
Abstract
FcαR, the Fc receptor for IgA, is essential for IgA-mediated immune responses. Previous studies have shown that IgA and IgA immune complexes can be rapidly endocytosed by FcαR. However, the underlying mechanism remains unclear. Here, we investigated the endocytic pathway of FcαR in monocytic cell line, U937, that naturally express FcαR and in transfected Chinese hamster ovary (CHO), COS-7 and Hela cells. By using selective chemical inhibitors of different endocytic pathways, overexpression of dominant-negative mutants of Eps15 and knockdown of clathrin heavy chain (CHC) via RNA interference, we demonstrated that endocytosis of FcαR was through a clathrin-mediated pathway. The endocytosed FcαR went into Rab5- and Rab11-positive endosomes. However, endocytosis of FcαR could not be blocked by a dominant-negative mutant of Rab5. We also demonstrated that endocytosis of FcαR was dynamin-dependent by overexpressing a dominant-negative mutant of dynamin. The potential endocytic motif for FcαR was also examined. Unexpectedly, we found that the entire cytoplasmic domain of FcαR was not required for the endocytic process of FcαR. We conclude that endocytosis of FcαR is clathrin- and dynamin-dependent, but is not regulated by Rab5, and the endocytic motif is not located in the cytoplasmic domain of FcαR.
Similar content being viewed by others


Introduction
In humans, IgA is the most abundant antibody in mucosal surfaces and the second most abundant antibody in blood 1, 2. IgA plays important roles in both mucosal and systemic immunity 3, 4. FcαR (CD89), the Fc receptor for IgA, plays crucial roles in IgA-mediated immune responses by coupling the innate and adaptive immune responses in effector cell activation. Binding of antigen-complexed IgA to FcαR initiates a variety of responses, and endocytosis is one of them 5, 6.
It has been known for a long time that IgA and IgA-complexed immune complex can be internalized by FcαR 7. However, the underlying mechanism remains unknown. Since FcαR is devoid of recognized signaling motifs in its cytoplasmic domain, it is believed that the immune responses elicited by antigen-complexed IgA are dependent on its functional association with the FcRγ-chain, which contains ITAM (immunoreceptor tyrosine-based activation motif), a signaling motif shared by other immune receptors, such as FcγRI, FcγRIII, BCR, TCR, etc 8, 9, 10. Although the γ-chain contains a well-characterized endocytic motif (YXXL), which plays important roles in endocytosis and antigen presentation 11, several studies have confirmed that γ-chain is not required for FcαR-mediated endocytosis of IgA and IgA immune complexes 9, 12. Instead, γ-chain is involved in the intracellular trafficking of FcαR after endocytosis 12, 13. Launay et al. 12 observed that internalized IgA was recycled back to the cell surface when FcαR was not associated with the γ-chain, whereas internalized IgA was delivered to lysosome for degradation after endocytosis when FcαR is associated with the γ-chain. Therefore, endocytosis of IgA by γ-chain-unassociated FcαR may play important roles in maintaining serum IgA concentration by protecting it from degradation.
Endocytosis of membrane receptors can occur through various pathways. Clathrin-dependent and clathrin-independent pathways represent the two major routes for internalization of cell surface receptors 14, 15, 16. Clathrin-mediated endocytosis, the most thoroughly studied endocytic pathway, is characterized by formation of clathrin-coated pits at the plasma membrane. Forming the coated pits requires several components including clathrin, AP-2, AP-180, Eps15, etc. Clathrin-dependent endocytosis requires the GTPase dynamin 17, 18, 19, which participates in the budding of clathrin-coated vesicles that are then destined for endosomal compartments. On the other hand, clathrin-independent endocytosis is less well understood until recently 20. Several endocytic pathways belong to this category, including caveolae-mediated endocytosis and lipid raft-mediated endocytosis, as well as pathways that are both clathrin- and caveolae/lipid raft independent 20. For example, a recent study has shown that the high-affinity IgE receptor (FcɛRI) is endocytosed by an AP-2/clathrin-independent mechanism, which appears to be lipid raft-mediated and regulated by dynamin 21.
Although there are different endocytic pathways for receptors to enter cells, the internalization, intracellular trafficking and membrane fusion of endocytic vesicles are generally regulated by a subfamily of Ras-like small GTPases (Rab GTPases) 22, 23. Approximately 40 members of Rab GTPases have been identified, and each is believed to be specifically associated with a particular organelle or pathway. Among them, Rab5 is localized in clathrin-coated vesicles and early endosomes, regulating internalization and early endosome fusion 24, 25; Rab11 is localized in recycling endosomes, regulating the endocytic recycling 26, 27, 28; Rab7 and Rab9 are localized in late endosomes, regulating the transport from early to late endosomes and late endsomes to the trans-Golgi, respectively 29, 30, 31, 32. It is known that endocytosis and intracellular trafficking of many G protein-coupled receptors are controlled by Rab GTPases 33.
In the present study, we demonstrate that FcαR is endocytosed through the clathrin-dependent pathway and the internalized receptor sequentially goes into Rab5- and Rab11-positive endosomes. Endocytosis of FcαR requires dynamin, but is not regulated by Rab5. Furthermore, we find that the cytoplasmic domain of FcαR is dispensable for its endocytosis.
Results
Endocytosis of FcαR in U937 cells
The U937 cell is a human cell line with monocytic characteristics and naturally expresses FcαR. Previous study shows that FcαR undergoes rapid endocytosis in U937 cells 12. Therefore, U937 cells were used to investigate the endocytic pathway(s) of FcαR. Several chemical inhibitors were used to selectively block clathrin- or caveolae/lipid raft-mediated endocytosis, which are two endocytic pathways commonly used by many membrane receptors. As shown in Figures 1A and 1B, blocking clathrin-mediated endocytosis by hypertonic sucrose (0.4 M), MDC (100 μM) and K+ depletion 34, 35, 36 dramatically inhibited FcαR internalization. In comparison, FcαR endocytosis was hardly inhibited when cells were treated with Filipin III (5 μg/ml) or nystatin (50 μg/ml), which is able to inhibit caveolae/lipid raft-mediated endocytosis 37, 38, 39.

Endocytosis of FcαR in U937 Cells. (A) U937 cells that grew on poly-lysine coated coverslip were treated with PMA (10−7 M) for 24 h. Then cells were treated with hypertonic sucrose (0.4 M), MDC (100 μM), Filipin III (5 μg/ml), nystatin (50 μg/ml), genistein (100 μg/ml) or herbimycin A (1 μM) for 60 min at 37 °C, or subjected to K+ depletion as described in Materials and Methods. Cells were then incubated with FITC-labeled MIP8a-F(ab′)2 in medium containing inhibitors for another 60 min at 4 °C, washed and transferred quickly to pre-warmed (37 °C) medium containing inhibitors and incubated for 30 min to allow endocytosis. Then, cells were fixed and nuclei were stained by Hoechst 33258 (blue). Endocytosis was examined by confocal laser-scanning microscope. Bar represents 7.5 μm. Data are representative of three independent experiments. (B) Quantitative analysis of the effect of chemical inhibitors on FcαR endocytosis by flow cytometry, see Materials and Methods. *P < 0.01.
Clathrin-dependent pathway does not but some clathrin-independent pathways require tyrosine kinase activity for ligand internalization 40, 41, 42. So, we examined whether tyrosine kinase activity was required for FcαR endocytosis by using tyrosine kinase inhibitors, genistein and herbimycin A. As shown in Figures 1A and 1B, treatment of cells by genistein (100 μg/ml) or herbimycin A (1 μM) had no influence on FcαR endocytosis, suggesting that tyrosine kinase activity was not required for FcαR endocytosis and further indicating that the endocytosis was mediated by a clathrin-dependent pathway.
Endocytosis of FcαR in transfected CHO cells
To study mechanisms of FcαR endocytosis, a stable transfectant of FcαR in CHO cells was established. Flow cytometry analysis showed that transfected CHO cells expressed FcαR on cell surface (Figure 2A) and were able to bind IgA (Figure 2B). The molecular mass of expressed FcαR was between 55 and 75 kDa (Figure 2C), which was similar to the naturally expressed receptor.

Endocytosis of FcαR in stably transfected CHO cells. (A) CHO cells stably expressing FcαR were incubated with MIP8a (black line) or isotype control MOPC21 (filled) for 60 min on ice, followed by incubation with FITC-conjugated goat anti-mouse IgG. (B) CHO cells (filled) or CHO cells stably expressing FcαR (black line) were incubated with FITC-conjugated human IgA2 for 60 min on ice. (C) 5×106 CHO cells or CHO cells stably expressing FcαR were lysed, FcαR were purified by anti-FcαR mAb-coupled beads and analyzed by western blot. (D) Chemical inhibitor tests were done the in the same manner as that in U937 cells, and the endocytosis was examined by confocal laser-scanning microscope. Bar represents 10 μm. Data are representative of at least three independent experiments. (E) Quantitative analysis of the effect of chemical inhibitors on FcαR endocytosis by flow cytometry, see Materials and Methods. *P < 0.05.
To investigate whether the endocytic pathway of FcαR in CHO cells is the same as that in U937 cells, which naturally express FcαR, we performed chemical inhibitor assays in CHO cells stably expressing FcαR. As shown in Figures 2D and 2E, both confocal microscopy and flow cytometry analysis showed that hypertonic sucrose (0.4 M), MDC (100 μM) and K+ depletion dramatically inhibited FcαR endocytosis, whereas Filipin III (5μg/ml) or nystatin (50 μg/ml) was unable to do so. Tyrosine kinase inhibitors, genistein (100 μg/ml) and herbimycin A (1 μM), were also unable to inhibit FcαR endocytosis in CHO cells. As a control, CHO cells that were not transfected with FcαR did not bind or internalize IgA or FITC-conjugated MIP8a, demonstrating that endocytosis of IgA or FITC-conjugated MIP8a in transfected CHO cells was mediated by FcαR, but not other unidentified receptors (data not shown). Taken together, these results suggested that endocytosis of FcαR in CHO cells followed the same pathway as that in U937 cells.
Dominant-negative mutants of Eps15 inhibit FcαR internalization
Eps15 is a protein that binds directly to the plasma membrane adaptor AP-2 and is required for clathrin-mediated endocytosis. Overexpression of dominant-negative mutants of Eps15 can selectively block clathrin-mediated endocytosis 43. So, we overexpressed dominant-negative mutants of Eps15 to further examine whether FcαR was internalized via clathrin-mediated endocytosis. EGFP-tagged Eps15-D3Δ2 (wild-type (WT) control, which has no influence on clathrin-mediated endocytosis) and two EGFP-tagged dominant-negative mutants, Eps15-DIII and Eps15-EH29, were transiently transfected into CHO cells stably expressing FcαR. The inhibitory effect of Eps15 mutants on clathrin-mediated endocytosis was examined by uptake of Texas Red-conjugated transferrin (Texas Red-Tfn). As shown in Figures 3A and 3B, uptake of Texas Red-Tfn was inhibited by up to 40%-60% when cells were transfected with EGFP-tagged Eps15-EH29 and Eps15-DIII. The WT control EGFP-tagged Eps15-D3Δ2 had no effect on Tfn uptake. Similar results were observed for FcαR endocytosis. Both Eps15-EH29 and Eps15-DIII but not Eps15-D3Δ2 inhibited internalization of FcαR (Figures 3C and 3D), indicating that FcαR was internalized via clathrin-mediated endocytosis.

Overexpression of dominant-negative mutants of Eps15 inhibited FcαR endocytosis. CHO cells stably expressing FcαR were transiently transfected with EGFP-tagged Eps15-D3Δ2 (WT control), Eps15-DIII or Eps15-EH29. At 48 h posttransfection, cells were allowed to internalize TRITC-conjugated MIP8a-F(ab′)2 for 30 min at 37 °C (C). For transferrin uptake (A), cells were serum starved for 1 h at 37 °C, and then incubated with 50 μg/ml Texas Red-Tfn in DMEM without serum at 37 °C for 30 min. At the end of endocytosis, cells were cooled to 4 °C quickly, washed and fixed. Nuclei were stained with Hoechst 33258 (blue). Endocytosis was analyzed by confocal laser-scanning microscope. (B) and (D) are quantitative analysis for Tfn uptake and FcαR endocytosis, respectively. *P < 0.01. Bars represent 10 μm. Data are representative of at least three independent experiments.
FcαR endocytosis is abolished when clathrin heavy chain is depleted by short-hairpin (shRNA)
The above mentioned inhibition experiments using chemical inhibitors and dominant-negative mutants of Eps15 indicated that endocytosis of FcαR was dependent on clathrin. To obtain direct evidence, RNA interference was used to knockdown clathrin heavy chain (CHC). We first tried to knockdown CHC in CHO cells, but failed (data not shown). Considering that our target sequence (GTA ATC CAA TTC GAA GAC C) against CHC is only conserved in humans and mice, but not in rats, we speculated that this target sequence might not be conserved in hamster-originated CHO cells either. So, we used human-originated Hela cells instead. FcαR was cotransfected with pSUPER vector (mock control) or pSUPER-shRNA CHC in Hela cells, and then endocytosis of FcαR was examined. The expression level of clathrin in Hela cells was obviously decreased 72 h posttransfection of pSUPER-shRNA CHC, as determined by western blot (Figures 4A and 4B). Immunofluorescent staining of CHC also showed that CHC was successfully depleted in pSUPER-shRNA CHC-transfected cells, but not in pSUPER-transfected cells (Figure 4C). As shown in Figures 4C and 4D, FcαR was able to be internalized when cells were cotransfected with pSUPER vector, but the endocytosis was abolished when pSUPER-shRNA CHC was cotransfected. These results demonstrated that FcαR was internalized via a clathrin-dependant pathway in Hela cells.

Depletion of clathrin heavy chain by shRNA abolished the endocytosis of FcαR in Hela cells. (A) CHC depletion was analyzed by western blot. Hela cells were transfected with pSUPER-shRNA CHC. At 72 h posttransfection, equal numbers of cells were lysed, separated by 5%-18% SDS-PAGE and transferred onto a nitrocellulose membrane. CHC was detected by mouse monoclonal Abs TD-1. (B) Relative expression of CHC was quantified by densitometry. Data are from three independent experiments. *P < 0.01. (C) Endocytosis of FcαR in Hela cells cotransfected with pcDNA3.1-FcαR and pSUPER-shRNA CHC. At 72 h posttransfection, cells were allowed to internalize TRITC-conjugated MIP8a-F(ab′)2 (red) for 30 min at 37 °C. At the end of endocytosis, cells were fixed, permeabilized and CHC was stained by rabbit anti-clathrin heavy chain polyclonal Abs followed by FITC-conjugated goat anti-rabbit IgG. Nuclei were stained with Hoechst 33258 (blue). Endocytosis of FcαR was examined by confocal laser-scanning microscope. Bar represents 10 μm. Data are representative of three independent experiments. (D) Quantitative analysis for FcαR endocytosis after depletion of CHC. *P < 0.01.
To further investigate whether clathrin is required for endocytosis of FcαR in U937 cells, which naturally express this receptor, we used a lentiviral shRNA vector pLVTHM to knockdown CHC in U937 cells. U937 cells were infected with viruses packed in 293T cells, and cells with high expression level of GFP were sorted by FACS. Western blot showed that CHC expression in shRNA CHC U937 cells decreased to 18% of mock shRNA cells (Figure 5A), which was in line with the result of immunofluorescent staining of CHC, showing that CHC staining in CHC knockdown cells was significantly weaker than in control cells (Figure 5C). Then, endocytosis of FcαR in CHC knockdown U937 cells was examined. As shown in Figure 5D, FcαR endocytosis in CHC knockdown cells decreased remarkably. Quantitative analysis by flow cytometry showed that endocytosis of FcαR in CHC knockdown U937 cells decreased to 28% of control (Figure 5B), demonstrating that endocytosis of FcαR in U937 cells was clathrin dependent.

Knockdown of CHC in U937 cells inhibits FcαR endocytosis. (A) CHC expression in U937 cells stably infected with pLVTHM or pLVTHM-shRNA CHC was analyzed by western blot. A total of 30 μg protein from each cell lysate was separated by 5%-18% SDS-PAGE, transferred onto a nitrocellulose membrane and CHC was detected by mouse monoclonal Abs TD-1. The number under each lane is the relative expression level of CHC quantified by densitometry. (B) Quantification of FcαR endocytosis in pLVTHM and pLVTHM-shRNA CHC cells by flow cytometry. Data are from three independent experiments. *P < 0.01. (C) Examination of CHC expression in U937 cells stably infected with pLVTHM or pLVTHM-shRNA CHC was by confocal laser-scanning microscope. Cells grew on coverslip were fixed, permeabilized, and stained by rabbit anti-clathrin heavy chain polyclonal Abs followed by FITC-conjugated goat anti-rabbit IgG. Nuclei were stained by Hoechst 33258 (blue). (D) Endocytosis of FcαR in U937 cells stably infected with pLVTHM or pLVTHM-shRNA CHC. Cells were incubated with TRITC-conjugated MIP8a-F(ab′)2 for 60 min at 4 °C, washed with cold HBSS and then incubated at 37 °C for 30 min to allow endocytosis. After endocytosis, cells were fixed and nuclei were stained by Hoechst 33258 (blue). Bar represents 15 μm. Data are representative of four independent experiments.
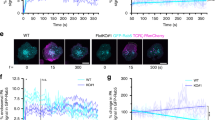
Endocytosed FcαR is localized in Rab5- and Rab11-positive endosomes, but endocytosis of FcαR is not regulated by Rab5
Small GTPases broadly regulate endocytosis, intracellular trafficking and membrane fusion of endocytic vesicles. Rab5, Rab11, Rab7 and Rab9 are recognized as markers for early endosomes, recycling endosomes and late endosomes. We examined whether FcαR was present in these small GTPase-containing endosomes during endocytosis and intracellular trafficking. To visualize the colocalization of FcαR with these Rab proteins, CHO cells stably expressing FcαR were transiently transfected with plasmids encoding EGFP-tagged Rab5, Rab11, Rab7 or Rab9. At 48 h posttransfection, FcαR expressed on the cell surface was labeled with TRITC-conjugated MIP8a-F(ab′)2, a monoclonal antibody (mAb) specifically against FcαR. Then cells were incubated at 37 °C to allow for endocytosis. Localization of internalized FcαR and EGFP-Rab proteins was determined by confocal microscopy. Figure 6A shows that internalized FcαR colocalized with Rab5 and Rab11 30 min after endocytosis. Analysis of the time course of colocalization showed that FcαR colocalized with Rab5 as early as 5 min after the cells were incubated at 37 °C (Supplementary information, Figure S1A). This colocalization reached a maximum at 30 min and then decreased slowly. Colocalization of FcαR with Rab11 was observed at 10 min and reached a maximum at 60 min, then decreased through the following 3 h (Supplementary information, Figure S1B). In comparison, little colocalization between FcαR and Rab7 or Rab9 was observed through the whole time course (Supplementary information, Figure S2). These data suggest that internalized FcαR went into Rab5-positive early endosomes, then were possibly recycled back to the cell surface through Rab11-positive recycling endosomes, but not delivered to late endosomes.

Colocalization of endocytosed FcαR with Rab5 and Rab11. CHO cells stably expressing FcαR were transiently transfected with EGFP-tagged Rab5, Rab11 or Rab5-S34N. At 48 h posttransfection, cells were incubated with TRITC-conjugated MIP8a-F(ab′)2 for 60 min at 4 °C. Excess ligands were washed and cells were transferred to pre-warmed medium (37 °C) to allow for endocytosis for 30 min. Nuclei were stained by Hoechst 33258 (blue). Endocytosis of FcαR was examined by confocal laser-scanning microscope. (A) Colocalization of internalized TRITC-conjugated MIP8a-F(ab′)2 with Rab5 (upper panel) or Rab11 (lower panel). (B) Effect of Rab5-S34N on FcαR endocytosis. Bars represent 10 μm. Data are representative of at least three independent experiments.
Rab5-S34N is a dominant-negative mutant of Rab5, which has been shown to be able to inhibit endocytosis of some receptors 33. Since FcαR was colocalized with Rab5, we next examined whether the endocytosis of FcαR was regulated by Rab5. At 48 h posttransfection of EGFP-tagged Rab5-S34N in CHO cells stably expressing FcαR, cells were allowed to internalize TRITC-conjugated MIP8a-F(ab′)2 for 30 min at 37 °C. As shown in Figure 6B, overexpression of dominant-negative mutant of Rab5 did not block the internalization of FcαR in CHO cells, suggesting that the endocytosis of FcαR was not regulated by Rab5.
FcαR endocytosis is dynamin dependent
Clathrin-dependent and a subset of clathrin-independent endocytosis require the activity of dynamin, a GTPase responsible for pinching vesicles from the plasma membrane and thereby driving cargo internalization into carrier vesicles 17, 18, 19. To determine if FcαR internalization was dynamin dependent, HA-tagged WT dynamin (Dyn-WT-HA) or a dominant-negative dynamin mutant K44A (Dyn-K44A-HA) was transiently transfected into CHO cells stably expressing FcαR. Similar to Tfn (Figures 7A and 7B), FcαR endocytosis was dramatically reduced in cells expressing dominant-negative dynamin mutant, but not in cells expressing the WT dynamin (Figures 7C and 7D), suggesting that dynamin was required for FcαR endocytosis.

FcαR endocytosis is dynamin dependent. CHO cells stably expressing FcαR were transiently transfected with HA-tagged wild-type dynamin (Dyn-WT-HA) or dominant-negative mutant dynamin K44A (Dyn-K44A-HA). At 48 h posttransfection, cells were allowed to internalize TRITC-conjugated MIP8a-F(ab′)2 for 30 min at 37 °C (C). For Tfn uptake (A), cells were serum starved for 1 h at 37 °C, then incubated with 50 μg/ml Texas Red-Tfn in DMEM without serum at 37 °C for 30 min. At the end of endocytosis, cells were cooled to 4 °C quickly, washed, fixed and nuclei were stained with Hoechst 33258 (blue). Endocytosis was analyzed by confocal laser-scanning microscope. (B) and (D) are quantitative analysis for Tfn uptake and FcαR endocytosis, respectively. *P < 0.01. Bars represent 10 μm. Data are representative of at least three independent experiments.
The endocytic motif of FcαR is not located in the cytoplasmic domain of FcαR
Sorting of transmembrane proteins to endosomes is mediated by consensus motifs present within the cytoplasmic domains of these proteins, the most common endocytic motifs are tyrosine-based motif YXXØ (X stands for any amino acid and Ø stands for an amino acid residue with a bulky hydrophobic side chain) and dileucine-based signals [DE]XXXL[LI] or DXXLL 44. As no such conserved motifs are found within the cytoplasmic domain of FcαR, we speculate that some unrecognized motif within the cytoplasmic domain might be responsible for FcαR endocytosis. To test this hypothesis, the entire cytoplasmic domain of FcαR (41 amino acids) was deleted. This tail-less FcαR was transiently transfected in COS-7 cells and stably transfected in CHO cells. Flow cytometry analysis showed that this tail-less FcαR was able to be expressed on cell surface, and bind IgA to the same extent as the full-length receptor (data not shown). Endocytosis results showed that, just like the full-length receptor, the tail-less FcαR was able to be internalized both in COS-7 (Figure 8A) and CHO cells (Figure 8B).

The cytoplasmic domain of FcαR is not required for its endocytosis. FcαR and tail-less FcαR were transiently transfected in COS-7 cells (A) or stably transfected in CHO cells (B). Cells were incubated with FITC-conjugated MIP8a-F(ab′)2 for 60 min at 4 °C, then washed and transferred quickly to pre-warmed (37 °C) medium and incubated for 30 min to allow for endocytosis. The endocytosis was examined by fluorescence microscope. (C) Endocytosis of tail-less FcαR stably transfected in CHO cells in the presence of sucrose (0.4 M), MDC (100 μM) or Filipin III (5 μg/ml). The endocytosis was examined by fluorescence microscope. (D) Endocytosis of tail-less FcαR in CHC knockdown Hela cells. Hela cells were cotransfected with pcDNA3.1-tail-less FcαR and pSUPER or pSUPER-shRNA CHC. At 72 h posttransfection, cells were allowed to internalize TRITC-conjugated MIP8a-F(ab′)2 for 30 min at 37 °C. At the end of endocytosis, cells were fixed, permeabilized and CHC was stained by rabbit anti-clathrin heavy chain polyclonal Abs followed by FITC-conjugated goat anti-rabbit IgG. Nuclei were stained by Hoechst 33258 (blue). The endocytosis was examined by confocal laser-scanning microscope. Bar represents 15 μm. Data are representative of three independent experiments. (E) Kinetic analysis of endocytosis of FcαR and tail-less FcαR in stably transfected CHO cells by flow cytometry. (F) Quantitative analysis of tail-less FcαR endocytosis in CHC knockdown Hela cells. *P < 0.01.
To test whether this tail-less FcαR is internalized through the same pathway as the full-length receptor, chemical inhibitors of clathrin- or caveolae/lipid raft-mediated endocytosis pathway were used. As shown in Figures 8C and 8E, hypertonic sucrose (0.4 M) and MDC (100 μM), but not Filipin III (5 μg/ml), inhibited endocytosis of tail-less FcαR, suggesting that this tail-less FcαR was also endocytosed through a clathrin-dependent pathway. Next, tail-less FcαR was cotransfected with pSUPER or pSUPER-shRNA CHC in Hela cells, as done for full-length FcαR. As expected, CHC was depleted in pSUPER-shRNA CHC-transfected cells, but not in pSUPER-transfected cells. In addition, endocytosis of tail-less FcαR in CHC knockdown Hela cells was also inhibited (Figures 8D and 8F). These data demonstrated that endocytosis of tail-less FcαR was, like full-length FcαR, also through the clathrin-dependent pathway.
We also compared the kinetics of endocytosis of FcαR and tail-less FcαR in stably transfected CHO cells, and the result showed no difference between them (Figure 8E). These data suggest that endocytosis of both full-length FcαR and tail-less FcαR follows the same clathrin-dependent pathway, but the cytoplasmic domain is not required for endocytosis of FcαR.
Discussion
Although endocytosis is a well-known feature of FcαR, the mechanism remains unknown. Previous studies showed that FcαR redistributed into lipid raft 30 s after being cross-linked, but this redistribution decreased to a basal level within 5 min before endocytosis occurred 9, 10. In addition, FcαR was not colocolized with the lipid raft marker, GM-1 10. These results suggested that endocytosis of FcαR was not lipid raft mediated. In the present study, we investigated the FcαR endocytic pathway and demonstrated that FcαR was internalized through a clathrin- and dynamin-dependent pathway.
Endocytosis of FcαR was blocked by hypertonic sucrose, MDC and K+ depletion, but not by nystatin and Filipin III, suggesting that FcαR underwent endocytosis via a clathrin-dependent pathway, but not the caveolae or lipid raft-mediated endocytosis. Furthermore, tyrosine kinases were not required for FcαR endocytosis, as genistein and herbimycin A did not inhibit the endocytosis of FcαR. This is very similar to FcγRI (CD64), which can undergo endocytosis in the absence of γ-chain in a tyrosine kinase-independent manner 45. In comparison, tyrosine kinase is required for the endocytosis of FcγRIIA 46. These data suggest that endocytosis of different Fc receptors is differentially regulated by tyrosine kinase.
To validate the results of chemical inhibitor experiments, dominant-negative mutants of Eps15 were used, which can specifically inhibit clathrin-mediated endocytosis 43. As shown in Figure 3, expression of both mutants, but not WT control Eps15, blocked the endocytosis of FcαR. This further demonstrated that FcαR was internalized through clathrin-mediated endocytosis. Moreover, FcαR endocytosis was abolished when clathrin heavy chain was depleted by shRNA, providing direct evidence that clathrin was required for endocytosis of FcαR. Taken together, these data demonstrate that endocytosis of FcαR is clathrin-mediated.
FcαR is expressed on various myeloid cells either as monomers or FcR-γ-chain-associated multimers 12. Previous studies using IIA1.6 and RBL-2H3 cells showed that FcR γ-chain was not required for FcαR-mediated endocytosis of IgA and IgA immune complexes 9, 12. Here, we demonstrated that FcαR was able to undergo endocytosis in transfected CHO, COS-7 and Hela cells, which are not myeloid cells and devoid of γ-chain. Therefore, endocytosis is an intrinsic characteristic of FcαR itself in various kinds of cells, regardless of the presence of FcR γ-chain.
Although FcR γ-chain is not required for FcαR endocytosis, it may affect the fate of FcαR after it enters the cell. Previous study showed that, in transfected RBL cells, internalized FcαR-R209L (a mutant FcαR where arginine 209 at the transmembrane region is replaced by lysine, therefore it cannot associate with the γ-chain) was colocalized with Tfn 12, a known marker for the endocytosis-recycling route. In agreement with these results, we found that internalized FcαR went into Rab5- and Rab11-positive early endosomes and recycling endosomes, and it hardly went to Rab7- and Rab9-positive late endosomes. In contrast, when FcαR was associated with γ-chain, the endocytosed ligand was sorted to lysosomes for degradation and further for antigen presentation 13. Therefore, γ-chain plays important roles in determining the intracellular trafficking routes of FcαR after endocytosis 12, 13.
Although Rab5 has been shown to be able to regulate the endocytosis of some membrane receptors 33, overexpression of a dominant-negative mutant of Rab5 (Rab5-S34N) had no influence on the endocytosis of FcαR in this study. This indicates that endocytosis of FcαR is not regulated by Rab5, similar phenomenon have been reported for other receptors 47, 48.
It is believed that signals for clathrin-mediated endocytosis lie in the cytoplasmic domain of membrane receptors. Because there are no such conserved endocytic motifs within the cytoplasmic domain of FcαR, we, as well as others 13, speculated that there might be some unrecognized motif within the cytoplasmic domain of FcαR, which could interact with endocytic machinery and mediate endocytosis of FcαR. However, we surprisingly found that FcαR could still be internalized when the entire cytoplasmic domain was deleted. What is more interesting is that endocytosis of this tail-less FcαR was inhibited by hypertonic sucrose, MDC and knockdown of CHC, demonstrating that endocytosis of tail-less FcαR was also clathrin dependent. Nevertheless, this finding is consistent with the fact that no conserved endocytic motifs are found within the cytoplasmic domain of FcαR. A few studies showed that signals for endocytosis could localize in the extracellular and/or transmembrane domain of a receptor 45, 49, 50. For example, it has been reported that endocytic motif of another Fc receptor, FcγRI (CD64), localizes within the extracellular domain of the receptor rather than the transmembrane or cytoplasmic domain 45. How FcαR could be internalized through clathrin-mediated endocytosis in the absence of its cytoplasmic domain is not clear at present.
First, it is possible that the endocytic motif for FcαR might also be localized within the extracellular domain of FcαR, just like FcγRI. The extracellular region of FcαR contains two Ig-like domains, EC1 and EC2. EC1 contains the binding site for IgA, and our mAb against FcαR also recognizes EC1. Deletion of EC1 will result in a receptor without the capacity to bind its natural ligand and our mAb will not recognize it. FcαR without the entire EC2 domain is actually an isoform of FcαR, which is generated through alternative splicing 5. This isoform of FcαR has been only detected at mRNA level, and no surface expression on natural cells has been reported 51. So, EC2 is important for the surface expression of FcαR. Therefore, to characterize the exact endocytic motif of FcαR, one would need to make site-directed mutations within its extracellular or transmembrane domain.
Second, we think that characterizing endocytic adaptors of FcαR is more important, because wherever the endocytic motif is localized, this motif must interact with the endocytic adaptors to accomplish endocytosis. So, we have tried to identify FcαR-interacting endocytic adaptors (or other transmembrane molecules) by CoIP. However, silver staining generated many weak bands (data not shown), which were difficult to characterize and might reflect non-specific bindings. Other techniques, such as yeast two-hybrid screening, might be able to identify FcαR-interacting proteins.
In conclusion, we demonstrate that FcαR is able to be internalized through clathrin-mediated endocytosis in the absence of its cytoplasmic domain. Our data shed new light on the mechanism of clathrin-mediated endocytosis by showing that the endocytic motif does not have to be localized in the cytoplasmic domain of the receptor. Further studies are needed for characterizing the exact endocytic motif of FcαR and endocytic adaptors that help tail-less FcαR accomplish endocytosis. Better understanding of the mechanisms of FcαR endocytosis will help us elucidate the role of this receptor in regulating serum IgA homeostasis and IgA-mediated immune responses.
Materials and Methods
Antibody and agents
The murine anti-FcαR mAb MIP8a, rabbit anti-FcαR polyclonal antibody, FITC or tetramethylrhodamine isothiocyanate (TRITC, USA)-labeled F(ab′)2 fragments of MIP8a and human IgA2 were prepared as described previously 52, 53, 54. Rabbit anti-clathrin heavy chain (CHC) polyclonal antibody (ab21679) was from Abcam (Cambridge, UK), mouse anti-CHC mAb (TD-1) was from Santa Cruz Biotechnology (Santa Cruz, USA). Goat anti-HA polyclonal antibody, HRP-conjugated goat anti-mouse IgG polyclonal antibody and FITC-conjugated goat anti-rabbit IgG polyclonal antibody were from Zhongshan Biotechnology Co (Beijing, China). Sucrose, monodansylcadaverine crystalline (MDC), Filipin III, nystatin, genistein, herbimycin A and phorbol-12-myristate-13-acetate (PMA) were from Sigma-Aldrich (St. Louis, MO, USA). Texas Red-conjugated Tfn was from Invitrogen (Carlsbad, USA).
Plasmids
The cDNA of FcαR was cloned from human peripheral white blood cells by reverse transcription PCR, and inserted into the BamHI and XhoI sites of pcDNA 3.1 vector (Invitrogen). The tail-less FcαR (the entire cytoplasmic domain of FcαR was deleted) was constructed by standard PCR procedure and inserted into pcDNA3.1 vector at the same sites. EGFP-tagged Eps15-EH29, DIII and D3Δ2 were provided by Dr Alexandre Benmerah 43. HA-tagged WT dynamin and its dominant-negative mutant K44A were provided by Dr Sandra Schmid 19. EGFP-tagged Rab7, Rab9 and Rab11 were gifts from Dr Richard E Pagano 55. EGFP-tagged Rab5 and Rab5-S34N were provided by Dr Feng Du (Tsinghua University, China).
Cell culture and transfection
U937 cells were grown in RPMI1640 containing 10% fetal bovine serum, and CHO, COS-7 and Hela cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum and antibiotics, at 37 °C in a humidified atmosphere of 5% CO2. Cells were transfected with plasmids encoding of FcαR using Lipofectamine 2000 (Invitrogen). Stably transfected CHO cells were selected with 800 μg/ml G418 and were evaluated for receptor expression by flow cytometry and western blot. Cells stably expressing FcαR were transiently transfected with EGFP-tagged WT and dominant-negative mutant of Eps15, HA-tagged dynamin WT and K44A or EGFP-tagged Rab GTPases using Lipofectamine™2000.
Manipulation of endocytic pathways by chemical inhibitors
Because the expression level of FcαR on U937 cells is relatively low, the signal of FcαR staining under laser-scanning confocal microscopy is weak. So, PMA was used to increase the expression level of FcαR 12. U937 cells that grew on poly-lysine-coated coverslip were treated with 10−7 M PMA for 24 h before experiments. CHO cells stably expressing FcαR were cultured as described above. 24 h prior to experiments, cells were seeded onto coverslip placed in 24-well plates and cultured for another 24-48 h. On the day of experiment, cells were pretreated with 0.4 M sucrose, 100 μM MDC, 5 μg/ml Filipin III, 50 μg/ml nystatin, 100 μg/ml genistein or 1 μM herbimycin A for 1 h at 37 °C. K+ depletion was done according to the procedure of Altankov and Grinnell 36. Briefly, cells were rinsed once with potassium-free buffer (140 mM NaCl, 20 mM Hepes, 1 mM CaCl2, 1 mM MgCl2, 1 mg/ml D-glucose, pH 7.4), then incubated in hypotonic medium (50% potassium-free buffer/50% H2O) for 5 min at 37 °C, followed by incubation in potassium-free buffer for 20 min at 37 °C. Cells were then cooled to 4 °C and incubated with FITC or TRITC-labeled MIP8a-F(ab′)2 in medium or potassium-free buffer at 4 °C for 30 min. Cells were washed with cold HBSS (or potassium-free buffer), then transferred quickly into pre-warmed (37 °C) medium supplemented with inhibitors or potassium-free buffer, followed by incubation at 37 °C for various times to allow for endocytosis. Controls constituted of equal volume of respective solvent added in medium. In K+ depletion assay, control was conducted with 10 mM KCl added in potassium-free buffer. At the end of endocytosis, cells were placed on ice, fixed with 4% paraformaldehyde and mounted. The inhibition of endocytosis was determined by confocal microscopy and quantified by flow cytometry.
Flow cytometry
To analyze the expression of FcαR on CHO cells, stably transfected cells were incubated with MIP8a or isotype control MOPC21 (Sigma-Aldrich) in 1% BSA/PBS followed by incubation with FITC-conjugated goat anti-mouse IgG. Then samples were analyzed by a flow cytometer (FACS Aria, USA). For IgA binding, CHO cells or FcαR stably transfected CHO cells were incubated with FITC-conjugated human IgA2 in 1% BSA/PBS for 60 min on ice, then washed and analyzed by flow cytometry.
Endocytosis of FcαR in U937 cells and stably transfected CHO cells was quantified by flow cytometry. Cells were incubated with MIP8a in medium (or medium containing inhibitors) for 60 min on ice, washed with cold HBSS thrice (for U937 cells, cells were centrifuged at 400 × g for 2 min after each wash), then cells were incubated in medium (or medium containing inhibitors) at 37 °C to allow for endocytosis for various times. The control sample was placed on ice all the time (Time 0). At the end of endocytosis, cells were cooled to 4 °C quickly by rinsing twice with cold HBSS; FcαR remaining on cell surface after endocytosis were stained with FITC-conjugated goat anti-mouse IgG by incubating in 1% BSA/PBS/AZ at 4 °C for 45 min. Then, cells were washed with cold 1% BSA/PBS/AZ thrice and analyzed by flow cytometry (CHO cells were detached by trypsin digestion before analysis). The percentage of FcαR endocytosis was calculated as: (MIP8a on cell surface at 4 °C (Time 0)−MIP8a on cell surface after 37 °C incubation)/ MIP8a on cell surface at 4 °C (Time 0) ×100%.
Immunofluoresence and confocal microscopy
CHO cells stably expressing FcαR were allowed to grow on coverslip as described above. 24 h later, cells were transfected with EGFP-tagged Eps15-EH29, DIII, D3Δ2, HA-tagged dynamin WT, dynamin K44A or EGFP-tagged Rab GTPases. In shRNA experiment, Hela cells were cotransfected with pcDNA3.1-FcαR and pSUPER-shRNA CHC at the ratio of 1:1. At 48 h posttransfection, cells were cooled to 4 °C and incubated with FITC or TRITC-labeled MIP8a-F(ab′)2 in medium at 4 °C for 60 min, then washed with cold HBSS and transferred quickly into pre-warmed (37 °C) medium, followed by incubation at 37 °C for various times. At the end of endocytosis, cells were placed on ice and fixed with 4% paraformaldehyde. To detect HA and clathrin, cells were permeabilized by 0.2% Triton X-100 for 10 min at room temperature, then blocked with 5% horse serum for 30 min. Cells were incubated with rabbit anti-clathrin heavy chain polyclonal antibody or goat anti-HA polyclonal antibody in 1% BSA/PBS for 60 min at room temperature, then washed with 1% BSA/PBS, followed by incubation with FITC-conjugated goat anti-rabbit IgG or rabbit anti-goat IgG for another 60 min at room temperature. Then, cells were washed and nuclei were stained with Hoechst 33258 (Merck & Co, USA). The colocalization of endocytosed MIP8a-F(ab′)2 and Rabs GTPases during various time points was determined by a confocal laser-scanning microscope system (Leica TCS SP2 SE, Germany) with 100×1.44 numerical aperture oil immersion lens. The effects of Eps15, dynamin and Rabs GTPases on FcαR endocytosis were also examined by confocal microscopy. Each image was a single confocal slice. For quantitative analysis, the fluorescence intensity of endocytosed TRITC-conjugated MIP8a-F(ab′)2 within cells were quantified by Image J software (NIH, USA). In each experiment, 100 untransfected cells and 100 transfected cells were quantified for their endocytosis of TRITC-conjugated MIP8a-F(ab′)2, and the results were expressed as mean ± SEM.
Transferrin endocytosis assay
First, cells were serum starved by incubation in medium without serum for 1 h at 37 °C. Then, cells were incubated with 50 μg/ml Texas Red-Tfn in medium without serum at 37 °C for various times. At the end of endocytosis, cells were cooled to 4 °C quickly and washed with cold HBSS. Then, cells were fixed, stained with Hoechst 33258, mounted and analyzed as described above.
RNA interference
Target sequence of shRNA duplexes against CHC (GTA ATC CAA TTC GAA GAC C) was obtained from a published study 56. For the expression of shRNA, oligonucleotides containing target sequence to CHC were synthesized and duplex oligo DNA was inserted into the pSUPER vector (Oligoengine, USA). This plasmid DNA was cotransfected with pcDNA3.1-FcαR in Hela cells. At 72 h posttransfection, the efficiency of CHC depletion and its influence on FcαR endocytosis were examined by confocal microscopy. To knockdown CHC in U937 cells, a lentiviral shRNA vector pLVTHM 57 was used. pLVTHM expresses GFP and shRNA simultaneously, therefore infected cells can be easily detected by fluorescent microscopy and also can be sorted by FACS. The same target sequence of CHC was cloned in pLVTHM. pLVTHM or pLVTHM-shRNA CHC was cotransfected with packaging vector psPAX2 and envelope vector pMD2.G into 293T cells. Then U937 cells were infected with supernatants from packing cells supplemented with 8 μg/ml Polybrene. After 3 infections, cells with high GFP expression level were sorted by FACS. The expression of CHC in GFP-positive U937 cells was determined by western blot and fluorescent staining.
Western blot
To validate FcαR stably expressed in CHO cells, FcαR from 5×106 cells was purified with anti-FcαR mAb-coupled beads as described previously 50, separated by 10% SDS-PAGE and transferred onto a nitrocellulose membrane. After blocking with 5% defatted milk for 1 h at room temperature, membranes were incubated with rabbit anti-FcαR polyclonal antibody (1: 500) overnight at 4 °C. Then, membranes were washed with PBS containing 0.05% Tween-20 and incubated with HRP-conjugated goat anti-rabbit IgG (1: 3 000) at room temperature for 1 h. Then membranes were visualized by SuperSignal West Pico chemiluminescent substrate (Pierce, USA). In RNA interference experiment, depletion of CHC was also examined by western blot. Briefly, 72 h posttransfection, equal number of pSUPER or pSUPER-shRNA CHC-transfected cells was lysed with loading buffer and separated by 5%-18% SDS-PAGE, then proteins were transferred onto a nitrocellulose membrane. Mouse monoclonal anti-CHC antibody (TD-1) was used to detect CHC (1: 200).
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
References
Conley ME, Delacroix DL . Intravascular and mucosal immunoglobulin A: two separate but related systems of immune defense? Ann Intern Med 1987; 106:892–899.
Mestecky J, Russell MW . Mucosal immunoglobulins and their contribution to defence mechanisms: an overview. Biochem Soc Trans 1997; 25:457–462.
Hellwig SM, van Spriel AB, Schellekens JF, et al. Immunoglobulin A-mediated protection against Bordetella pertussis infection. Infect Immun 2001; 69:4846–4850.
van Egmond M, van Garderen E, van Spriel AB, Damen CA et al. FcαRI-positive liver Kupffer cells: reappraisal of the function of immunoglobulin A in immunity. Nat Med 2000; 6:680–685.
Monteiro RC, van de Winkel JG . IgA Fc receptors. Annu Rev Immunol 2003; 21:177–204.
Ravetch JV, Bolland S . IgG Fc receptors. Annu Rev Immunol 2001; 19:275–290.
Stewart WW, Mazengera RL, Shen L, et al. Unaggregated serum IgA binds to neutrophil FcαR at physiological concentrations and is endocytosed but cross-linking is necessary to elicit a respiratory burst. J Leukoc Biol 1994; 56:481–487.
Morton HC, van den Herik-Oudijk IE, Vossebeld P, et al. Functional association between the human myeloid immunoglobulin A Fc receptor (CD89) and FcR γ chain. J Biol Chem 1995; 270:29781–29787.
Lang ML, Shen L, Wade WF . γ chain dependent recruitment of tyrosine kinases to membrane rafts by the human IgA receptor FcαR. J Immunol 1999; 163:5391–5398.
Lang ML, Chen YW, Shen L, et al. IgA Fc receptor (FcαR) cross-linking recruits tyrosine kinases, phosphoinositide kinases and serine/threonine kinases to glycolipid rafts. Biochem J 2002; 364:517–525.
Amigorena S, Salamero J, Davoust J, et al. Tyrosine-containing motif that transduces cell activation signals also determines internalization and antigen presentation via type III receptors for IgG. Nature 1992; 358:337–341.
Launay P, Patry C, Lehuen A, et al. Alternative endocytic pathway for immunoglobulin A Fc receptors (CD89) depends on the lack of FcR γ association and protects against degradation of bound ligand. J Biol Chem 1999; 274:7216–7225.
Shen L, van Egmond M, Siemasko K, et al. Presentation of ovalbumin internalized via the immunoglobulin-A Fc receptor is enhanced through Fc receptor γ chain signaling. Blood 2001; 97:205–213.
Conner SD, Schmid SL . Regulated portals of entry into the cell. Nature 2003; 422:37–44.
Parton RG, Richards AA . Lipid rafts and caveolae as portals for endocytosis: new insights and common mechanisms. Traffic 2003; 4:724–738.
Kirkham M, Parton RG . Clathrin-independent endocytosis: new insights into caveolae and non-caveolar lipid raft carriers. Biochim Biophy Acta 2005; 1746:349–363.
Hill E, van Der Kaay J, Downes CP, et al. The role of dynamin and its binding partners in coated pit invagination and scission. J Cell Biol 2001; 152:309–323.
Sever S, Damke H, Schmid SL . Dynamin: GTP controls the formation of constricted coated pits, the rate limiting step in clathrin-mediated endocytosis. J Cell Biol 2000; 150:1137–1148.
Damke H, Baba T, Warnock DE, et al. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J Cell Biol 1994; 127:915–934.
Mayor S, Pagano RE . Pathways of clathrin-independent endocytosis. Nat Rev Mol Cell Biol 2007; 8:603–612.
Fattakhova G, Masilamani M, Borrego F, et al. The high-affinity immunoglobulin-E receptor (FcɛRI) is endocytosed by an AP-2/clathrin-independent, dynamin-dependent mechanism. Traffic 2006; 7:673–685.
Somsel Rodman J, Wandinger-Ness A . Rab GTPases coordinate endocytosis. J Cell Sci 2000; 113:183–192.
Mohrmann K, van der Sluijs P . Regulation of membrane transport through the endocytic pathway by rab GTPases. Mol Membr Biol 1999; 16:81–87.
Nielsen E, Severin F, Backer JM, et al. Rab5 regulates motility of early endosomes on microtubules. Nat Cell Biol 1999; 1:376–382.
Simonsen A, Lippé R, Christoforidis S, et al. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature 1998; 394:494–498.
Chen W, Feng Y, Chen D, et al. Rab11 is required for trans-Golgi network-toplasma membrane transport and a preferential target for GDP dissociation inhibitor. Mol Biol Cell 1998; 9:3241–3257.
Hamelin E, Thériault C, Laroche G, et al. The intracellular trafficking of the G protein-coupled receptor TPbeta depends on a direct interaction with Rab11. J Biol Chem 2005; 280:36195–36205.
Moore RH, Millman EE, Alpizar-Foster E, et al. Rab11 regulates the recycling and lysosome targeting of beta2-adrenergic receptors. J Cell Sci 2004; 117:3107–3117.
Feng Y, Press B, Wandinger-Ness A . Rab 7: an important regulator of late endocytic membrane traffic. J Cell Biol 1995; 131:1435–1452.
Bucci C, Thomsen P, Nicoziani P, et al. Rab7: a key to lysosome biogenesis. Mol Biol Cell 2000; 11:467–480.
Lombardi D, Soldati T, Riederer MA, et al. Rab9 functions in transport between late endosomes and the trans-Golgi network. EMBO J 1993; 12:677–682.
Barbero P, Bittova L, Pfeffer SR . Visualization of Rab9-mediated vesicle transport from endosomes to the trans-Golgi in living cells. J Cell Biol 2002; 156:511–518.
Seachrist JL, Ferguson SS . Regulation of G protein-coupled receptor endocytosis and trafficking by Rab GTPases. Life Sci 2003; 74:225–235.
Heuser JE, Anderson RG . Hypertonic media inhibit receptor-mediated endocytosis by blocking clathrin-coated pit formation. J Cell Biol 1989; 108:389–400.
Phonphok Y, Rosenthal KS . Stabilization of clathrin coated vesicles by amantadine, tromantadine and other hydrophobic amines. FEBS Lett 1991; 281:188–190.
Altankov G, Grinnell F . Depletion of intracellular potassium disrupts coated pits and reversibly inhibits cell polarization during fibroblast spreading. J Cell Biol 1993; 120:1449–1459.
Ros-Baro A, Lopez-Iglesias C, Peiro S, et al. Lipid rafts are required for GLUT4 internalization in adipose cells. Proc Natl Acad Sci USA 2001; 98:12050–12055.
Orlandi PA, Fishman PH . Filipin-dependent inhibition of cholera toxin: evidence for toxin internalization and activation through caveolae-like domains. J Cell Biol 1998; 141:905–915.
Singh RD, Puri V, Valiyaveettil JT, et al. Selective caveolin-1-dependent endocytosis of glycosphingolipids. Mol Biol Cell 2003; 14:3254–3265.
Minshall RD, Tiruppathi C, Vogel SM, et al. Endothelial cell-surface gp60 activates vesicle formation and trafficking via G(i)-coupled Src kinase signaling pathway. J Cell Biol 2000; 150:1057–1070.
Sharma DK, Brown JC, Choudhury A, et al. Selective stimulation of caveolar endocytosis by glycosphingolipids and cholesterol. Mol Biol Cell 2004; 15:3114–3122.
Damm EM, Pelkmans L, Kartenbeck J, et al. Clathrin- and caveolin-1-independent endocytosis: entry of simian virus 40 into cells devoid of caveolae. J Cell Biol 2005; 168:477–488.
Benmerah A, Bayrou M, Cerf-Bensussan N, et al. Inhibition of clathrin-coated pit assembly by an Eps15 mutant. J Cell Sci 1999; 112:1303–1311.
Bonifacino JS, Traub LM . Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem 2003; 72:395–447.
Davis W, Harrison PT, Hutchinson MJ, et al. Two distinct regions of FcγRI initiate separate signalling pathways involved in endocytosis and Phagocytosis. EMBO J 1995; 14:432–441.
Mero P, Zhang CY, Huang ZY, et al. Phosphorylation-independent ubiquitylation and endocytosis of FcγRIIA. J Biol Chem 2006; 281:33242–33249.
Dinneen JL, Ceresa BP . Expression of dominant negative rab5 in HeLa cells regulates endocytic trafficking distal from the plasma membrane. Exp Cell Res 2004; 294(2):509–522.
Seachrist JL, Laporte SA, Dale LB, et al. Rab5 association with the angiotensin II type 1A receptor promotes Rab5 GTP binding and vesicular fusion. J Biol Chem 2002; 277:679–685.
Leser GP, Ector KJ, Ng DT, et al. The signal for clathrin-mediated endocytosis of the paramyxovirus SV5 HN protein resides at the transmembrane domain-ectodomain boundary region. Virology 1999; 262:79–92.
Guo Y, Smith K, Lee J, et al. Identification of methionine-rich clusters that regulate copper-stimulated endocytosis of the human Ctr1 copper transporter. J Biol Chem 2004; 279:17428–17433.
Patry C, Sibille Y, Lehuen A, et al. Identification of Fcα receptor (CD89) isoforms generated by alternative splicing that are differentially expressed between blood monocytes and alveolar macrophages. J Immunol 1996; 156:4442–4448.
Zhang W, Bi B, Oldroyd RG, et al. Neutrophil lactoferrin release induced by IgA immune complexes differed from that induced cross-linking of fcalpha receptors (FcαR) with a monoclonal antibody, MIP8a. Clin Exp Immunol 2000; 121:106–111.
Yin N, Peng M, Xing Y, et al. Intracellular pools of FcαR (CD89) in human neutrophils are localized in tertiary granules and secretory vesicles, and two FcαR isoforms are found in tertiary granules. J Leukoc Biol 2007; 82:551–558.
Brüggemann M, Williams GT, Bindon CI, et al. Comparison of the effector functions of human immunoglobulins using a matched set of chimeric antibodies. J Exp Med 1987;166:1351–1361.
Choudhury A, Dominguez M, Puri V, et al. Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. J Clin Invest 2002; 109:1541–1550.
Enari M, Ohmori K, Kitabayashi I, et al. Requirement of clathrin heavy chain for p53-mediated transcription. Genes Dev 2006; 20:1087–1099.
Wiznerowicz M, Trono D . Conditional suppression of cellular genes: lentivirus vector-mediated drug-inducible RNA interference. J Virol 2003; 77:8957–8961.
Acknowledgements
This research was supported by grants from the National Natural Science Foundation of China (30170878 and 30571693). The authors thank Jane Brill for her language review of this manuscript. We greatly appreciate the gift of EGFP-tagged Rab7, Rab9 and Rab11 from Dr Richard E Pagano, EGFP-tagged Rab5 from Dr Feng Du, EGFP-tagged Eps15 from Dr Alexandre Benmerah and HA-tagged dynamin from Dr Sandra Schmid. We also thank Dr Didier Trono for providing the lentiviral shRNA vector pLVTHM.
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary information, Figure S1
Time course of colocalization of internalized FcαR with Rab5 and Rab11. (PDF 1493 kb)
Supplementary information, Figure S2
Internalized FcαR were not colocalized with Rab7 and Rab9. (PDF 1232 kb)
Rights and permissions
About this article
Cite this article
Peng, M., Yin, N. & Zhang, W. Endocytosis of FcαR is clathrin and dynamin dependent, but its cytoplasmic domain is not required. Cell Res 20, 223–237 (2010). https://doi.org/10.1038/cr.2009.120
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2009.120
