
Abstract
Nitric oxide (NO) produced by endothelial NO synthase (eNOS) plays an important role in vascular functions, including vasorelaxation. We here investigated the pharmacological effect of the natural product syringaresinol on vascular relaxation and eNOS-mediated NO production as well as its underlying biochemical mechanism in endothelial cells. Treatment of aortic rings from wild type, but not eNOS-/- mice, with syringaresinol induced endothelium-dependent relaxation, which was abolished by addition of the NOS inhibitor NG-monomethyl-L-arginine. Treatment of human endothelial cells and mouse aortic rings with syringaresinol increased NO production, which was correlated with eNOS phosphorylation via the activation of Akt and AMP kinase (AMPK) as well as elevation of intracellular Ca2+ levels. A phospholipase C (PLC) inhibitor blocked the increases in intracellular Ca2+ levels, AMPK-dependent eNOS phosphorylation, and NO production, but not Akt activation, in syringaresinol-treated endothelial cells. Syringaresinol-induced AMPK activation was inhibited by co-treatment with PLC inhibitor, Ca2+ chelator, calmodulin antagonist, and CaMKKβ siRNA. This compound also increased eNOS dimerization, which was inhibited by a PLC inhibitor and a Ca2+-chelator. The chemicals that inhibit eNOS phosphorylation and dimerization attenuated vasorelaxation and cGMP production. These results suggest that syringaresinol induces vasorelaxation by enhancing NO production in endothelial cells via two distinct mechanisms, phosphatidylinositol 3-kinase/Akt- and PLC/Ca2+/CaMKKβ-dependent eNOS phosphorylation and Ca2+-dependent eNOS dimerization.
Similar content being viewed by others


Introduction
Nitric oxide (NO) is produced from L-arginine by the reaction of three isoforms of NO synthases, including the constitutive neuronal (nNOS or NOS1) and endothelial (eNOS or NOS3) isoforms and the inducible isoform (iNOS or NOS2) (Albrecht et al., 2003). The catalytic activity of the two constitutive enzymes is generally regulated by intracellular Ca2+ levels. Both enzymes generate small quantities of NO sufficient for activating soluble guanylyl cyclase (sGC) to generate cGMP, cGMP plays a role of cellular signaling mediators, such as a neurotransmitter and vasodilator, in neuronal and endothelial cells under most circumstances (Hobbs et al., 1999). A third isoform, iNOS, typically expressed after exposure of immune cells and hepatocytes to inflammatory stimuli (e.g. cytokines and microbial products), produces large quantities of NO eliciting cytotoxicity against tumor cells and pathogens (Hobbs et al., 1999). Therefore, the level of NO production and the functional role of NO can vary among cell types.
NO generated by eNOS in endothelial cells plays an important role in vasorelaxation, inhibition of platelet aggregation, endothelial cell survival, and angiogenesis, resulting in protection of the vasculature against various pathological conditions (Dudzinski and Michel, 2007; Namkoong et al., 2008). As such endothelial cells play an important role in regulating vascular functions via eNOS-dependent NO production. In fact, endothelial dysfunction contributes to the pathogenesis and progression of vascular disorders due to reduced synthesis of NO from eNOS and/or increased NO scavenging by superoxide anion (Beckman et al., 1990; Cai and Harrison, 2000). In addition, mice deficient in eNOS have been shown to develop vascular diseases, including hypertension and vascular inflammation (Mashimo and Goyal, 1999). Thus, chemical or biological molecules that regulate eNOS activity can be used as a potential therapeutic drug for vascular diseases by increasing NO production and bioavailability.
The catalytic activity of eNOS is associated with its homodimerization and/or phosphorylation of serine 1177 by increasing intracellular Ca2+ levels and activating the phosphatidylinositol 3-kinase (PI3K)/Akt pathway (Fulton et al., 1999). Shear stress and some growth factors, including vascular endothelial growth factor, promote eNOS phosphorylation and NO production, resulting in an increase in vasodilation (Scotland et al., 2002; Hambrecht et al., 2003). Estrogen can stimulate eNOS activity in pulmonary artery endothelial cells via the elevation of intracellular Ca2+ level, suggesting that estrogen may be involved in NO-mediated pulmonary vasodilation at birth (Lantin-Hermoso et al., 1997). Recent studies revealed additional modes of eNOS activity regulation involving its protein/protein interactions and subcellular localization (Fulton et al., 2001; Albrecht et al., 2003).
Lignan compounds are considered as the representatives of the phytoestrogens, a group of compounds that have shown to act as modulator of mammalian hormonal system (Adlercreutz, 1995). The lignan compound syringaresinol has been shown to inhibit inflammation in lipopolysaccharide-treated macriohages (Jung et al., 2003), gastric ulcer (Fujikawa et al., 1996), and oxidative injury of endothelial cells (Liu et al., 2009). Our previous results showed that another lignan compound sesamine promoted eNOS phosphorylation and NO production, which plays an important role in endothelial cell survival and angiogenesis (Chung et al., 2010), indicating that lignan compounds can increase NO production via eNOS phosphorylation. These evidences suggest that syringaresinol can potentially cause vascular relaxation by elevating NO production in endothelial cells; however, this possibility has not been investigated. Therefore, we examined the pharmacological effect of syringaresinol on vasorelaxation and eNOS-dependent NO production and its underlying mechanism upon post-translation activation of eNOS.
We found that syringaresinol elicited vascular relaxation in aortic rings pre-contracted with phenylephrine (PhE), which was abolished by NOS inhibitor. Syringaresinol also increased in vitro and ex vivo NO production by activating two different signal pathways, Akt/eNOS- and phospholipase C (PLC)/Ca2+/CaMKKβ-dependent eNOS phosphorylation and Ca2+-dependent eNOS dimerization. Altogether, our findings demonstrate that syringaresinol promotes vascular relaxation by increasing endothelial NO production through phosphorylation and dimerization of eNOS.
Results
Syringaresinol induces vascular relaxation by endothelium-dependent NO production
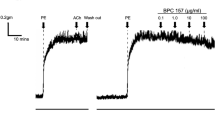
To examine whether syringaresinol regulates vascular relaxation, endothelium-intact and -denuded aortic rings pre-contracted with PhE were treated with the indicated concentrations of syringaresinol and the relaxation was determined. Treatment with syringaresinol caused relaxation in endothelium-intact aortic rings in a concentration-dependent manner, and endothelium denudation completely abolished the syringaresinol-induced vasorelaxation (Figure 1A). Treatment with syringaresinol promoted relaxation in endothelium-intact aortic rings in a time-dependent manner, and this relaxation was significantly inhibited by the NOS inhibitor NG-monomethyl-L-arginine (L-NMA) (Figure 1B). However, we did not find any cytotoxicity in aortic rings treated with syringaresinol in the presence of L-NMA for 40 min, as determined by confirming their normal relaxant responses to the exogenous NO donor sodium nitroprusside (SNP, 0.1 µM) (data not shown). These data indicate that syringaresinol elicits vasorelaxation by regulating NO production in endothelium. Since NO produced by endothelium diffuses to smooth muscle cells and promotes vasorelaxation by producing cGMP via the activation of sGC (Hobbs et al., 1999), we determined cGMP levels in aortic rings treated with syringaresinol. Syringaresinol increased cGMP levels, which was completely blocked by L-NMA (Figure 1C). Since three isotypes of NOS have been identified, we next examined the regulatory effect of syringaresinol on vasorelaxation in aortic rings isolated from knockout mouse of each NOS isotype (Figure 1D). Syringaresinol exerted vasorelaxation in aortic rings from nNOS-/- and iNOS-/- mice, but not in those from eNOS-/- mice, indicating that syringaresinol increases eNOS-mediated NO production. Similar effects were obtained in syringaresinol-induced cGMP production (Figure 1E). These findings indicate that syringaresinol induces vascular relaxation by activation of the eNOS/NO/cGMP signal pathway.

Syringaresinol causes relaxation in aortic rings pre-contracted with PhE. (A) Syringaresinol (SY)-induced relaxation was determined in endothelium-intact (closed circle) and -denuded (open circle) aortic rings pre-contracted with PhE. Data are the mean ± SD (n ≥ 8). **P < 0.01 versus endothelium-denuded aortic rings. (B) Syringaresinol-induced relaxation was determined in endothelium-intact (closed circle and rectangle) and -denuded (open circle) aortic rings in the presence (rectangle) or absence (circles) of 1 mM L-NMA for 40 min. Data are the mean ± SD (n ≥ 8). *P < 0.05 and **P < 0.01 versus endothelium-denuded aortic rings. #P < 0.01 versus endothelium-intact aortic rings treated with SY anole. (C) The level of cGMP was determined in the tissue homogenates from endothelium-intact aortic rings, which were incubated with 30 µM syringaresinol in the presence or absence of 1 mM L-NMA for 40 min. Data are the mean ± SD (n ≥ 5). **P < 0.01 versus untreated control. #P < 0.01 versus endothelium-intact aortic rings treated with SY. (D) Vascular relaxation was determined in NOS-deficient aortic rings treated with 30 µM syringaresinol for 40 min following pre-contraction with PhE. Data shown are the mean ± SD (n ≥ 8). **P < 0.01 versus control. (E) Aortic rings were treated with 30 µM syringaresinol and cGMP level was determined as shown in (C). Data shown are the mean ± SD (n ≥ 5). **P < 0.01 versus untreated control.
Syringaresinol stimulates NO production in human umbilical vein endothelial cells (HUVECs) and mouse carotid artery
When HUVECs were incubated with 30 µM syringaresinol, NO production was significantly increased in vitro, as determined by the intensity of fluorescent DAF-FM (Figure 2A). The level of NO produced by 30 µM of syringaresinol was relatively comparable to that of vascular endothelial growth factor (VEGF, 20 ng/ml), a well-known growth factor that stimulates NO production in endothelial cells (Figure 2A). The syringaresinol-induced NO production was effectively inhibited to basal levels by the addition of the NOS inhibitor L-NMA (Figure 2A), indicating that NO production was due to eNOS activation. However, syringaresinol exhibited no cytotoxicity in the same experimental condition as determined by crystal violet staining (data not shown). We further determined whether syringaresinol increases NO production in an ex vivo vessel model using isolated mouse aortas. Syringaresinol-treated mouse aortic rings significantly increased NO production when compared with control, and this increase was also inhibited to basal levels by the addition of L-NMA (Figure 2B).

Syringaresinol stimulates in vivo and ex vivo NO production. (A) HUVECs were incubated with or without VEGF, syringaresinol, and L-NMA for 1 h. The intracellular levels of NO were determined by confocal laser microscopy using DAF-FM diacetate. (B) Mouse aortic rings were incubated with or without syringaresinol and L-NMA, and the NO level was determined by real-time fluorescence microscopy using DAF-FM diacetate. Data shown are the mean ± SD (n ≥ 5). *P < 0.05 and **P < 0.01 versus untreated control.
Syringaresinol increases NO production by activating PI3K/Akt- and AMPK-dependent eNOS phosphorylation
Previous studies have demonstrated that specific protein kinases, Akt and AMPK, are involved in eNOS phosphorylation and NO production (Dimmeler et al., 2009; Fisslthaler and Fleming, 2009). We examined the effect of syringaresinol on activation of these kinases and phosphorylation of eNOS. Syringaresinol treatment resulted in significant increases in phosphorylation-dependent activation of Akt and AMPK in dose-dependent manners, leading to eNOS phosphorylation in HUVECs (Figure 3A). To investigate the functional involvement of Akt and AMPK-dependent pathways in syringaresinol-induced eNOS phosphorylation, we examined eNOS phosphorylation in HUVECs co-treated with syringaresinol and Wortmannin (upstream inhibitor of Akt) or Compound C (AMPK inhibitor). Syringaresinol-induced phosphorylation of eNOS was significantly inhibited by Wortmannin and partially reduced by Compound C (Figure 3B). To further examine a role of AMPK in eNOS phosphorylation, eNOS phosphorylation was examined in HUVECs treated with the AMPK activator aminoimidazole carboxamide ribonucleotide (AICAR) in the presence or absence of Compound C (Figure 3C). Treatment with AICAR or syringaresinol resulted in increases in AMPK and eNOS phosphorylation, which were decreased by Compound C. We next examined the inhibitory effects of Wortmannin and Compound C on syringaresinol-induced NO production in vitro and ex vivo by confocal microscopy. Syringaresinol-induced increases in NO production were effectively inhibited by Wortmannin and partially reduced by Compound C, in both HUVECs and mouse aortic rings (Figures 3D and 3E). These results suggest that syringaresinol increased NO production via phosphorylation-dependent activation of eNOS by activating both Akt and AMPK.

Syringaresinol increases NO production by activating PI3K/Akt- and AMPK-dependent eNOS phosphorylation. (A) HUVECs were treated with the indicated concentrations of syringaresinol for 30 min, and the levels of phosphorylated proteins were determined by Western blot analysis. (B) The cells were treated with syringaresinol for 30 min following pretreatment with Wortmannin (Wort, 100 nM) and Compound C (Comp C, 10 µM) for 30 min. The level of p-eNOS was determined by Western blot analysis (C) HUVECs were treated with AICAR (1 mM) or syringaresinol (30 µM) for 30 min following pretreatment with Compound C (Comp C, 10 µM) for 30 min. The levels of p-AMPK and p-eNOS were determined by Western blot analysis. (D) HUVECs were treated with or without syringaresinol, Wortmannin, and Compound C. The levels of NO production were determined by the same methods as described in Figure 2. (E) Mouse aortic rings treated with the same as described in (D), and the levels of ex vivo NO production were determined by the same method as described in Figure 2. Data shown in (D) and (E) are the mean ± SD (n ≥ 4). *P < 0.05 and **P < 0.01 versus syringaresinol alone.
Syringaresinol elevates NO production via PLC/Ca2+-dependent eNOS phosphorylation
Mobilization of intracellular Ca2+ levels is a critical event in eNOS-mediated NO production, a calmodulin (CaM) dependent process, which can be associated with the CaMKKβ pathway (Ramirez-Sanchez et al., 2010) and subsequently conformational change of the enzyme (Dudzinski and Michel, 2007). We next determined whether syringaresinol regulates intracellular Ca2+ levels in HUVECs. Treatment with syringaresinol increased intracellular Ca2+ mobilization compared with control, and this increase was inhibited to basal levels by the addition of the Ca2+ chelator BAPTA-AM (Figure 4A). To confirm the functional involvement of Ca2+ mobilization in syringaresinol-mediated activation of Akt and AMPK, which are responsible for eNOS phosphorylation, we examined the regulatory effect of BAPTA-AM on Akt- and AMPK-dependent activation of eNOS in endothelial cells treated with syringaresinol. Treatment with BAPTA-AM inhibited syringaresinol-induced phosphorylation of AMPK and eNOS, without affecting Akt phosphorylation (Figure 4B), indicating that intracellular Ca2+ mobilization is critically involved in syringaresinol-induced eNOS phosphorylation and NO production in HUVECs. Since VEGF has been shown to phosphorylate eNOS through Ca2+/CaM-dependent kinase and PKC, which are activated by intracellular Ca2+ release and diacyl glycerol production upon PLC activation (Wu et al., 1999), we examined the involvement of these signal mediators in syringaresinol-mediated eNOS activation. Treatment of HUVECs with the PLC inhibitor U73122 and the CaM antagonist W-7 abolished the effect of syringaresinol on AMPK and eNOS phosphorylation (Figure 4C), whereas the PKC inhibitors Genistein and GF109203X had no effect (Figure 4D). We next examined the possible involvement of CaMKKβ in syringaresinol-stimulated eNOS phosphorylation following knockdown of CaMKKβ by transfection with specific siRNA. Knockdown of CaMKKβ suppressed AMPK phosphorylation and subsequently reduced eNOS activation in HUVECs treated with syringaresinol (Figure 4E). Syringaresinol-induced NO production was inhibited by treatment with U73122, W-7, and CaMKKβ siRNA (Figure 4F). These results demonstrate that syringaresinol promotes eNOS phosphorylation and subsequent NO production by CaMKKβ-dependent AMPK activated by PLC-mediated intracellular Ca2+ mobilization.

Syringaresinol elevates NO production via PLC/Ca2+-dependent eNOS phosphorylation. (A) HUVECs were preincubated with Fluo-4 (5 µM) for 30 minutes at 37℃ and stimulated with syringaresinol (30 µM) in the presence or absence of BAPTA-AM (5 µM). The intracellular Ca2+ levels were captured using a confocal laser microscope. Relative levels of intracellular Ca2+ were calculated from fluorescence intensities of Fluo-4. Data are the mean ± SD (n ≥ 3). **P < 0.01 versus control. (B-D) Cells were treated with syringaresinol (30 µM) in the presence or absence of BAPTA-AM (5 µM), U73122 (5 µM), W-7 (50 µM), Genistein (25 µM), and GF109203X (2 µM). The levels of the indicated target proteins were determined by Western blot analysis. (E) HUVECs transfected with control or CaMKKβ siRNAs (100 nM) were cultured in fresh medium for 47 h and then treated with syringaresinol for 1 h. The levels of target proteins were analyzed by Western blotting. (F) HUVECs were treated with the same conditions as above, and the intracellular levels of NO were determined by confocal microscopy. Data are the mean ± SD (n ≥ 3). *P < 0.05 and **P < 0.01 versus syringaresinol alone.
Syringaresinol promotes NO production via eNOS dimerization
The active form of NOS appears to be a homodimer, which is regulated by intracellular levels of Ca2+, required for CaM binding to the linker region between reductase and oxygenase domains of the enzyme (Hellermann and Solomonson, 1997). We investigated the effect of syringaresinol on eNOS dimerization in HUVECs, as determined by LT-PAGE and Western blot analysis. Treatment with syringaresinol resulted in a significant increase in the dimer form of eNOS and a concomitant decrease in the monomer form, and eNOS dimerization was abolished by treatment with U73122 and BAPTA-AM (Figures 5A and 5B). In addition, treatment with BAPTA-AM inhibited syringaresinol-induced NO production (Figure 5C). These results indicate that syringaresinol promotes NO production via eNOS dimerization promoted by the elevation of PLC-dependent intracellular Ca2+ mobilization.

Syringaresinol increases NO production via eNOS dimerization. (A) HUVECs were treated with syringaresinol in the presence or absence of U73122 and BAPTA-AM. Monomers and dimers of eNOS were determined by LT-PAGE and Western blot analysis. Boiled sample was used as control for eNOS monomer. (B) The relative ratios of eNOS dimer and monomer were quantitated using a densitometer. Data are the mean ± SD (n ≥ 3). **P < 0.01 versus control. (C) The levels of intracellular NO production were determined in HUVECs treated with or without syringaresinol and BAPTA-AM by the same method as described in Figure 2. Data are the mean ± SD (n ≥ 3). *P < 0.05 versus syringaresinol alone.
Syringaresinol-induced vasorelaxation is associated with the activation of PLC/Ca2+/AMPK and PI3/Akt pathways
To verify whether the PLC/Ca2+ and PI3/Akt pathways play a critical role in syringaresinol-induced vascular relaxation, endothelium-intact arterial rings pretreated with inhibitors of PLC, AMPK, and PI3K as well as a Ca2+ chelator, followed by treatment with syringaresinol. Pretreatment with U73122, Compound C, Wortmannin, and BAPTA-AM significantly attenuated syringaresinol-induced vasorelaxation in aortic rings pre-contracted with PhE (Figure 6A). We further examined the effects of these inhibitors on syringaresinol-induced cGMP production in endothelium-intact aortic rings. Pretreatment with these inhibitors significantly decreased syringaresinol-induced increase in cGMP production (Figure 6B). These findings indicate that syringaresinol induces vascular relaxation by elevating eNOS-dependent NO production via the activation of PI3/Akt- and PLC/Ca2+-dependent pathways.

Syringaresinol-induced vasorelaxation is attenuated by the inhibition of PLC/Ca2+/AMPK and PI3/Akt pathways. Endothelium-intact aortic rings pre-contracted with PhE (1 µM) were incubated with U73122 (U, 5 µM), Compound C (CC, 10 µM), Wortmannin (Wt, 100 nM), and BAPTA-AM (BA, 5 µM) for 30 min, followed by treatment with syringaresinol (SY, 30 µM) for 40 min. (A) The vascular relaxation was determined by the same method as described in Figure 1. (B) The level of cGMP was determined in the tissue homogenates using a cGMP assay kit. All data shown are the mean ± SD (n ≥ 5). *P < 0.05 and **P < 0.01 versus treatment with syringaresinol alone.
Discussion
This study was undertaken to examine the pharmacological and biological effects of syringaresinol on vascular relaxation and NO production, as well as to investigate its underlying mechanism upon post-translational activation of eNOS in primary HUVECs and in ex vivo mouse aortic rings. We here found that syringaresinol effectively elicited vascular relaxation in aortic rings pre-contracted with PhE and significantly promoted in vitro and ex vivo NO production by both phosphorylation-dependent and conformation-based eNOS activation via PI3K/Akt- and PLC/CaMKKβ/AMPK-dependent pathways and Ca2+-mediated eNOS dimerization.
Endothelial cells respond to various physiological stimuli by releasing endothelium-dependent vasodilators, including NO (Ignarro et al., 1987) and prostacyclin (Moncada et al., 1978). Of them, NO is produced by eNOS in the endothelium and diffuses out of endothelial cells into surrounding cells; some enters smooth muscle cells where it activates sGC to catalyze the conversion of GTP to cGMP, which induces vascular relaxation by activating protein kinase G. In this study, we showed that syringaresinol results in vasorelaxation in endothelium-intact, but not in endothelium-denuded, mouse aortic rings, indicating that vasorelaxation is elicited by vasodilators produced by intact endothelium. In addition, we found that the NOS inhibitor L-NMA completely abolished syringaresinol-induced vasorelaxation and cGMP production. These results indicate that endothelium-dependent component of the relaxant action of syringaresinol is due to enhanced endothelial NO synthesis or bioavailability and subsequent activation of NO-cGMP pathway.
NO rapidly reacts with superoxide anion and forms peroxynitrite, which is considered to be a toxic compound (Beckman et al., 1990). It is also possible that this reaction inhibits the cellular effects of NO, such as vasodilation and platelet aggregation, probably by decreasing NO bioavailability and cGMP production (Oury et al., 1996). Antioxidants that scavenge superoxide anion increase NO bioavailability and promote vasorelaxation. Syringaresinol has been suggested to possess antioxidant activity (Lee et al., 1999). Our data demonstrate that syringaresinol-induced relaxation in aortic rings from wild type mice, which was inhibited by L-NMA. However, syringaresinol did not alter 0.1 µM SNP-induced relaxation in both wild-type aortic rings in the presence of L-NMA and eNOS-deficient aortic rings (data not shown), indicating that this compound did not affect NO bioavailability. These results suggest that syringaresinol-induced vasorelaxation is associated with endothelial NO synthesis, but not with its antioxidant activity.
Endothelial-derived NO production is linked not only to post-translational modulation of eNOS activity (Hellermann and Solomonson, 1997; Fulton et al., 2001; Albrecht et al., 2003), but also to increased transcriptional expression (MacRitchie et al., 1997). Although eNOS has been known to be constitutively expressed under quiescent conditions, this enzyme can be upregulated at the level of gene transcription by the female gonadal hormone estrogen (MacRitchie et al., 1997). We found that syringaresinol increased endothelial NO production without changing the levels of eNOS protein and mRNA in HUVECs as determined by Western blot (Figures 3 and 4) and real-time reverse transcriptase-polymerase chain reaction (data not shown), indicating that this compound increased endothelial NO synthesis by regulating eNOS activity without changing its expression and stability. Although eNOS activity was initially identified to be regulated by Ca2+/CaM-dependent protein dimerization, recent studies have revealed additional modes of post-translational eNOS regulation involving phosphorylation, protein/protein interactions, and subcellular localization (Fulton et al., 2001; Albrecht et al., 2003). Evidences provided herein demonstrate that syringaresinol increases in vitro and ex vivo NO production by two distinctive mechanisms, phosphorylation-dependent eNOS activation and Ca2+-dependent eNOS dimerization (Figure 7).

Schematic proposal for the mechanism responsible for syringaresinol-induced vasorelaxation. Syringaresinol activates both PLC and PI3K, which induce intracellular Ca2+ mobilization and Akt, respectively. Increased intracellular Ca2+ level promotes eNOS dimerization and CaMKKβ/AMPK responsible for eNOS phosphorylation. In addition, the PI3K/Akt pathway promotes eNOS phosphorylation. These biochemical events result in an increase in eNOS-dependent NO synthesis and subsequent activation of sGC in smooth muscle cells to synthesize cGMP from GTP, leading to vascular relaxation.
The eNOS activity can be stimulated by phosphorylation at Ser1177 (human)/1179 (bovine) in the reductase domain (Dimmeler et al., 1999; Fulton et al., 1999; Michell et al., 2001). Phosphorylation of Ser1177 is one of the critical steps for eNOS activation in response to several stimuli, such as shear stress, VEGF, statins, and thrombin, which are known to increase endothelial NO production (Dimmeler et al., 1999; Harris et al., 2004; Stahmann et al., 2006). These stimuli activate a number of protein kinases, including Akt, AMPK, PKA, and CaMKKβ, which in turn phosphorylate Ser1177 (Fulton et al., 2001; Michell et al., 2001; Dudzinski and Michel, 2007). We here demonstrate that syringaresinol promoted activation of the eNOS-phosphorylating kinases Akt and AMPK. Syringaresinol-induced eNOS phosphorylation and NO production were suppressed by treatment with the PI3K inhibitor (Wortmannin) and AMPK inhibitor (Compound C). These inhibitors also showed suppressive effects on syringaresinol-induced cGMP production and vasorelaxation in aortic rings. On the other hand, the PKA inhibitor H89 did not affect syringaresinol-induced eNOS phosphorylation and vasorelaxation (data not shown). These results suggest that syringaresinol caused vascular relaxation by increasing phosphorylation-dependent activation of eNOS and subsequent NO production via activation of both PI3K/Akt- and AMPK-dependent signal pathways.
Ca2+ mobilization in the cytosol is a critical determinant of eNOS activity through dimerization-based activation (Hellermann and Solomonson, 1997) and phosphorylation-dependent enzyme modification (Stahmann et al., 2006). A recent study demonstrated that thrombin induced eNOS phosphorylation and activation by increasing intracellular Ca2+ mobilization through PLC activation (Stahmann et al., 2006). Thrombin-stimulated eNOS activation is directly associated with a previously known Ca2+/CaM-dependent mechanism (eNOS dimerization) and a PKCδ sensitive, but PI3K/Akt-independent, pathway responsible for bovine eNOS phosphorylation at Ser1177. Our data showed that syringaresinol-induced eNOS phosphorylation was not affected by PKC inhibitors Genistein and GF109203X, indicating that PKC is not involved in phosphorylation of eNOS by syringaresinol. However, we found that treatment with PLC inhibitor, Ca2+ chelator, CaM antagonist, AMPK inhibitor, and CaMKKβ siRNA inhibited eNOS phosphorylation and NO production via the inhibition of AMPK. Our data also showed that AICAR induced AMPK activation and eNOS phosphorylation, which were suppressed by the AMPK inhibitor Compound C. These data indicate that syringaresinol promotes eNOS phosphorylation by activating AMPK via the activation of PLC/Ca2+/CaMKKβ axis, which represents a novel signaling mechanism for NO production in HUVECs and subsequent induction of vasodilation (Figure 7).
The eNOS enzyme is only fully functional in a homodimeric form (List et al., 1997). Dimerization of eNOS is dependent on the number of cofactor molecules bound, such as tetrahydrobiopterin and heme (Fulton et al., 2001; Albrecht et al., 2003). Dimerization of eNOS starts with the binding of heme; without heme, the eNOS enzyme only exists as a monomer. The initial formation of a dimer by binding heme to monomeric eNOS makes it possible for tetrahydrobiopterin to bind to the eNOS dimer, which leads to the stabilization of eNOS protein in the active homodimer form (List et al., 1997; Venema et al., 1997). Elevation of cytosolic Ca2+ levels also promotes dimerization of the oxygenase domain of human eNOS, as well as increases catalytic function of eNOS by facilitating electron transfer between the enzyme's reductase and oxygenase domains (Hellermann and Solomonson, 1997; Dudzinski and Michel, 2007). We found that syringaresinol significantly increased eNOS dimerization, which was inhibited by the PLC inhibitor U73122 and the Ca2+ chelator BAPTA-AM. Furthermore, these inhibitors attenuated syringaresinol-induced vasorelaxation and cGMP production. These results indicate that syringaresinol increases NO production via eNOS dimerization by promoting a PLC-dependent increase in cytosolic Ca2+ levels, resulting in vascular relaxation.
Decreased NO production in endothelial cells promotes the pathogenesis of various vascular diseases, such as hypertension, atherosclerosis, and delayed wound healing (Hobbs et al., 1999; Albrecht et al., 2003). Our results demonstrate that syringaresinol promoted vasorelaxation via the elevation of NO-dependent vascular cGMP production by two distinct mechanisms, such as phosphorylation-dependent eNOS activation via the PI3K/Akt- and PLC/CaMKKβ/AMPK-dependent pathways and conformation-based eNOS activation via Ca2+-mediated eNOS dimerization (Figure 7). Thus, these findings suggest that syringaresinol can be considered as a potential drug for hypertension by activating the vascular eNOS/NO/cGMP pathway.
Methods
Materials
Heparin, penicillin, streptomycin, and M199 were purchased from Invitrogen Life Technologies (Carlsbad, CA). VEGF was purchased from Upstate Biotechnology (Lake Placid, NY). All antibodies used in the study were purchased from Cell Signaling Technology (Danvers, MA) and BD Transduction Laboratories (Franklin Lakes, NJ). Wortmannin, Compound C, U73122, and BAPTA-AM were purchased from Calbiochem Inc. (San Diego. CA). W-7 and CaMKKβ siRNA were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Syringaresinol was isolated from dried stems of Acanthopanax divaricatus, and purity was more than 98% as determined by HPLC analysis. All other reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless indicated otherwise.
Vascular function analysis
C57BL/6J mice (wild type, iNOS-/-, eNOS-/-, and nNOS-/-) obtained from Jackson Laboratory were sacrificed by cervical dislocation. The carotid arteries from mice were carefully dissected from surrounding fat and connective tissues. The dissected artery was cut into 2 mm long circular segments that were immediately placed in ice-cold Krebs-Ringer solution (118 mM NaCl, 4.7 mM KCl, 1.1 mM MgCl2, 1.2 mM KH2PO4, 1.5 mM CaCl2, 25 mM NaHCO3, and 10 mM glucose, pH 7.4) and aerated with 95% O2 and 5% CO2. All dissecting procedures were done with extreme care to protect the endothelium from inadvertent damage. In some vessel rings, the endothelial layer was mechanically removed by gently rubbing the luminal surface of the ring back and forth several times with a human hair. Endothelial integrity or functional removal was verified by the presence or absence of the relaxant response to acetylcholine (1 µM) in contracted vessels. Isomeric vascular tone was measured as described previously (Chataigneau et al., 1999). Each carotid arterial ring was suspended by means of two wires inserted into the ring lumen in an organ bath containing Krebs-Ringer solution at 37℃, while being continuously supplied with mixed gas composed of 95% O2 and 5% CO2. The baseline load placed on the arterial ring was 2 g and the changes in isometric tension were recorded using a force-displacement transducer with a computerized recording system (DMT320M, AD Instruments, Denmark). In the first set of experiments, the arterial rings were contracted with PhE, 1 µM to obtain maximal response. The response to acetylcholine (1 µM) was tested as a positive control in endothelium-intact aortic rings contracted by PhE. After each test, the arterial rings were washed three times with fresh Krebs-Ringer solution and allowed 30 min to equilibrate. Once the maximal response to PhE had been obtained, the indicated concentrations or 30 µM of syringaresinol were added to the bath and the relaxation was recorded. To further explore the possible mechanism of relaxation response caused by syringaresinol, several chemical inhibitors were pre-incubated for 20 min in endothelium intact aortic rings and response caused by syringaresinol was obtained after treatment with 1 µM PhE.
Measurement of cGMP levels in mouse aorta
Strips of mouse aortic rings were incubated at 37℃ for 40 min in 12-well culture plates containing Krebs solution gassed with 95% O2 and 5% CO2 and incubated for an additional 5 min in the presence of 3-isobutyl-1-methylxanthine (100 µM). The aortic rings were incubated with syringaresinol in the presence or absence of the nitric oxide inhibitor L-NMA for 30 min and frozen quickly in liquefied N2. Tissue lysates were prepared by homogenization with a Polytron homogenizer and centrifugation at 12,000×g for 20 min. The level of cGMP was measured using an enzyme immunoassay kit (Cayman Chemical Company, Ann Arbor, MI) in accordance to the instructions of the manufacturer.
Cell culture
HUVECs were isolated from human umbilical veins by collagenase, as previously described (Lee et al., 2006) and used in passages 3-8. The cells were grown in M199 supplemented with 20% fetal bovine serum, 100 µg/ml streptomycin, 100 U/ml penicillin, 3 ng/ml basic fibroblast growth factor, and 5 U/ml heparin at 37℃ in humidified 5% CO2/95% air.
Cell viability assay
HUVECs (2×105 cells/well) were plated onto 6-well plates in 2 ml of M199 containing 20% fetal bovine serum until cell density was reached to 70-80% confluence. Cells were treated with syringaresinol for 1 h, and cell viability was determined by the crystal violet staining method, as described previously (Lee et al., 2005).
NO measurement
The level of cellular NO production was measured by using 4-amino-5-methylamino-2,7-difluorofluorescein (DAF-FM) diacetate (Molecular Probes, Eugene, OR), as described previously (Lee et al., 2006). Briefly, after treatment with or without 20 ng VEGF, 30 µM syringaresinol, and 1 mM L-NMA for 1 h, HUVECs were washed twice with serum-free M199 and then incubated with 5 µM DAF-FM diacetate for 30 min at 37℃. After removing the excess probe, relative levels of intracellular NO were determined from the fluorescence intensity of DAF-FM via confocal microscopy. ex vivo NO production was measured in mouse aortic rings incubated with 5 µM DAF-FM diacetate and syringaresinol (30 µM) using a real-time fluorescence microscope with optimized excitation and emission wavelength (470/525 nm).
Western blot analysis
Cell lysates were separated by electrophoresis and transferred to polyvinylidene difluoride membranes. The membranes were incubated with antibodies against target proteins for 2 h following pre-hybridization with bovine serum albumin. The signal intensities of immunoreactive bands were visualized by an enhanced chemiluminescence system, as described previously (Lee et al., 2007).
Intracellular Ca2+ mobilization
The levels of intracellular Ca2+ were measured in situ by confocal microscopy using the Ca2+ probe Fluo-4-AM (Molecular Probes, Eugene, OR). HUVECs were incubated in serum-free M199 containing 5 µM Fluo-4 for 30 min at 37℃. After removing the excess dye, cells were incubated with syringaresinol in Hanks balanced salt solution (Sigma-Aldrich). Intracellular fluorescence was analyzed using confocal microscopy and results were expressed as the relative fluorescence intensity.
Transfection with siRNA
CaMKKβ-specific siRNA (sc-38955) and control siRNA (5'-GGCUACGTCCAGGAGCGCA-3') were obtained from Santa Cruz (Santa Cruz, CA) and Samchully Pharm (Seoul, Korea), respectively. HUVECs were grown to 70% confluence and transfected with 40 nM siRNA CaMKKβ using Lipofectamine Plus reagent (Invitrogen) according to the manufacturer's instructions. The levels of CaMKKβ were analyzed by Western blot at 48 h after transfection.
Determination of eNOS dimerization
Dimers and monomers of eNOS were separated using low-temperature SDS-PAGE (LT-PAGE) as previously described (Yang et al., 2009; Woo et al., 2010). Briefly, HUVECs were treated with syringaresinol for 30 min following pretreatment with U73122 (5 µM) and BAPTA-AM (5 µM) for 30 min. Cell lysates were loaded on SDS-gel, which was equilibrated at 4℃ before electrophoresis, and the buffer tank was placed in an ice bath during electrophoresis to maintain the temperature of the gel < 10℃. Subsequent to LT-PAGE, eNOS proteins were determined by Western blotting analysis.
Statistical analysis
All data are presented as the mean ± standard deviation (S.D.) of at least three independent experiments. One-way analysis of variance (ANOVA) was used to compare each parameter. Student's t-test was also performed to identify which group difference accounted for the significant overall analysis of variance. P < 0.05 was considered statistically significant.
Abbreviations
- AICAR:
-
aminoimidazole carboxamide ribonucleotide
- CaM:
-
calmodulin
- DAF-FM:
-
4-amino-5-methylamino-2,7-difluorofluorescein
- eNOS:
-
endothelial nitric oxide synthase
- L-NMA:
-
NG-monomethyl-L-arginine
- nNOS:
-
neuronal nitric oxide synthase
- PhE:
-
phenylephrine
- sGC:
-
soluble guanylyl cyclase
- SNP:
-
sodium nitroprusside
References
Adlercreutz H . Phytoestrogens: Epidemiology and a possible role in cancer protection . Environ Health Perspect 1995 ; 103 : 103 - 112
Albrecht EW, Stegeman CA, Heeringa P, Henning RH, Goor H . Protective role of endothelial nitric oxide synthase . J Pathol 2003 ; 199 : 8 - 17
Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA . Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide . Proc Natl Acad Sci USA 1990 ; 87 : 1620 - 1624
Cai H, Harrison DG . Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress . Circ Res 2000 ; 87 : 840 - 844
Chataigneau T, Félétou M, Huang PL, Fishman MC, Duhault J, Vanhoutte PM . Acetylcholine-induced relaxation in blood vessels from endothelial nitric oxide synthase knockout mice . Br J Pharmacol 1999 ; 126 : 219 - 226
Chung BH, Lee JJ, Kim JD, Jeoung D, Lee H, Choe J, Ha KS, Kwon YG, Kim YM . Angiogenic activity of sesamin through the activation of multiple signal pathways . Biochem Biophys Res Commun 2010 ; 391 : 254 - 260
Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM . Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation . Nature 1999 ; 399 : 601 - 605
Dudzinski DM, Michel T . Life history of eNOS: partners and pathways . Cardiovasc Res 2007 ; 75 : 247 - 260
Fisslthaler B, Fleming I . Activation and signaling by the AMP-activated protein kinase in endothelial cells . Circ Res 2009 ; 105 : 114 - 127
Fujikawa T, Yamaguchi A, Morita I, Takeda H, Nishibe S . Protective effects of Acanthopanax senticosus harms from Hokkaido and its components on gastric ulcer in restrained cold water stressed rat . Biol Pharm Bull 1996 ; 19 : 1227 - 1230
Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC . Regulation of endothelium-derived nitric oxide production by the protein kinase Akt . Nature 1999 ; 399 : 597 - 601
Fulton D, Gratton JP, Sessa WC . Post-translational control of endothelial nitric oxide synthase: why isn't calcium/calmodulin enough ? J Pharmacol Exp Ther 2001 ; 299 : 818 - 824
Hambrecht R, Adams V, Erbs S, Linke A, Kränkel N, Shu Y, Baither Y, Gielen S, Thiele H, Gummert JF, Mohr FW, Schuler G . Regular physical activity improves endothelial function in patients with coronary artery disease by increasing phosphorylation of endothelial nitric oxide synthase . Circulation 2003 ; 107 : 3152 - 3158
Harris MB, Blackstone MA, Sood SG, Li C, Goolsby JM, Venema VJ, Kemp BE, Venema RC . Acute activation and phosphorylation of endothelial nitric oxide synthase by HMG-CoA reductase inhibitors . Am J Physiol Heart Circ Physiol 2004 ; 287 : H560 - H566
Hellermann GR, Solomonson LP . Calmodulin promotes dimerization of the oxygenase domain of human endothelial nitric-oxide synthase . J Biol Chem 1997 ; 272 : 12030 - 12034
Hobbs AJ, Higgs A, Moncada S . Inhibition of nitric oxide synthase as a potential therapeutic target . Annu Rev Pharmacol Toxicol 1999 ; 39 : 191 - 220
Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G . Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide . Proc Natl Acad Sci USA 1987 ; 84 : 9265 - 9269
Jung HJ, Park HJ, Kim RG, Shin KM, Ha JH, Choi JW, Kim HJ, Lee YS, Lee KT . In vivo anti-inflammatory and antinociceptive effects of liriodendrin isolated from the stem bark of Acanthopanax senticousus . Planta Med 2003 ; 69 : 610 - 616
Lantin-Hermoso RL, Rosenfeld CR, Yuhanna IS, German Z, Chen Z, Shaul PW . Estrogen acutely stimulates nitric oxide synthase activity in fetal pulmonary artery endothelium . Am J Physiol 1997 ; 273 : L119 - L126
Lee SJ, Yun YS, Lee IK, Ryoo IJ, Yun BS, Yoo ID . An antioxidant lignan and other constituents from the root bark of Hibiscus syriacus . Planta Med 1999 ; 65 : 658 - 660
Lee SJ, Kim KM, Namkoong S, Kim CK, Kang YC, Lee H, Ha KS, Han JA, Chung HT, Kwon YG, Kim YM . Nitric oxide inhibition of homocysteine-induced human endothelial cell apoptosis by down-regulation of p53-dependent Noxa expression through the formation of S-nitrosohomocysteine . J Biol Chem 2005 ; 280 : 5781 - 5788
Lee SJ, Namkoong S, Kim YM, Kim CK, Lee H, Ha KS, Chung HT, Kwon YG, Kim YM . Fractalkine stimulates angiogenesis by activating the Raf-1/MEK/ERK- and PI3K/Akt/eNOS-dependent signal pathways . Am J Physiol Heart Circ Physiol 2006 ; 291 : H2836 - H2846
Lee SJ, Namkoong S, Ha KS, Nam WD, Kwon YG, Lee H, Yoon EY, Chang DJ, Kim SO, Kim YM . Colchicine-derived compound CT20126 promotes skin allograft survival by regulating the balance of Th1 and Th2 cytokine production . Exp Mol Med 2007 ; 39 : 230 - 238
List BM, Klosch B, Volker C, Gorren AC, Sessa WC, Werner ER, Kukovetz WR, Schmidt K, Mayer B . Characterization of bovine endothelial nitric oxide synthase as a homodimer with down-regulated uncoupled NADPH oxidase activity: tetrahydrobiopterin binding kinetics and role of haem in dimerization . Biochem J 1997 ; 323 : 159 - 165
Liu C, Zhong SM, Chen RY, Wu Y, Zhu XJ . Two new compounds from the dried tender stems of Cinnamomum cassia . J Asian Nat Prod Res 2009 ; 11 : 845 - 849
MacRitchie AN, Jun SS, Chen Z, German Z, Yuhanna IS, Sherman TS, Shaul PW . Estrogen upregulates endothelial nitric oxide synthase gene expression in fetal pulmonary artery endothelium . Circ Res 1997 ; 81 : 355 - 362
Mashimo H, Goyal RK . Lessons from genetically engineered animal models. IV. Nitric oxide synthase gene knockout mice . Am J Physiol 1999 ; 277 : G745 - G750
Michell BJ, Chen ZP, Tiganis T, Stapleton D, Katsis F, Power DA, Sim AT, Kemp BE . Coordinated control of endothelial nitric-oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase . J Biol Chem 2001 ; 276 : 17625 - 17628
Moncada S, Korbut R, Bunting S, Vane JR . Prostacyclin is a circulating hormone . Nature 1978 ; 273 : 767 - 768
Namkoong S, Chung BH, Ha KS, Lee H, Kwon YG, Kim YM . Microscopic technique for the detection of nitric oxide-dependent angiogenesis in an animal model . Methods Enzymol 2008 ; 441 : 393 - 402
Oury TD, Day BJ, Crapo JD . Extracellular superoxide dismutase: a regulator of nitric oxide bioavailability . Lab Invest 1996 ; 75 : 617 - 636
Ramirez-Sanchez I, Maya L, Ceballos G, Villarreal F . (-)-epicatechin activation of endothelial cell endothelial nitric oxide synthase, nitric oxide, and related signaling pathways . Hypertension 2010 ; 55 : 1398 - 1405
Scotland RS, Morales-Ruiz M, Chen Y, Yu J, Rudic RD, Fulton D, Gratton JP, Sessa WC . Functional reconstitution of endothelial nitric oxide synthase reveals the importance of serine 1179 in endothelium-dependent vasomotion . Circ Res 2002 ; 90 : 904 - 910
Stahmann N, Woods A, Carling D, Heller R . Thrombin activates AMP-activated protein kinase in endothelial cells via a pathway involving Ca2+/calmodulin-dependent protein kinase kinase β . Mol Cell Biol 2006 ; 26 : 5933 - 5945
Venema RC, Ju H, Zou R, Ryan JW, Venema VJ . Subunit interactions of endothelial nitric-oxide synthase, Comparisons to the neuronal and inducible nitric-oxide synthase isoforms . J Biol Chem 1997 ; 272 : 1276 - 1282
Woo A, Min B, Ryoo S . Piceatannol-3'-O-beta-D-glucopyranoside as an active component of rhubarb activates endothelial nitric oxide synthase through inhibition of arginase activity . Exp Mol Med 2010 ; 42 : 524 - 532
Wu HM, Yuan Y, Zawieja DC, Tinsley J, Granger HJ . Role of phospholipase C, protein kinase C, and calcium in VEGF-induced venular hyperpermeability . Am J Physiol 1999 ; 276 : H535 - H542
Yang YM, Huang A, Kaley G, Sun D . eNOS uncoupling and endothelial dysfunction in aged vessels . Am J Physiol Heart Circ Physiol 2009 ; 297 : H1829 - H1836
Acknowledgements
We thank Mrs. Elaine Por for helpful comments and critical reading of this manuscript. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (No. 20110028790) and the Regional Core Research Program/Medical & Bio-Material Research Center funded by MEST.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Chung, BH., Kim, S., Kim, JD. et al. Syringaresinol causes vasorelaxation by elevating nitric oxide production through the phosphorylation and dimerization of endothelial nitric oxide synthase. Exp Mol Med 44, 191–201 (2012). https://doi.org/10.3858/emm.2012.44.3.014
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3858/emm.2012.44.3.014
Keywords
This article is cited by
-
Modulation of gut microbiota and delayed immunosenescence as a result of syringaresinol consumption in middle-aged mice
Scientific Reports (2016)
-
Traditional Chinese medicine’s intervention in endothelial nitric oxide synthase activation and nitric oxide synthesis in cardiovascular system
Chinese Journal of Integrative Medicine (2015)
