence derived from amplification of the junction fragment. The positions of the 5′ and 3′ flanking sequences and the segment of the GALT gene consisting of exon 8 and intron 8 are indicated underneath the electropherogram. In addition, the 12 bp of sequence of unknown origin is also indicated with the question mark.

Predicted restriction digestion patterns are consistent with observed restriction digest patterns from an individual homozygous for the deletion allele. Schematic illustrating the restriction digestion patterns predicted for EcoRI, Hind III and BamHI with both a wild-type allele and the GALT deletion allele. The restrictions patterns predicted are consistent with the restriction digestion patterns observed in Figure 1.
Discordance between GALT biochemical phenotype and genotype
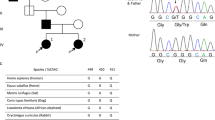
Two cases of discordance between the biochemical phenotype and the GALT genotype are illustrated in Figure 4. Alleles of the GALT gene can be defined by biochemical testing either as G (galactosemia), D (Duarte variant), LA (Los Angeles variant) or N (normal). G alleles have zero or very low activity GALT enzymatic activity, D alleles have approximately 50% activity, LA alleles have normal or slightly elevated GALT activity and N alleles have 100% activity. The proband in Figure 4A tested as a homozygote for the N314D variant, a genotype associated with the Duarte allele. This patient would be predicted to have approximately half the normal GALT enzyme activity. However, the patient's GALT activity was 35% of the normal control – a value more consistent with a D/G rather than a D/D phenotype. Isoelectric focusing analysis also yielded a result more consistent with a D/G rather than D/D phenotype (data not shown).

Testing for GALT deletion by amplification of the junction fragment. Pedigrees of families segregating the deletion allele and the results of the molecular and biochemical tests for each individual. The presence of the deletion was scored for by PCR amplification of the junction fragment.
We tested the parents by molecular and biochemical analyses to resolve the discordance between the proband's genotype and biochemical phenotype. Molecular analysis indicated that the father was homozygous for the LA variant (N314D-L218L), which is associated with mildly elevated rather than impaired activity when compared to wild type alleles. However, the father's GALT enzymatic activity was 51% of wild type, indicating the presence of a G allele, another discrepancy between genotype and biochemical phenotype. The mother of the proband had a genotype of N314D/Neg, consistent with her biochemical phenotype of D/N.
The proband in the second family (Fig. 4B) also appeared to be homozygous for N314D. Again, this genotype would predict a biochemical phenotype of D/D. Like the first proband, this patient had a D/G biochemical phenotype (data not shown). Analysis of the parents indicated that the father had a genotype of N314D/Neg, consistent with his biochemical phenotype of D/N. Molecular analysis of the mother failed to identify a mutation in the panel of nine common variants. The mother's biochemical phenotype was G/N, indicating that she was a carrier of a rare null GALT mutation.
The presence of the GALT deletion allele in each of these families could potentially resolve the discrepancies observed between the genotypes and biochemical phenotypes. To determine whether these families did carry the GALT deletion allele, we amplified the expected junction fragment. As shown in Figure 4A, the proband and the father in the first family carry the GALT deletion allele. Unfortunately, the amount of DNA from the mother was insufficient for analysis. Since the proband carried the deletion, his genotype is corrected to N314D/ DEL, consistent with his biochemical phenotype of D/G. Since the father also carried the deletion his genotype would be corrected to N314D-L218L/DEL, also consistent with his biochemical phenotype. In family 2 (Fig. 4B), the proband and the mother carry the deletion. The probands genotype would be corrected to N314D/ DEL and the mother's genotype would be DEL/NEG. The father in this family, who does not carry the deletion, serves as an important negative control illustrating the specificity of the GALT deletion PCR product.
Development and validation of a PCR assay for the GALT deletion
To determine if the breakpoints were the same for all patients previously determined to carry the GALT deletion by Southern analysis, we amplified and sequenced the junction fragment in all available samples. In all cases tested, the breakpoints were identical, suggesting a common origin for the deletion (data not shown). In developing a diagnostic test for this deletion we added a third primer to the PCR reaction, which hybridizes to the deleted segment, to score for the presence of a full-length allele (Fig. 5). In this PCR reaction, the deletion yielded an 500-bp product whereas a nondeleted allele yields a 680-bp product. A representative gel showing a normal, a deletion carrier, and a patient homozygous for the deletion is shown in Figure 5.

PCR assay for the GALT deletion. Schematic diagram illustrating the location of the three primers used in the assay. The sizes of the PCR products for the nondeleted allele and the deleted allele are indicated. A representative gel testing a normal, a deletion carrier and homozygous deletion affected individual.
Detection rate of the GALT deletion in the galactosemia population
We have encountered this deletion of the GALT gene numerous times during our galactosemia testing. To assess the frequency of the deletion, we screened DNA samples from patients referred over a 12-month period (331 total patients) whose genotype and biochemical phenotype were suggestive of the deletion. Like the probands in Figure 4, there were four patients who were homozygous for N314D but had a biochemical phenotype of D/G. One of the four had the deletion. In addition, we screened 21 carriers of an unknown G allele (negative for one of the G allele mutations in our panel). We found that two were positive for the deletion. We also identified one patient whose GALT DNA was unable to be amplified for genotyping. This individual was found to be homozygous for the deletion. Thus, over a 12 month period of galactosemia testing we identified five deletion alleles. This frequency is similar, or even greater than, that of GALT mutations such as L195P, Y209C and the IVS2-2A>G, which were found at the same or even lower frequencies over the same time period.
DISCUSSION
Simultaneous assessment of the GALT genotype and biochemical phenotype in patients being tested for galactosemia led to the identification of this deletion mutation. This combined approach also enables identification of variants that may otherwise go undetected or lead to erroneous diagnoses. Preliminary screening of our collection of galactosemia patient samples with discrepancies between apparent genotype and biochemical phenotype indicates that this deletion is a relatively common GALT mutation.
The deletion described by Berry et al. is different than the one we describe here.3 We expect that the primer pairs Af -Ar and Df -Dr listed in Berry et al., which yield PCR products in their deletion homozygous patient, would fail to produce PCR products in the homozygous deletion that we characterized. Though the Af primer binding site is located upstream of the 5′ deletion boundary, the Ar primer binding site is located downstream of the breakpoint and therefore would not yield a PCR product. The Df and Dr primer binding sites are both located within the deletion and would not yield a PCR product.
The deletion described here includes the GALT promoter. A fusion transcript containing sequence from both the GALT gene and the downstream Interleukin-11 Receptor α-Chain gene (IL-11Rα) has been described.10 This fusion transcript initiates from the GALT promoter, proceeds through the GALT gene, and continues into the downstream IL-11Rα gene. Portions of the GALT sequence are spliced out, but the entire IL-11Rα protein is retained in the fusion protein. The fusion protein produced from this transcript has no known function, and did not possess any GALT activity when over expressed in tissue culture cells. The deletion we describe would eliminate the promoter of the GALT gene resulting in loss of both transcripts. Therefore, homozygous deletion patients would lack both the GALT specific transcript and the GALT/IL-11Rα fusion transcript.
The deletion is unusual in that it is not a contiguous deletion, but actually consists of two deleted segments that flank a 117-bp segment of the GALT gene containing portions of exon 8 and intron 8. In addition, there is an insertion of 12 bp of sequence of unknown origin. In this allele, there are a total of 5,458 bp of GALT sequence removed. These findings are consistent with Southern analysis using EcoR1 that demonstrates the loss of 5.5kb of GALT DNA. Furthermore, the sizes of the restriction fragments predicted for a DEL/DEL homozygote after BamHI and HindIII digestion are observed in the Southern analysis (Figs. 1 and 3). Retention of a segment of exon 8 explains how the deletion is able to be detected by Southern analysis using a GALT cDNA probe. To our knowledge, this is the only deletion of this type that has been described and there is no obvious mutational mechanism to explain how this deletion occurred.
The complexity of the deletion suggests that it occurred as a single event and has been maintained in the population through a founder effect. We originally noted this deletion in three unrelated Ashkenazi Jewish patients.3 Ethnic information on the other patients identified in this study was not always available and we have identified individuals carrying the deletion with Hispanic surnames. We are therefore unable to comment on whether this deletion is seen exclusively in Ashkenazi Jewish individuals. A more thorough study is ongoing to estimate the frequency and ethnic distribution of this mutation in galactosemia patients.
References
Elsas LJ, Lai K . The molecular biology of galactosemia. Gen Med 1998; 1: 40–48.
Tyfield L, Reichardt J, Fridovich-Keil J, Croke DT, et al. Classical galactosemia and mutations at the galactose-1-phosphate uridyl transferase (GALT) gene. Hum Mutat 1999; 13: 417–430.
Berry GT, Leslie N, Reynolds R, Yager CT, et al. Evidence for alternate galactose oxidation in a patient with deletion of the galactose-1-phosphate uridyltransferse gene. Mol Genet Metab 2001; 72: 316–321.
Barbouth D, Slepak T, Klapper H, Lai K, et al. Prevention of a molecular misdiagnosis in galactosemia. Genet Med 2006; 8: 178–182.
Miller SA, Dykes DD, Polesky HF . A simple salting out procedure for extracting DNA from human nucleated cells. Nuc Acids Res 1988; 16: 1215.
Mellman WJ, Tedesco TA . Am improved assay of erythrocyte galactose-1-phosphate uridyl transfease: stabilization of the enzyme by a thiol protective reagent. J Lab Clin Med 1965; 66: 980–986.
Ng WG, Bergren WR, Donnel GN . An improved procedure for the assay of hemolysate galactose-1-phosphate. Clin Chim Acta 1967; 15: 489–492.
Lee JES, Ng WG . Semi-micro techniques for the genotyping of galactokinase and galactose-1-phosphate uridyl transferase. Clin Chem Acta 1982; 124: 351–356.
Elsas LJ, Dembure PP, Langley S, Paulk EM, et al. A common mutation associated with the Duarte galactosemia allele. Am J Hum Genet 1994; 54: 1030–1036.
Magrangeas F, Pitiot G, Dubois S, Bragado-Nilsson E, et al. Cotranscription and intergenic splicing of human galactose-1-phosphate uridylyltransferase and interleukin-11 receptor alpha-chain genes generate a fusion mRNA in normal cells. Implication for the production of multidomain proteins during evolution. J Biol Chem 1998; 273: 16005–16010.
Acknowledgements
We would like to thank Dr. Lora Bean and Dr. Judy Fridovich-Kiel for careful review of the manuscript.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Coffee, B., Hjelm, L., Delorenzo, A. et al. Characterization of an unusual deletion of the galactose-1-phosphate uridyl transferase (GALT) gene. Genet Med 8, 635–640 (2006). https://doi.org/10.1097/01.gim.0000237720.78475.fb
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1097/01.gim.0000237720.78475.fb
Keywords
This article is cited by
-
Biochemical changes and clinical outcomes in 34 patients with classic galactosemia
Journal of Inherited Metabolic Disease (2018)
-
Expanding preconception carrier screening for the Jewish population using high throughput microfluidics technology and next generation sequencing
BMC Medical Genomics (2016)
-
Novel GALTvariations and mutation spectrum in the Korean population with decreased galactose-1-phosphate uridyltransferase activity
BMC Medical Genetics (2014)
