
Abstract
A characteristic cellular feature of the mammalian liver is the progressive polyploidization of the hepatocytes, where individual cells acquire more than two sets of chromosomes. Polyploidization results from cytokinesis failure that takes place progressively during the course of postnatal development. The proportion of polyploidy also increases with the aging process or with cellular stress such as surgical resection, toxic stimulation, metabolic overload, or oxidative damage, to involve as much as 90% of the hepatocytes in mice and 40% in humans. Hepatocyte polyploidization is generally considered an indicator of terminal differentiation and cellular senescence, and related to the dysfunction of insulin and p53/p21 signaling pathways. Interestingly, the high prevalence of hepatocyte polyploidization in the aged mouse liver can be reversed when the senescent hepatocytes are serially transplanted into young mouse livers. Here we review the current knowledge on the mechanism of hepatocytes polyploidization during postnatal growth, aging, and liver diseases. The biologic significance of polyploidization in senescent reversal, within the context of new ways to think of liver aging and liver diseases is considered.
Similar content being viewed by others


Facts
-
The hepatocyte is a main contributor to liver functions, including in metabolic homeostasis, synthesis, storage, distribution, and detoxification of xenobiotic compounds.
-
Hepatocyte polyploidization is a characteristic feature of mature mammalian hepatocytes and an indicator of hepatocyte senescence.
-
Waves of hepatocyte polyploidization occur successively during postnatal development and again upon advanced ageing.
-
The frequency and extent of hepatic polyploidy are increased in liver injury by partial hepatectomy, radiation or oxidative stress, but are reduced in hepatocellular carcinoma.
-
The senescent mouse hepatocyte is rejuvenated and exhibits polyploidy reversal upon immersion into the microenvironment of a young liver.
Open questions
-
What is the significance of the waves of hepatocyte polyploidization in normal development and ageing?
-
What are the roles of altered hepatocyte polyploidization status with different pathologic conditions?
-
What is the causal relationship between polyploidy reversal and the rejuvenation of senescent hepatocytes?
The liver is the largest solid organ endowed with unique regenerative capacity in the mammalian body, and accounts for about 2% of the total body weight in humans and 5% in mice. It is an essential organ with multiple complex functions in metabolic homeostasis, synthesis, and distribution of nutrients and serum proteins, metabolism and storage of amino acids, vitamins, lipids, and carbohydrates, and detoxification of xenobiotic compounds.1, 2 Most of these functions are carried out primarily by hepatocytes that account for 70% of all liver cells.3 Hepatocytes have unique functions depending on their location within the liver lobule.4 The peripotal hepatocytes mediate amino acid and energy metabolism, lipid oxidation, and gluconeogenesis. In contrast, glycolysis, lipogenesis, and glutamine synthase are mediated by pericentral hepatocytes. Although hepatocytes are quiescent in the adult and turnover slowly with a mean life span of 200–300 days,5 they are able to proliferate rapidly in response to liver injury.
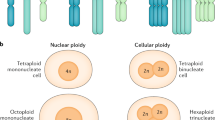
A characteristic feature of hepatocytes is polyploidy, which is an increase in the numbers of chromosome sets per cell. Liver polyploidy occurs during postnatal development as an age-dependent process, as well as during repair in response to injury from various stress stimuli. Polyploidization in the liver has been studied widely and summarized in excellent reviews.6, 7 Hepatocyte ploidy depends on the DNA content of each nucleus (for example, diploid, tetraploid, octoploid, and so on) plus the number of nuclei per cell.8 For example, polyploid hepatocytes can be tetraploid (for example, binucleate with two diploid nuclei or mononucleate with a single tetraploid nucleus), or octoploid (for example, binucleate with two tetraploid nuclei or mononucleate with a single octoploid nucleus). Enlarged cell size is the most obvious consequence of liver polyploidization. Different studies have reported that the volume of hepatocytes in human and mouse livers is approximately doubled with the doubling of DNA content.9 However, there is no significant difference in the volume of binucleated tetraploid (2 × 2n) and that of mononucleated tetraploid (4n) hepatocytes or between binucleated (2 × 4n) and mononucleated (8n) octoploid hepatocytes. Although polyploid hepatocytes were documented over a century ago, the physiologic significance of polyploidy status in liver homeostasis, regeneration, and disease is poorly understood. In this review, we will explore the possible mechanism underlying liver ployploidization, and discuss how polyploidization is regulated during liver physiological development, and its roles in aging, pathological processes, and senescent reversal.
Hepatocyte Polyploidization During the Growth and Development of the Postnatal Liver
All hepatocytes are diploid in the newborn liver. During postnatal growth, the liver parenchyma undergoes dramatic changes with gradual polyploidization, with the emergence of hepatocytes of several ploid classes.8, 10 Polyploidization begins from the first 3 weeks postnatal, generating binucleated tetraploid (2 × 2n) or mononucleated tetraploid (4n) hepatocytes. Octoploid (binucleated 2 × 4n and mononucleated 8n) hepatocytes begin to accumulate in significant numbers during the second and third months.8, 11 Hepatocyte polyploidization reaches a plateau upon animal maturity. The degree of liver polyploidization varies between mammals (Figure 1).12, 13, 14, 15, 16, 17, 18, 19, 20 80–90% of hepatocytes in adult C57BL mice are polyploid,13, 14, 15, 16 and 70–80% of adult rat hepatocytes are polyploid.17, 18 In the adult human liver, the percentage of polyploidy is more than 20%.19, 20

The distribution of ploidy classes in different mammals. The graphs show the percentage of diploid, tetraploid, and octoploid hepatocytes in newborn, adult, and aged human, mice and rat. Adult mice and rats were 8–12 weeks old; adult human were 20–40 years old. Aged mice were over 18 months, and aged human were over 60 years old. The data were compiled from published studies (Duncan AW, et al.;13 Wang MJ, et al.;14 Chipchase MD, et al.;15 Pandit SK, et al.;16 Margall-Ducos G, et al.;17 Gandillet A, et al.;18 Kudryavtsev BN, et al.;19 Duncan AW, et al.20)
Cytokinesis failure is the main mechanism of polyploidization during postnatal liver development (Figure 2). In successful cytokinesis, a diploid hepatocyte gives rise to two diploid hepatocytes, but an incomplete cytokinesis generates one tetraploid hepatocyte with two diploid nuclei.8, 17 The binucleated tetraploid hepatocyte retains the capability to undergo future DNA replication with successful cyrokinesis, generating two mononucleated tetraploid cells. However, if cytokinesis fails again during mitosis of a mononucleated tetraploid cell, a binucleated octoploid hepatocyte results. In this way, progressive polyploidization with the formation of one or two nucleated tetraploid and octoploid cells appears successively in the liver parenchyma. Cytokinesis failure is reported to be regulated by insulin signaling that may act via the phosphoinositide 3-kinase (PI3K)-protein kinase B (Akt)-cytoskeleton regulation pathway.11, 21, 22 These studies demonstrate that reduced circulating insulin levels resulted in the decreased generation of binucleated tetraploid hepatocytes, while elevated insulin levels increased the formation of binucleated tetraploid hepatocytes. Inhibition of PI3K/Akt phosphorylation blocked these failed cytokinetic event, and complete cytokinesis occurs followed by actin cytoskeleton polarization, cytoskeleton reorganization, and RhoA recruitment.

Polyploidization during postnatal liver growth. From the postnatal growth, a diploid hepatocyte can give rise to two diploid hepatocytes with successful cytokinesis or follows with cytokinesis failure and generates a tetraploid hepatocyte with two diploid nuclei. The binucleated tetraploid hepatocyte then follows a new round cell cycle, generating two mononucleated tetraploids or one binucleated octoploidy. In the adult liver parenchyma, it consists of diploid, tetraploid and octoploid hepatocytes. Insulin signaling, E2F transcription factors including E2F8 and E2F1, and miR-122 have been reported to regulate cytokinesis failure and be crucial for liver polyploidization during postnatal development
A number of other factors in addition to the insulin-signaling pathway may contribute also to liver polyploidization (Table 1). E2F transcription factors have been implicated as crucial for liver polyploidization during postnatal development.16, 23, 24 E2F8 deficiency induces the expression of E2F target genes, promoting cytokinesis and preventing liver polyploidization. In contrast, E2F1 deficiency inhibits cytokinesis and enhances liver polyploidization. In mouse models, silencing of cell-cycle-regulated factors CDK1, Skp2, Ccne2, p21, p53, pRb, survivin, Ssu72, and nucleotide excision repair gene ERCC1 led to enhanced liver polyploidization, but silence of Ccne1 repressed liver polyploidy.15, 25, 26, 27, 28, 29, 30, 31, 32, 33 Importantly, a recent study concluded that miR-122 is not only necessary but also sufficient for hepatic polyploidization.34 Downregulating miR-122 expression decreases polyploid hepatocytes, and this trend is reversible by miR-122 over-expression. MiR-122 antagonizes the expression of the pro-cytokinesis effectors Cux1, Rhoa, Mapre1, Iqgap1, Nedd4l, and Slc25a34, leading to cytokinesis failure and expansion of binucleated hepatocytes.
Hepatocyte Polyploidization in the Aged Liver and its Reversal Phenomenon
A second wave of high polyploidization occurs as an aging-dependent process (Figure 1).14, 15, 19 Most hepatocytes are diploid in young individuals,20 where the relative number of polyploidy cells generally does not exceed 15% in 20-year-old adult humans.19 However, the fraction of polyploid hepatocytes increases to 42% in the liver of an 80-year-old adult.19 Our previous study,14 corroborating this aging-related trend in the mouse, documented that the percentage of octoploid hepatocytes in mouse liver increases from 16.8±5.1% at 2 months to 34.06±1.8% at 18 months.
Ageing-related hepatocyte polyploidy has been regarded as a manifestation of hepatic cellular senescence. The liver regenerative capacity after a 70% hepatectomy is decreased in older animals with greater proportions of polyploid hepatocytes.35, 36 It has been shown previously that hepatocytes are formed adjacent to the portal zone and stream toward the terminal hepatic vein as they age.37 The DNA content of hepatocytes also increases with their age.37, 38 As reported in our previous study, diploid, tetraploid, and octoploid hepatocytes from 2-month mice rarely expressed senescent markers, including p16ink4α, p21 and p53, and have no significant difference in liver repopulation after transplanting into the recipient livers. However, the expression of p16ink4α, p21 and p53 in diploidy, tetraploidy, and octoploidy from the 18-month mice were higher than from the 2-month mice. Moreover, the expression levels of p16ink4a, p21, and p53 in the 18-month mice were significantly higher in the octoploid than in the diploid and tetraploid hepatocytes. Importantly, after transplantation, the proliferative capacity and liver repopulation of octoploid hepatocytes were lower than diploid and tetraploid donor hepatocytes.14 Similar to in vivo hepatocytes, cultured primary hepatocytes from the 18-month-old mice contained more SA-β-gal positive cells and fewer bromodeoxyuridine (BrdU)-positive cells as compared with the 2-month mice.14 Our study correlated increased polyploid hepatocytes in older mice with senescence, suggesting that polyploidy may induce senescence-type changes during aging and possible association with liver disease.
The p16ink4α and p53-p21 pathways regulating hepatocyte senescence may be a mechanism of polyploidization during aging. The level of p16ink4α was also increased in fibroblasts as they approached senescence, accompanied with formation of polyploidy.39 The cyclin-denpendent kinase (CDK) inhibitor p21, which is regulated by p53 at the transcriptional level, has been reported to affect S-phase progression, G2/M arrest, and polyploidization.26, 40, 41 As shown in our previous study,14 the expression levels of p16ink4α, p21, and p53 were higher in octoploid hepatocytes of 18-month age mice, and the levels were lowered with rejuvenation and ploidy reversal.
Consistent with our report, increased polyploidy after two-thirds partial hepatectomy in rats or mice exhibit senescence-type changes with increased β-galactosidase activity and accumulation of p21.22, 36 Moreover, with increased senescent polyploidy, the proliferative capacity of partial hepatectomy-induced hepatocytes was significantly attenuated compared to hepatocytes from normal rats.36 However, ploidy reversal with multipolar mitosis exists during hepatocyte proliferation.13 As shown in our previous study,14 repopulated hepatocytes collected from 10-week post-transplantation of 18-month donor cells by flow cytometry and found that the ratio of diploid, tetraploid, and octoploid cells became similar to that seen in the liver of 2-month mice. The percentage of octoploid hepatocytes decreased from 34.06±1.8% before transplantation to 17.4±0.96%, while the percentage of diploid hepatocytes increased from 13.6±0.66% to 21.96±1.78%. Furthermore, isolated octoploid hepatocytes from 18-month-old mice would completely repopulate in young Fah−/− recipients at 10 weeks post-transplantation. Analysis of repopulated hepatocytes showed that pure octoploid cells produced daughter cells with 2c and 4c DNA content. And mitotic structures with multipolar spindles or tripolar division were detected during hepatocyte proliferation in vivo. In addition, F-actin and Hoechst 33342 co-staining in the repopulated livers indeed revealed the existence of ploidy reversal with reduction in DNA content after proliferation. Accompanied by polyploidy reversal, senescent polyploid hepatocytes became rejuvenated as represented by increased proliferative capacity, decreased β-galactosidase activity and expression of p16ink4α, p21, and p53 (Figure 3). According to these data, ploidy reversal with rejuvenation of proliferative capacity may provide a potential clue to develop promising therapeutic strategies for age-dependent diseases.

Senescent polyploid hepatocytes are rejuvenated accompanied by ploidy converyor. In the aged liver, senescent polyploid hepatocytes reenter cell cycles after transplantation. These proliferating tetraploid or octoploid hepatocytes can generate mononucleated diploid and tetraploid hepatocytes, as well as binucleated tetraploidy with triploar or double mitosis. Accompanied with senescent hepatocytes reversal, the percentage of octoploid hepatocytes decreases while the percentage of diploid hepatocytes increases. The p16ink4α and p53-p21 pathway regulating hepatocyte senescence may be a mechanism of polyploidization during aging
Hepatocyte Polyploidization and Liver Dysfunction
Differentiated hepatocytes retain a high proliferative capacity. Upon tissue injury such as partial hepatectomy (PHx), toxin, and drug-induced liver disease, the quiescent hepatocytes will reenter into the cell cycle and contribute to the recovery of the injured liver. Mononucleated and binucleated polyploid hepatocytes accumulated during two-thirds partial hepatectomy-induced liver regeneration.42 Although all hepatocytes entered into the cell cycle after 70% partial hepatectomy, only a few continue through the M phase. Hepatic polyploidy can also be modified by metabolic overload, DNA damage, and chemical-induced liver injury. It was reported that in the long-evans cinnamon (LEC) rat, an animal model of Wilson’s Disease, with copper and iron overloads in the liver, hepatocytes had increased polyploidy with delayed mitotic progression.43 Furthermore, polyploidy was further increased following exposure to radiation or oxidative stress.44 In contrast, hepatocellular carcinoma was shown to have a lower overall polyploidy compared to the normal liver.45, 46 Diploid mononucleated hepatocytes increased in frequency with the decrease of polyploidy hepatocytes in both rat models induced by chemical carcinogens (diethylnitrosamine (DEN) and 2-acetyl-aminofluorene (2-AAF)) and human hepatocellular carcinoma (HCC).45, 46 Reduced ploidy in HCC may reflect an inverse correlation between growth capacity and ploidy, since it has been reported that HCC’s hepatocytes proliferate as diploid cells.12 In addition, diploid cells are less protected against mutagenic change than polyploid cells. Thus, rapidly dividing diploid tumor cells are more easily mutated than the polyploid cells, resulting in increasing malignancy.47, 48 HCC development is accompanied by decreased expression of miR-122. MiR-122, frequently the most specific miRNA in the liver, is considered necessary and sufficient for liver polyploidization, in addition to being an important tumor suppressor in hepatocellular carcinoma.49, 50, 51 Thus, the data implicate decreased miR-122 level as key to suppressed polyploidization in mouse or rat models of DEN-induced HCC and human HCC. Additional factors, such as survivin, may also play a role. Survivin is overexpressed in human tumor and survivin deficiency induces polyploidy and cell cycle arrest in hepatocellular carcinoma cells,52 which suggest deletion of survivin promotes liver polyploidization. Additional studies have demonstrated increased polyploidization in cancer cells treated with mitotic kinase inhibitors or spindle inhibitor nocodazole. Intriguingly, polyploid cancer cells are more sensitive to genotoxic stress and anticancer agents.53, 54, 55 Together, these observations suggest that manipulation of polyploidization may be an important strategy to induce cell death in cancer therapies.
Recently, Gentric et al. examined hepatic ploidy in a nonalcoholic fatty liver disease (NAFLD) model of ob/ob mice and wild-type (WT) mice fed with methionine-choline-deficient diet (MCD) or high-fat diet (HFD), and found a large proportion of highly polyploid hepatocytes in the parenchyma of the fatty liver.56 Most importantly, a similar phenotype was also observed in patients with nonalcoholic steatohepatitis. Hepatocytes of NAFLD in primary cultures progressed through G1 and entered S-phase, but they had delayed exit from S phase and accumulated/arrested in G2, suggesting that endoreplication might be a mechanism for polyploidy (Figure 4). Moreover, the NAFLD hepatocytes in S-phase and G2 displayed a high level of oxidative stress accompanied by robust phosphorylation of ATR (an indicator of DNA damage response) and markers for cell-cycle arrest (phosphorylation of p53 and increased p21 expression). Together, these observations indicate that pathological polyploidization is promoted with oxidative stress by activating the ATR/p53/p21 signaling axis.56, 57, 58

Pathological polyploidization in the liver. In the pathological liver, hepatocytes progress through G1 and enter S-phase, but fail with cell mitosis, leading to formation of mononucleated polyploid hepatocytes. The G2/M arrest is controlled by p53/p21 signaling pathway
Biological Significance of Hepatocyte Polyploidization in the Liver
The biological significance of liver polyploidization remains enigmatic, but a number of hypotheses have been considered. One hypothesis suggests that hepatic polyploidy is associated with hepatocyte maturity and terminal differentiation.7, 18, 35, 59, 60 On the basis of the blood vessel location and blood flow direction, the liver lobule can be subdivided into an upstream ‘periportal’ and a downstream ‘pericentral’ zone. Hepatocytes are believed to mature as they transit, or ‘stream’ from the periportal to pericentral region.35, 59, 60 A number of studies have indicated that diploid hepatocytes are located in the periportal region, while downstream, and older pericentral hepatocytes exhibit greater ploidy. However, a more recent study failed to support these observations, and it indicates that diploidy and polyploidy are distributed randomly throughout the hepatic lobule.17 Importantly, polyploid hepatocytes are likely not the terminal form of differentiation, since there was no significant difference in regenerative capacity between the liver of E2f8−/− mice with predominant diploid hepatocytes and the adult wild-type liver.16 Competitive repopulation of co-transplanted diploid and octoploid hepatocytes isolated from 8-week-old mice into fumarylacetoacetate hydrolase (Fah)-deficient mice shows that the proliferative capacity was similar between the ploidy classes.13 In addition, isolated diploid, tetraploid, and octoploid hepatocytes that were separately transplanted all could complete repopulation with similar proliferative capacity.14 The data indicate that polyploidization in the young normal liver is not an accurate hallmark for terminal differentiation and has no obvious impact on proliferation in physiological process. However, in the aging or diseased liver, there appears an association of senescence with polyploidization. In our study, we observed that polyploid hepatocytes from old mice had less proliferative capacity and also expressed senescent markers.14 During liver regeneration after partial hepatectomy, polyploidy increased with senescence-type changes and the rate of proliferation of polyploid hepatocytes was lower than that of diploid hepatocytes.36
A second hypothesis suggests that liver ploidy may serve to enhance hepatocyte function. The liver participates in a wide array of activities related to protein synthesis/secretion, metabolism, and detoxification. Polyploidy may allow two- or four-fold increased expression of some genes/proteins and thereby enhance particular metabolic functions. However, comparison of gene expression profiles of isolated diploids, tetraploids and octoploids by microarray analysis showed that only 50 candidate genes from a wide range of different biological processes were differently expressed.61 Whether or not liver polyploidization enhances, hepatocytes function remains debatable. However, a recent study used comparative genome-scale analysis to demonstrate that polyploidy boost tissue-specific functions. Polyploidy-activated genes are present in all essential liver-specific functions, including nitrogenous metabolism, blood protein synthesis, redox state maintance, xenobiotic metabolism, and immunity. Moreover, the study revealed a tendency of the polyploidy liver to an increased anaerobic energy production and obtained ATP from carbohydrates rather than from fatty acids, suggesting polyploidy is associated with the switch of liver-specific functions to economy saving mode, instead of investing it into cell division.62, 63
A third hypothesis suggests that polyploidy confers protection of hepatocytes against oxidative stress and genotoxic damage. Acquiring multiple sets of chromosomes could function to buffer against gene-inactivating mutations by DNA damaging agents. For instance, early tumor lesions in the liver are characterized by the increase of diploid cells, which are less protected against mutations than polyploid liver cells.64 Notably, genome-scale comparison of diploid and polyploid hepatocytes indicates that polyploidy induces genes fighting against pathogens, DNA lesions and oxidative stress, and inhibits genes promoting apoptosis.62, 63 In addition, the progressive polyploidization observed during the liver aging process, implying polyploidization allow the liver to adapt to aging-related cellular loss or may be a protective response to the accumulation of damaged DNA. Under pathological condition, the injury liver with loss of liver function may try to compensate the loss of its mass by polyploidization.
Conclusion
Polyploid cells, occurring with aging, oxidative stress, and DNA damage, are frequently found in various mammalian organs and tissues, including the skeletal muscle, heart, placenta, brain, liver, and blood cells. In the liver, the first wave of polyploidy is seen at postnatal growth, and the second wave of ploidy elevation is observed at senescence. Many previous studies have indicated that polyploid cells can proliferate as well as diploid hepatocytes in the developmental liver, suggesting that polyploidization is not required for hepatocyte differentiation. Polyploid hepatocytes become senescent and decrease their proliferative capacity with aging and pathological process, but senescent polyploid hepatocytes can rejuvenate with ploidy reversal. The molecular cues controlling polyploidization and reversal remain unknown, and a better understanding of ploidy reversal is needed to approach clinical research. Under pathological conditions, liver polyploidization mainly indicates the severity of the damage: the higher the observed polyploidization rate, the greater the injury that has occurred. However, increase of diploid cells is a main characteristic feature in hepatocellular carcinoma, owing to their heightened proliferative capacity and their susceptibility to further mutations. Induced polyploidization could be an underlying strategy for cancer therapies. Finally, further work is also needed to unravel what diseases are associated with pathological polyploidization, how polyploidy is regulate during pathological progression, and whether these new insights will provide more clues for therapeutic strategies in liver diseases.
References
Duncan AW, Dorrell C, Grompe M . Stem cells and liver regeneration. Gastroenterology 2009; 137: 466–481.
Michalopoulos GK, DeFrances MC . Liver regeneration. Science 1997; 276: 60–66.
Si-Tayeb K, Lemaigre FP, Duncan SA . Organogenesis and development of the liver. Dev Cell 2010; 18: 175–189.
Jungermann K, Kietzmann T . Zonation of parenchymal and nonparenchymal metabolism in liver. Annu Rev Nutr 1996; 16: 179–203.
Bucher NLR, Malt RA Regeneration of Liver and Kidney. Little, Brown: Boston, MA, USA, 1971.
Gentric G, Celton-Morizur S, Desdouets C . Polyploidy and liver proliferation. Clin Res Hepatol Gastroenterol 2012; 36: 29–34.
Gupta S . Hepatic polyploidy and liver growth control. Semin Cancer Biol 2000; 10: 161–171.
Guidotti JE, Bregerie O, Robert A, Debey P, Brechot C, Desdouets C . Liver cell polyploidization: a pivotal role for binuclear hepatocytes. J Biol Chem 2003; 278: 19095–19101.
Martin NC, McCullough CT, Bush PG, Sharp L, Hall AC, Harrison DJ . Functional analysis of mouse hepatocytes differing in DNA content: volume, receptor expression and effect of IFN gamma. J Cell Physiol 2002; 191: 138–144.
Epstein CJ . Cell size, nuclear content and the development of polyploidy in the mammalian liver. Proc Natl Acad Sci USA 1967; 57: 327–334.
Celton-Morizur S, Merlen G, Couton D, Desdouets C . Polyploidy and liver proliferation: central role of insulin signaling. Cell Cycle 2010; 9: 460–466.
Celton-Morizur S, Desdouets C . Polyploidization of liver cells. Adv Exp Med Biol 2010; 676: 123–135.
Duncan AW, Taylor MH, Hickey RD, Hanlon Newell AE, Lenzi ML, Olson SB et al. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature 2010; 467: 707–710.
Wang MJ, Chen F, Li JX, Liu CC, ZhangHB, Xia Y et al. Reversal of hepatocytes senescence after continuous in vivo cell proliferation. Hepatology 2014; 60: 349–361.
Chipchase MD, O'Neill M, Melton DW . Characterization of premature liver polyploidy in DNA repair (Ercc1)-deficient mice. Hepatology 2003; 38: 958–966.
Pandit SK, Westendorp B, Nantasanti S, van Liere E, Tooten PC, Cornelissen PW et al. E2F8 is essential for polyploidization in mammalian cells. Nat Cell Biol 2012; 14: 1181–1191.
Margall-Ducos G, Celton-Morizur S, Couton D, Brégerie O, Desdouets C . Liver tetraploidization is controlled by a new process of incomplete cytokinesis. J Cell Sci 2007; 120: 3633–3639.
Gandillet A, Alexandre E, Holl V, Royer C, Bischoff P, Cinqualbre J et al. Hepatocyte ploidy in normal young rat. Comp Biochem Physiol A Mol Integr Physiol 2003; 134: 665–673.
Kudryavtsev BN, Kudryavtseva MV, Sakuta GA, Stein GI . Human hepatocyte polyploidization kinetics in the course of life cycle. Virchows Arch B Cell Pathol Incl Mol Pathol 1993; 64: 387–393.
Duncan AW, Hanlon Newell AE, Smith L, Wilson EM, Olson SB, Thayer MJ et al. Frequent aneuploidy among normal human hepatocytes. Gastroenterology 2012; 142: 25–28.
Celton-Morizur S, Merlen G, Couton D, Margall-Ducos G, Desdouets C . The insulin/Akt pathway controls a specific cell division program that leads to generation of binucleated tetraploid liver cells in rodents. J Clin Invest 2009; 119: 1880–1887.
Celton-Morizur S, Desdouets C . Polyploidy and liver proliferation. Adv Exp Med Biol 2010; 676: 123–135.
Wong JV, Dong P, Nevins JR, Mathey-Prevot B, You L . Network calisthenics: control of E2F dynamics in cell cycle entry. Cell Cycle 2011; 10: 3086–3094.
Chen HZ, Ouseph MM, Li J, Pecot T, Chokshi V, Kent L et al. Canonical and atypical E2Fs regulate the mammalian endocycle. Nat Cell Biol 2012; 14: 1192–1202.
Nevzorova YA, Tschaharganeh D, Gassler N, Geng Y, Weiskirchen R, Sicinski P et al. Aberrant cell cycle progression and endoreplication in regenerating livers of mice that lack a single E-type cyclin. Gastroenterology 2009; 137: 691–703.
Sheahan S, Bellamy CO, Treanor L, Harrison DJ, Prost S . Additive effect of p53, p21 and Rb deletion in triple knockout primary hepatocytes. Oncogene 2004; 23: 1489–1497.
Kurinna S, Stratton SA, Coban Z, Schumacher JM, Grompe M, Duncan AW et al. p53 Regulates a mitotic transcription program and determines ploidy in normal mouse liver. Hepatology 2013; 57: 2004–2013.
Mayhew CN, Bosco EE, Fox SR, Okaya T, Tarapore P, Schwemberger SJ et al. Liver-specific pRB loss results in ectopic cell cycle entry and aberrant ploidy. Cancer Res 2005; 65: 4568–4577.
Diril MK, Ratnacaram CK, Padmakumar VC, Du T, Wasser M, Coppola V et al. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc Natl Acad Sci USA 2012; 109: 3826–3831.
Minamishima YA, Nakayama K . Recovery of liver mass without proliferation of hepatocytes after partial hepatectomy in Skp2-deficient mice. Cancer Res 2002; 62: 995–999.
Kim SH, Jeon Y, Kim HS, Lee JK, Lim HJ, Kang D et al. Hepatocyte homeostasis for chromosome ploidization and liver function is regulated by Ssu72protein phosphatase. Hepatology 2016; 63: 247–259.
Hagemann S, Wohlschlaeger J, Bertram S, Levkau B, Musacchio A, Conway EM et al. Loss of Survivin influences liver regeneration and is associated with impaired Aurora B function. Cell Death Differ 2013; 20: 834–844.
Li D, Cen J, Chen X, Conway EM, Ji Y, Hui L . Hepatic loss of survivin impairs postnatal liver development and promotes expansion of hepatic progenitor cells in mice. Hepatology 2013; 58: 2109–2121.
Hsu SH, Delgado ER, Otero PA, Teng KY, Kutay H, Meehan KM et al. MicroRNA-122 regulates polyploidization in the murine liver. Hepatology 2016; 64: 599–615.
Gerlyng P, Abyholm A, Grotmol T, Erikstein B, Huitfeldt HS, Stokke T et al. Binucleation and polyploidization patterns in developmental and regenerative rat liver growth. Cell Prolif 1993; 26: 557–565.
Sigal SH, Rajvanshi P, Gorla GR, Erikstein B, Huitfeldt HS, Stokke T et al. Partial hepatectomy-induced polyploidy attenuates hepatocyte replication and activates cell aging events. Am J Physiol 1999; 276: G1260–G1272.
Turner R, Lozoya O, Wang Y, Cardinale V, Gaudio E, Alpini G et al. Human hepatic stem cell and maturational liver lineage biology. Hepatology 2011; 53: 1035–1045.
Schwartz-Arad D, Zajicek G, Bartfeld E . The streaming liver IV: DNA content of the hepatocyte increases with its age. Liver 1989; 9: 93–99.
Zindy F, Quelle DE, Roussel MF, Sherr CJ . Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene 1997; 15: 203–211.
Mirzayans R, Andrais B, Scott A, Paterson MC, Murray D . Single-cell analysis of p16(INK4a) and p21(WAF1) expression suggests distinct mechanisms of senescence in normal human and Li-Fraumeni Syndrome fibroblasts. J Cell Physiol 2010; 223: 57–67.
Ullah Z, Lee CY, Depamphilis ML . Cip/Kip cyclin-dependent protein kinase inhibitors and the road to polyploidy. Cell Div 2009; 4: 10.
Miyaoka Y, Ebato K, Kato H, Arakawa S, Shimizu S, Miyajima A . Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Curr Biol 2012; 22: 1166–1175.
Yamada T, Sogawa K, Kim JK, Izumi K, Suzuki Y, Muramatsu Y et al. Increased polyploidy, delayed mitosis and reduced protein phosphatase-1 activity associated with excess copper in the Long Evans Cinnamon rat. Res Commun Mol Pathol Pharmacol 1998; 99: 283–304.
Gorla GR, Malhi H, Gupta S . Polyploidy associated with oxidative injury attenuates proliferative potential of cells. J Cell Sci 2001; 114: 2943–2951.
Schwarze PE, Saeter G, Armstrong D, Cameron RG, Laconi E, Sarma DS et al. Diploid growth pattern of hepatocellular tumours induced by various carcinogenesis treatments. Carcinogenesis 1991; 12: 325–327.
Anti M, Marra G, Rapaccini GL, Rumi C, Bussa S, Fadda G et al. DNA ploidy pattern in human chronic liver diseases and hepatic nodular lesions. Flow cytometric analysis on echo-guided needle liver biopsy. Cancer 1994; 73: 281–288.
Seglen PO . DNA ploidy and autophagic protein degradation as determinants of hepatocellular growth and survival. Cell Biol Toxicol 1997; 13: 301–315.
Tormos AM, Taléns-Visconti R, Sastre J . Regulation of cytokinesis and its clinical significance. Crit Rev Clin Lab Sci 2015; 52: 159–167.
Simerzin A, Zorde-Khvalevsky E, Rivkin M, Adar R, Zucman-Rossi J, Couchy G et al. The liver-specific microRNA-122*, the complementary strand of microRNA-122, acts as a tumor suppressor by modulating the p53/mouse double minute 2 homolog circuitry. Hepatology 2016; 64: 1623–1636.
Bandiera S, Pfeffer S, Baumert TF, Zeisel MB . miR-122—a key factor and therapeutic target in liver disease. J Hepatol 2015; 62: 448–457.
He J, Zhao K, Zheng L, Xu Z, Gong W, Chen S et al. Upregulation of microRNA-122 by farnesoid X receptor suppresses the growth of hepatocellular carcinoma cells. Mol Cancer 2015; 14: 163.
Dai D, Liang Y, Xie Z, Fu J, Zhang Y, Zhang Z . Survivin deficiency induces apoptosis and cell cycle arrest in HepG2 hepatocellular carcinoma cells. Oncol Rep 2012; 27: 621–627.
Aihara A, Tanaka S, Yasen M, Matsumura S, Mitsunori Y, Murakata A et al. The selective Aurora B kinase inhibitor AZD1152 as a novel treatment for hepatocellular carcinoma. J Hepatol 2010; 52: 63–71.
Hau PM, Siu WY, Wong N, Lai PB, Poon RY . Polyploidization increases the sensitivity to DNA-damaging agents in mammalian cells. FEBS Lett 2006; 580: 4727–4736.
Marxer M, Foucar CE, Man WY, Chen Y, Ma HT, Poon RY . Tetraploidization increases sensitivity to Aurora B kinase inhibition. Cell Cycle 2012; 11: 2567–2577.
Gentric G, Maillet V, Paradis V, Couton D, L’Hermitte A, Panasyuk G et al. Oxidative stress promotes pathologic polyploidization in nonalcoholic fatty liver disease. J Clin Invest 2015; 125: 981–992.
Hsu SH, Duncan AW . Pathological polyploidy in liver disease. Hepatology 2015; 62: 968–970.
Gentric G, Desdouets C . Liver polyploidy: Dr Jekyll or Mr Hide? Oncotarget 2015; 6: 8430–8431.
Schmucker DL . Hepatocyte fine structure during maturation and senescence. J Electron Microsc Tech 1990; 14: 106–125.
Asahina K, Shiokawa M, Ueki T, Yamasaki C, Aratani A, Tateno C et al. Multiplicative mononuclear small hepatocytes in adult rat liver: their isolation as a homogeneous population and localization to periportal zone. Biochem Biophys Res Commun 2006; 342: 1160–1167.
Lu P, Prost S, Caldwell H, Tugwood JD, Betton GR, Harrison DJ . Microarray analysis of gene expression of mouse hepatocytes of different ploidy. Mamm Genome 2007; 18: 617–626.
Anatskaya OV, Vinogradov AE . Genome multiplication as adaptation to tissue survival: evidence from gene expression in mammalian heart and liver. Genomics 2007; 89: 70–80.
Anatskaya OV, Vinogradov AE . Somatic polyploidy promotes cell function under stress and energy depletion: evidence fromtissue-specific mammal transcriptome. Funct Integr Genomics 2010; 10: 433–446.
Pandit SK, Westendorp B, de Bruin A . Physiological significance of polyploidization in mammalian cells. Trends Cell Biol 2013; 23: 556–566.
Acknowledgements
This work was funded by National Key Basic Research and Development Program of China (2010CB945600), National Natural Science Foundation of China (31601101), Shanghai Committee of Science and Technology of China (15JC1403900), Science and Research Fund of Shanghai Municipal Commission of Health and Family Planning (20164Y0013), Shanghai Sailing Program(17YF1424400). JTYL was supported by National Institutes of Health grant R01AI056082.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Agostini
Rights and permissions
Cell Death and Disease is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wang, MJ., Chen, F., Lau, J. et al. Hepatocyte polyploidization and its association with pathophysiological processes. Cell Death Dis 8, e2805 (2017). https://doi.org/10.1038/cddis.2017.167
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2017.167
This article is cited by
-
Polyploidy and mTOR signaling: a possible molecular link
Cell Communication and Signaling (2024)
-
Phenotypic characterization of liver tissue heterogeneity through a next-generation 3D single-cell atlas
Scientific Reports (2024)
-
A neuroprotective dose of trehalose is harmless to metabolic organs: comprehensive histopathological analysis of liver, pancreas, and kidney
DARU Journal of Pharmaceutical Sciences (2023)
-
Rapid single-cell physical phenotyping of mechanically dissociated tissue biopsies
Nature Biomedical Engineering (2023)
-
Hepatitis, testicular degeneration, and ataxia in DIDO3-deficient mice with altered mRNA processing
Cell & Bioscience (2022)
